A Comparison of the Maximum Entropy Principle Across Biological Spatial Scales


Abstract
:1. Introduction
2. Maximum Entropy Principle: Preliminaries and Fundamentals
2.1. Forward versus Inverse Problems
2.1.1. Forward Modeling
2.1.2. Inverse Modeling
2.2. Maximum Entropy Principle: Definitions and Methods
2.2.1. State Space, Observables, and Average Values
2.2.2. Entropy Maximization under Constraints
3. Examples at Different Spatial Scales
3.1. Amino Acid Interactions in Proteins
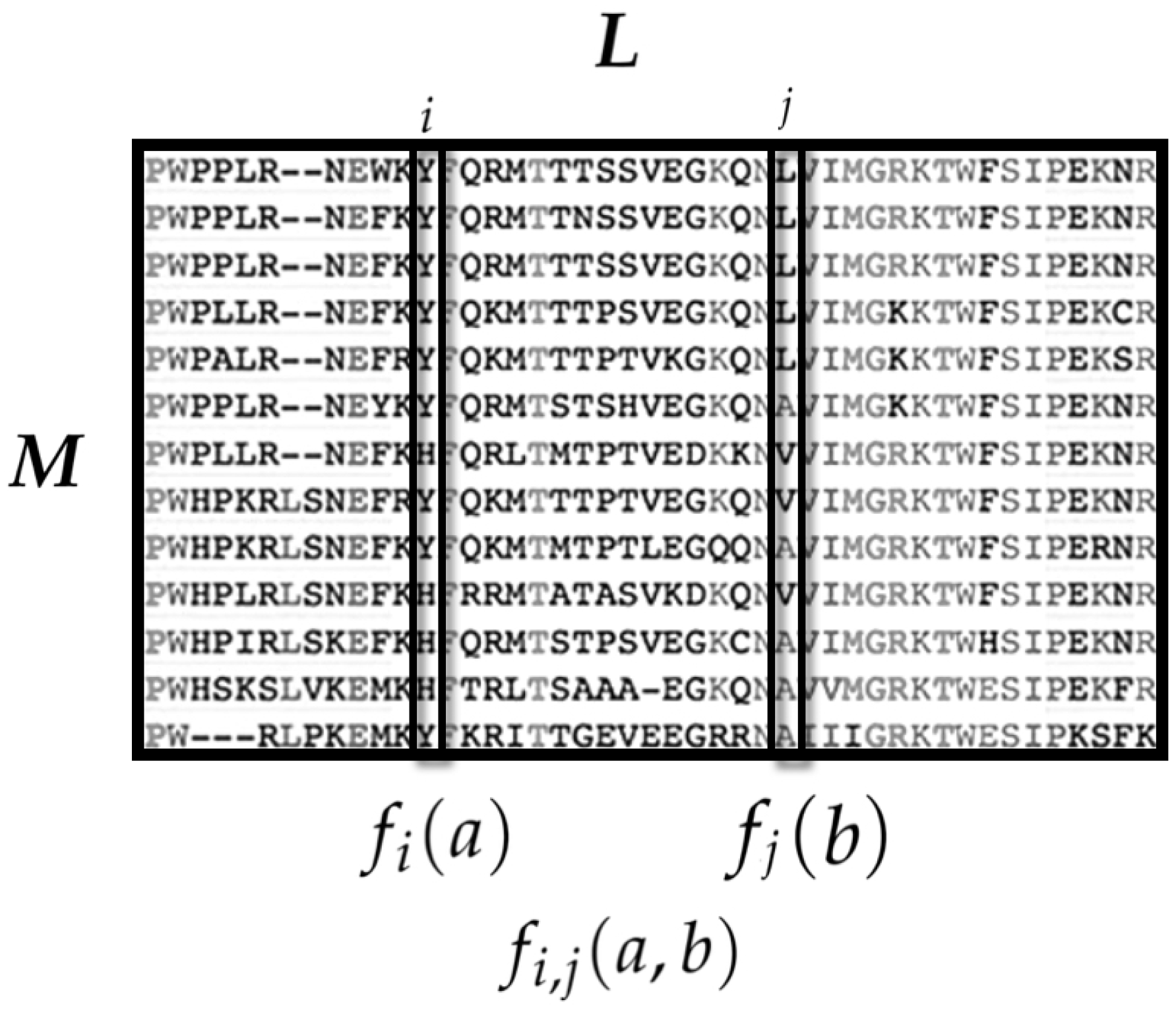
3.1.1. State Space
3.1.2. Observables and Average Values
- is the average occurrence of the amino acid a at the ith sequence site.
- is the average co-occurrence of the amino acids a at the ith site and b in the jth site.
3.1.3. Inferred Information
- Co-evolving site pairs: The interaction strength between site i and site j can be obtained as a function of the model parameters , i.e., the interaction between amino acid a in site i with the amino acid b in site j. This coupling strength can be used to identify evolutionary constraints on the site-interactions of the protein family.
- Contact Prediction: The protein tertiary structure is associated with a topology of contacts between far amino acids residues. Interestingly, this topology can be inferred from the coefficients. For predicting the tertiary structure of proteins, interactions between sites with a minimum separation of five sites on the linear sequence are usually studied—which is equivalent to one turn in an -helix. The MEP approach outperforms the pairwise site contact prediction given by correlation-based methods (e.g., mutual information) [25,26,42], illustrating the power of considering interactions instead of mere correlations.
- Protein Design and the Effect of Mutations: According to the energy landscape theory of protein folding [43], proteins conserved along evolution tend to minimize their free energy in their folded state. Using the MEP, the energy of each amino acid sequence can be computed, which allows to score each sequence according to its energy. This results in a set of non-naturally occurring proteins that minimizes the energy and, possibly, preserves the same functions as the original protein family. This inferred information has been applied to test and predict the effect of mutations [44,45].
3.2. Retina
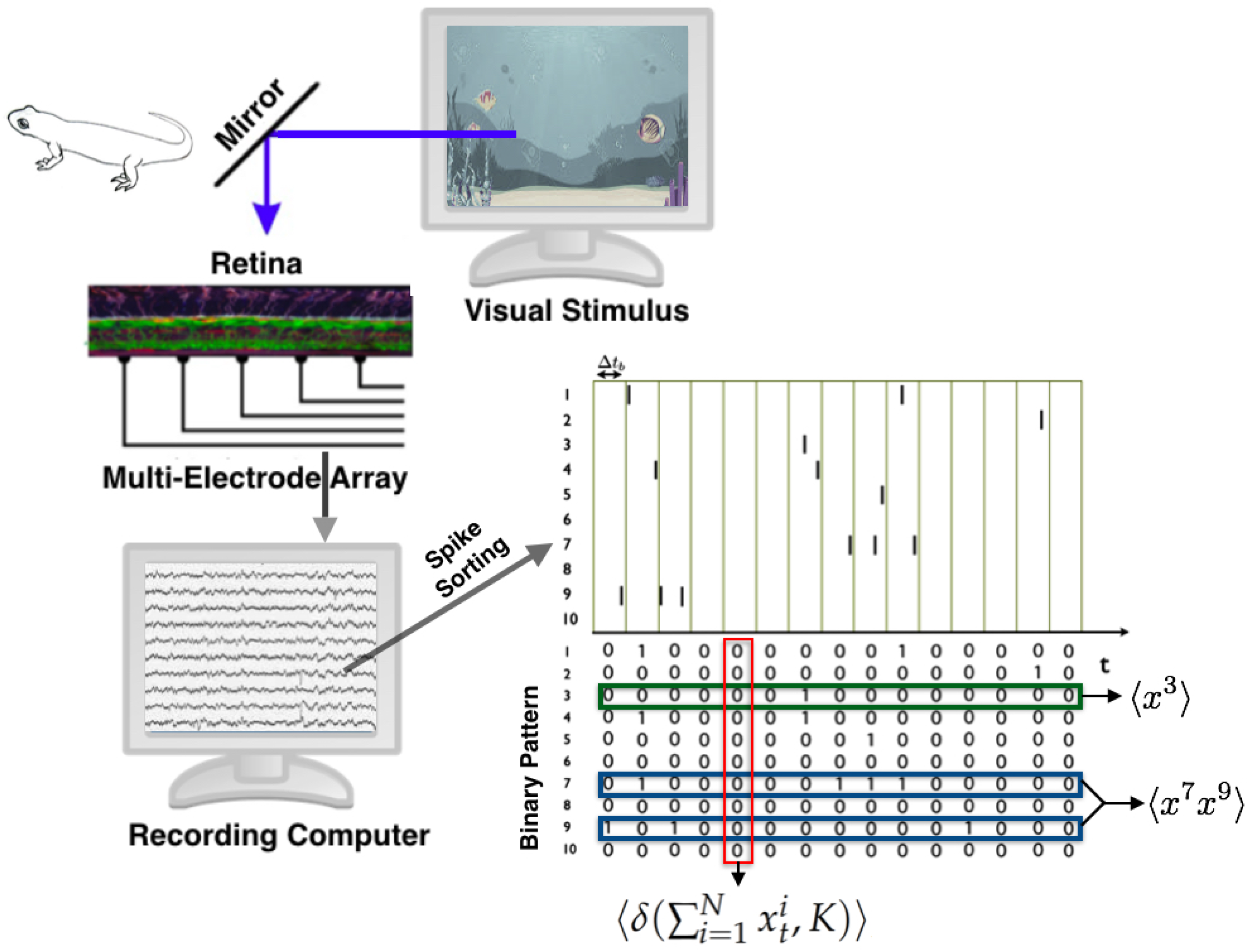
3.2.1. State Space
3.2.2. Observables and Average Values
- : The firing rate of neuron i, for all neurons.
- : The synchronous pairwise correlation between neuron i and neuron j, for all pairs of neurons.
- , for .
3.2.3. Inferred Information
- Joint Shannon entropy: To characterize the size of the neural vocabulary, the effective number of configurations is reduced to (1). The entropy represents the ability of the system to explore these available states, and hence assesses the capacity of the neural population to represent visual information. In this case, a low entropy shown that the expected frequency of spike patterns are extremely inhomogeneous.
- Classification of activity patterns into meta-stable collective modes: The energy landscape inferred from the maximum entropy method presents valleys, which resembles a “clustering of patterns” of neural activity, but obtained without a particular metric for similarity among patterns.
- Redundancy: From the inferred joint distribution , the authors computed the conditional marginal distributions , where means all except i. They showed that the state of individual neurons is highly predictable from the rest of the population, characterizing in this way the level of redundancy in the neural population. This property is suggested to allow error correction capabilities.
3.3. Resting State Networks in the Human Brain
3.3.1. State Space
3.3.2. Observables and Average Values
- : The activation rate of region i, 12 for the DMN and 11 for the FPN.
- : The synchronous pairwise correlation between region i and region j, for all pairs of regions of the DMN and FPN.
3.3.3. Inferred Information
3.4. Plant Communities Relative Abundances
3.4.1. State Space
3.4.2. Observables
3.4.3. Inferred Information
3.5. Macroecology and Biodiversity
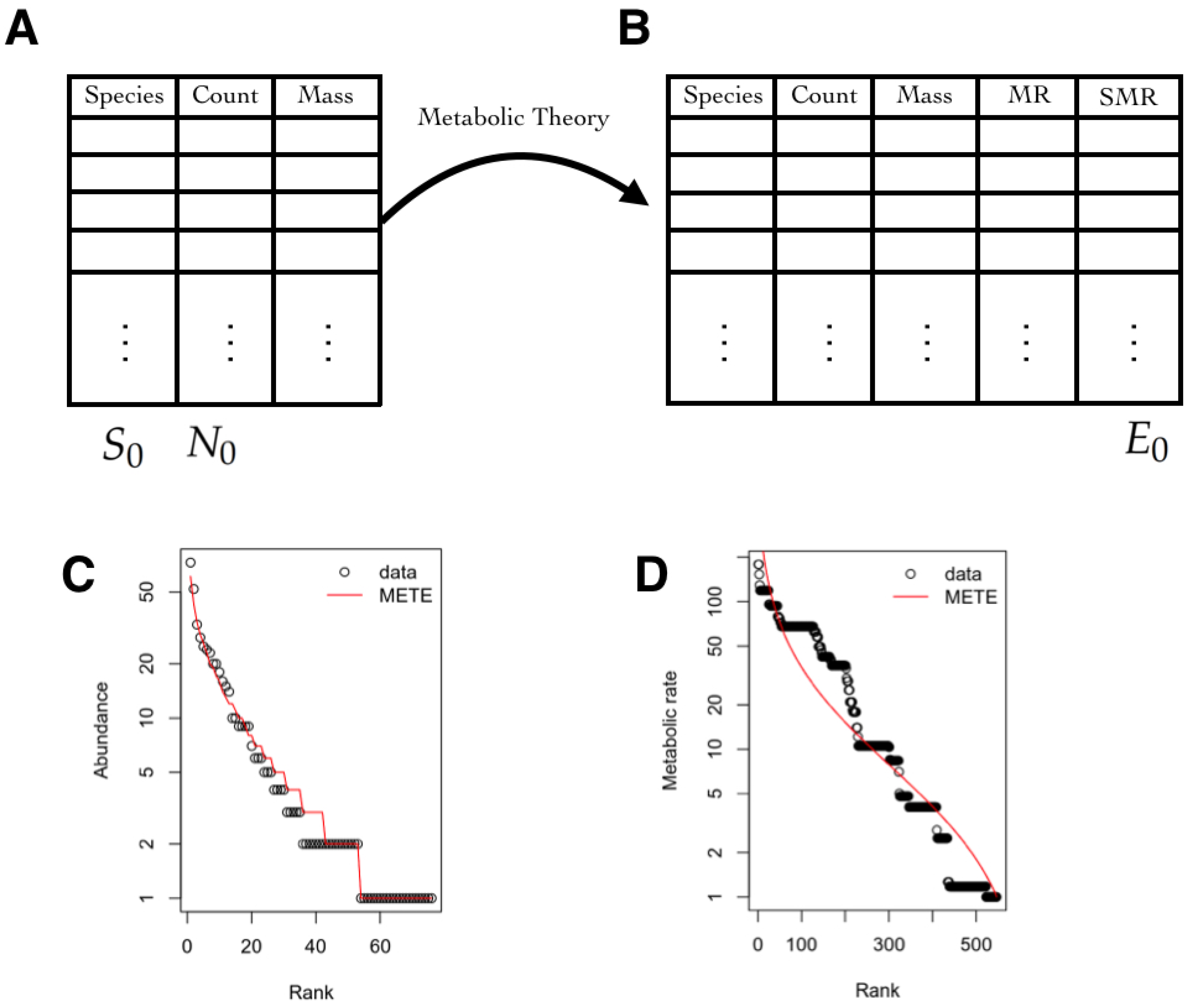
3.5.1. State Space
3.5.2. Observables
3.5.3. Inferred Information
3.6. Human Voting Interactions in the US Supreme Court
3.6.1. State Space
3.6.2. Observables
3.6.3. Inferred Information
4. Discussion
4.1. Lessons from the Case Studies
- (i)
- The MEP can be applied to a wide range of systems. The flexibility of the MEP allows its application to biological systems. Moreover, the range of application not only spans spatial scales, but also includes technically diverse scenarios. In effect, in some of the considered case studies, the observables are directly related to causal/mechanistic interactions, while in others they are not. Moreover, the averages of these observables are in some cases temporal, while in other cases spatial. The fact that the same formalism can be adapted to such different contexts highlights the flexibility of the MEP approach [19].
- (ii)
- It is critical (and highly non-trivial) to choose an appropriate state space and observables. While different applications of the MEP do not require conceptual changes to the basic method, the results rely entirely on the chosen state variables and observables, which are both determined by the modeler. For this reason, the researcher needs to double-check that these choices are adequate, i.e., if the model is capable of predicting (with some degree of accuracy) average values of observables not included in the MEP to fit the data, and if the model is capable of addressing the questions that one want to ask. It is crucial not to lose perspective on this issue; as the MEP is based on a concave maximization problem, it will always finds a unique solution, which might be useless if the state space and observables are not chosen appropriately.
- (iii)
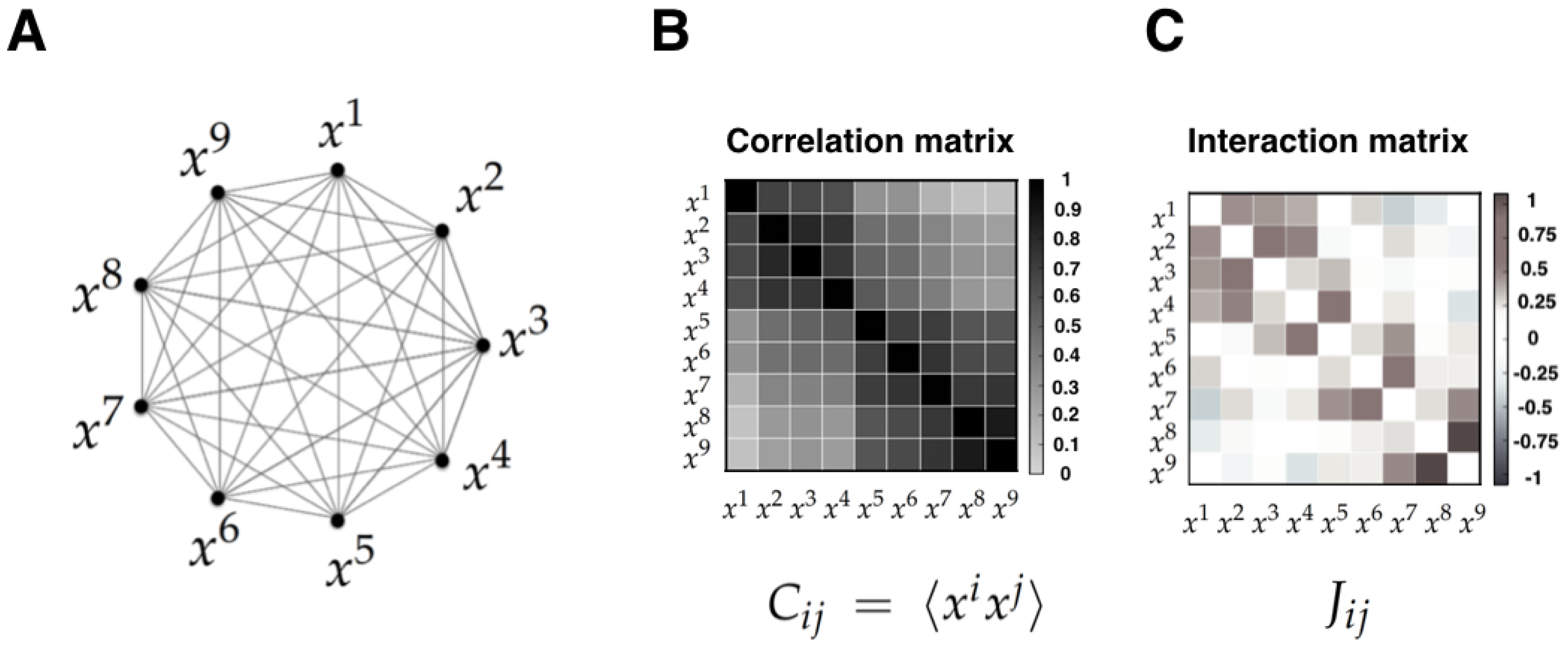
- Correlations versus interactions. It is important to note that the MEP makes a strong distinction between interactions and correlations. Indeed, correlations are statistical dependencies between variables, while interactions are the local rules of the system from which correlations and collective phenomena emerge. Importantly, each interaction term depends on all the correlation terms, and hence there exists no simple mapping between the correlation and the interaction between two sub-units. Furthermore, it has been shown that the interactions inferred using the MEP give a more useful account of the physical topology of some system than correlations. Examples of this include neural structural connectivity [51,80], and contacts between proteins sites [42]. In these examples, the inferred interactions obtained from the MEM parameters outperform linear or nonlinear correlation when predicting physical interactions between system variables. We believe that the key advantage of interactions over correlations is the fact that they faithfully reflect conditions of conditional independency, which are key in many statistical causal frameworks [81]. This crucial property might be behind the success of MEM in assessing emerging behavior in networks of interacting agents in biology [4,21,82,83,84].
4.2. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MEP | Maximum entropy principle |
| MEM | Maximum entropy model |
| DNA | Deoxyribonucleic Acid |
| MSA | Multiple sequence alignment |
| LFP | Local field potential |
| fMRI | Functional magnetic resonance imaging |
| BOLD | Blood Oxygen Level-Dependent signals |
| RSN | Resting state network |
| DMN | Default mode network |
| FPN | Fronto-parietal network |
| METE | Maxent Theory of Ecology |
| SCOTUS | Supreme Court of the United States |
| BIEN | Botanical Information and Ecology Network |
| Symbol List | |
| State of kth variable at time t | |
| State of ith variable on x | |
| Configuration of the N-elements of a system | |
| State space of a random variable | |
| State space of a N-elements system | |
| Set of real numbers | |
| Set of probability distributions that match | |
| Probability distribution that maximizes entropy and match | |
| Entropy of the probability measure p | |
| Number of observables | |
| Observable k | |
| Empirical average value of observable k | |
| Expected value of with respect to q | |
| Set of probability measures | |
| Lagrange multiplier associated to the constraint | |
| Energy function | |
| Z | Partition Function |
References
- Waldrop, M.M. Complexity: The Emerging Science at the Edge of Order and Chaos; Simon and Schuster: New York, NY, USA, 1993. [Google Scholar]
- Stein, R.B. More Is Different. Science 1972, 177, 393–396. [Google Scholar]
- Rosas, F.; Mediano, P.A.; Gastpar, M.; Jensen, H.J. Quantifying high-order interdependencies via multivariate extensions of the mutual information. Phys. Rev. E. 2019, 100, 032305. [Google Scholar] [CrossRef] [Green Version]
- Tkačik, G.; Marre, O.; Amodei, D.; Schneidman, E.; Bialek, W.; Berry, M.J. Searching for collective behavior in a large network of sensory neurons. PLoS Comput. Biol. 2014, 10, e1003408. [Google Scholar] [CrossRef] [PubMed]
- Nasser, H.; Cessac, B. Parameter Estimation for Spatio-Temporal Maximum Entropy Distributions: Application to Neural Spike Trains. Entropy 2014, 16, 2244–2277. [Google Scholar] [CrossRef] [Green Version]
- Jaynes, E. Information theory and statistical mechanics. Phys. Rev. 1957, 106. [Google Scholar] [CrossRef]
- Santolini, M.; Mora, T.; Hakim, V. A General Pairwise Interaction Model Provides an Accurate Description of In Vivo Transcription Factor Binding Sites. PLoS ONE 2014, 9, e99015. [Google Scholar] [CrossRef] [PubMed]
- Weigt, M.; White, R.A.; Szurmant, H.; Hoch, J.A.; Hwa, T. Identification of direct residue contacts in protein–protein interaction by message passing. Proc. Natl. Acad. Sci. USA 2009, 106, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301. [Google Scholar] [CrossRef]
- Barton, J.; Chakraborty, A.K.; Cocco, S.; Jacquin, H.; Monasson, R. On the Entropy of Protein Families. J. Stat. Phys. 2015, 162. [Google Scholar] [CrossRef]
- Mora, T.; Walczak, A.M.; Bialek, W.; Callan, C.G. Maximum entropy models for antibody diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 5405–5410. [Google Scholar] [CrossRef] [Green Version]
- Elhanati, Y.; Murugan, A.; Callan, C.G., Jr.; Mora, T.; Walczak, A.M. Quantifying selection in immune receptor repertoires. Proc. Natl. Acad. Sci. USA 2014, 111, 9875–9880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidman, E.; Berry, M.; Segev, R.; Bialek, W. Weak pairwise correlations imply string correlated network states in a neural population. Nature 2006, 440, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Jackson, D.; Hobbs, J.; Chen, W.; Smith, J.; Patel, H.; Prieto, A.; Petrusca, D.; Grivich, M.; Sher, A.; et al. A Maximum Entropy Model Applied to Spatial and Temporal Correlations from Cortical Networks. In Vitro J. Neurosci. 2008, 28, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Tkačik, G.; Mora, T.; Marre, O.; Amodei, D.; Berry, M., II; Bialek, W. Thermodynamics for a network of neurons: Signatures of criticality. Proc. Natl. Acad. Sci. USA 2015, 112, 11508–11513. [Google Scholar] [CrossRef] [PubMed]
- Marre, O.; El Boustani, S.; Frégnac, Y.; Destexhe, A. Prediction of spatiotemporal patterns of neural activity from pairwise correlations. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef] [PubMed]
- Cofré, R.; Cessac, B. Exact computation of the maximum entropy potential of spiking neural networks models. Phys. Rev. 2014, 107, 368. [Google Scholar] [CrossRef]
- Cofré, R.; Maldonado, C. Information Entropy Production of Maximum Entropy Markov Chains from Spike Trains. Entropy 2018, 20, 34. [Google Scholar] [CrossRef]
- Cofré, R.; Videla, L.; Rosas, F. An Introduction to the Non-Equilibrium Steady States of Maximum Entropy Spike Trains. Entropy 2019, 21, 884. [Google Scholar] [CrossRef]
- Bialek, W.; Cavagna, A.; Giardina, I.; Mora, T.; Silvestri, E.; Viale, M.; M Walczak, A. Statistical mechanics for natural flocks of birds. Proc. Natl. Acad. Sci. USA 2012, 109, 4786–4791. [Google Scholar] [CrossRef] [Green Version]
- Cavagna, A.; Giardina, I.; Ginelli, F.; Mora, T.; Piovani, D.; Tavarone, R.; M Walczak, A. Dynamical maximum entropy approach to flocking. Phys. Rev. Stat. Nonlinear Soft Matter Phys. 2014, 89, 042707. [Google Scholar] [CrossRef] [Green Version]
- Shemesh, Y.; Sztainberg, Y.; Forkosh, O.; Shlapobersky, T.; Chen, A.; Schneidman, E. High-order social interactions in groups of mice. eLife 2013, 2, e00759. [Google Scholar] [CrossRef] [PubMed]
- Harte, J. Maximum Entropy and Ecology. A Theory of Abundance, Distribution, and Energetics; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Harte, J.; Newman, E. Maximum information entropy: A foundation for ecological theory. Trends Ecol. Evol. 2014, 29, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Marks, D.; Sander, C. Inferring Pairwise Interactions from Biological Data Using Maximum-Entropy Probability Models. PLoS Comput. Biol. 2015, 11, e1004182. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.C.; Zecchina, R.; Berg, J. Inverse statistical problems: From the inverse Ising problem to data science. Adv. Phys. 2017, 66, 197–261. [Google Scholar] [CrossRef]
- De Martino, A.; Martino, D. An introduction to the maximum entropy approach and its application to inference problems in biology. Heliyon 2018, 4, e00596. [Google Scholar] [CrossRef] [Green Version]
- Natale, J.L.; Hofmann, D.; Hernández, D.G.; Nemenman, I. Reverse-engineering biological networks from large data sets. arXiv 2017, arXiv:1705.06370. [Google Scholar]
- Battistin, C.; Dunn, B.; Roudi, Y. Learning with unknowns: Analyzing biological data in the presence of hidden variables. Curr. Opin. Syst. Biol. 2017, 1, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Tkačik, G. From statistical mechanics to information theory: Understanding biophysical information processing systems. arXiv 2010, arXiv:1006.4291. [Google Scholar]
- Marquet, P.A.; Allen, A.P.; Brown, J.H.; Dunne, J.A.; Enquist, B.J.; Gillooly, J.F.; Gowaty, P.A.; Green, J.L.; Harte, J.; Hubbell, S.P.; et al. On theory in ecology. BioScience 2014, 64, 701–710. [Google Scholar] [CrossRef]
- Cessac, B.; Kornprobst, P.; Kraria, S.; Nasser, H.; Pamplona, D.; Portelli, G.; Vieville, T. PRANAS: A New Platform for Retinal Analysis and Simulation. Front. Neuroinform. 2017, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Kazama, J.; Tsujii, J. Maximum Entropy Models with Inequality Constraints: A Case Study on Text Categorization. Mach. Learn. 2005, 60, 159–194. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory, 2nd ed.; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Bowen, R. Equilibrium States and the Ergodic Theory of Anosov Diffeomorphisms; Lecture Notes in Mathematics; Springer: Berlin/Heidelberg, Germany, 2008; Volume 470. [Google Scholar]
- Jebara, T. Machine Learning: Discriminative and Generative; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Soding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Katta, H.Y.; Mojica, A.; Chen, I.M.A.; Kyrpides, N.C.; Reddy, T. Genomes OnLine database (GOLD) v.7: Updates and new features. Nucleic Acids Res. 2018, 47, D649–D659. [Google Scholar] [CrossRef]
- Cocco, S.; Feinauer, C.; Figliuzzi, M.; Monasson, R.; Weigt, M. Inverse Statistical Physics of Protein Sequences: A Key Issues Review. Rep. Prog. Phys. 2017, 81. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Wolynes, P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004, 14, 70–75. [Google Scholar] [CrossRef]
- Cheng, R.R.; Nordesjö, O.; Hayes, R.L.; Levine, H.; Flores, S.C.; Onuchic, J.N.; Morcos, F. Connecting the Sequence-Space of Bacterial Signaling Proteins to Phenotypes Using Coevolutionary Landscapes. Mol. Biol. Evol. 2016, 33, 3054–3064. [Google Scholar] [CrossRef] [Green Version]
- Hopf, T.; B Ingraham, J.; Poelwijk, F.; P I Schärfe, C.; Springer, M.; Sander, C.; Marks, D. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 2017, 35, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Pillow, J.W.; Shlens, J.; Paninski, L.; Sher, A.; Litke, A.M.; Chichilnisky, E.J.; Simoncelli, E.P. Spatio-temporal correlations and visual signaling in a complete neuronal population. Nature 2008, 454, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Ganmor, E.; Segev, R.; Schneidman, E. Sparse low-order interaction network underlies a highly correlated and learnable neural population code. Proc. Natl. Acad. Sci. USA 2011, 108, 9679–9684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, J.; Palacios, A.; Marre, O.; Berry II, M.; Cessac, B. Gibbs distribution analysis of temporal correlation structure on multicell spike trains from retina ganglion cells. J. Physiol. Paris 2012, 106, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Huang, D.; Singer, W.; Nikolic, D. A Small World of Neuronal Synchrony. Cereb. Cortex 2008, 18, 2891–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohiorhenuan, I.E.; Mechler, F.; Purpura, K.P.; Schmid, A.M.; Hu, Q.; Victor, J.D. Sparse coding and high-order correlations in fine-scale cortical networks. Nature 2010, 466, 617–621. [Google Scholar] [CrossRef]
- Watanabe, T.; Hirose, S.; Wada, H.; Imai, Y.; Machida, T.; Shirouzu, I.; Konishi, S.; Miyashita, Y.; Masuda, N. A pairwise maximum entropy model accurately describes resting-state human brain networks. Nat. Commun. 2013, 4, 1370. [Google Scholar] [CrossRef] [Green Version]
- Buckner, R.; Andrews-Hanna, J.; Schacter, D. The brain’s default network: Anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 2008, 1124, 1–38. [Google Scholar] [CrossRef]
- MacArthur, R.H. Geographical Ecology: Patterns in the Distribution of Species; Princeton University Press: Princeton, NJ, USA, 1984. [Google Scholar]
- Verberk, W. Explaining general patterns in species abundance and distributions. Nat. Educ. Knowl. 2011, 3, 38. [Google Scholar]
- Hubbell, S. The Unified Neutral Theory of Biodiversity and Biogeography; Princeton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Volkov, I.; Banavar, J.R.; Hubbell, S.P.; Maritan, A. Neutral theory and relative species abundance in ecology. Nature 2003, 424, 1035. [Google Scholar] [CrossRef]
- Shipley, B.; Vile, D.; Garnier, É. From plant traits to plant communities: A statistical mechanistic approach to biodiversity. Science 2006, 314, 812–814. [Google Scholar] [CrossRef]
- Sonnier, G.; Shipley, B.; Navas, M.L. Plant traits, species pools and the prediction of relative abundance in plant communities: A maximum entropy approach. J. Veg. Sci. 2010, 21, 318–331. [Google Scholar] [CrossRef]
- Kattge, J.; Diaz, S.; Lavorel, S.; Prentice, I.C.; Leadley, P.; Bönisch, G.; Garnier, E.; Westoby, M.; Reich, P.B.; Wright, I.J.; et al. TRY–a global database of plant traits. Glob. Chang. Biol. 2011, 17, 2905–2935. [Google Scholar] [CrossRef]
- Violle, C.; Navas, M.L.; Vile, D.; Kazakou, E.; Fortunel, C.; Hummel, I.; Garnier, E. Let the concept of trait be functional! Oikos 2007, 116, 882–892. [Google Scholar] [CrossRef]
- Maitner, B.S.; Boyle, B.; Casler, N.; Condit, R.; Donoghue, J.; Durán, S.M.; Guaderrama, D.; Hinchliff, C.E.; Jørgensen, P.M.; Kraft, N.J.; et al. The bien r package: A tool to access the Botanical Information and Ecology Network (BIEN) database. Methods Ecol. Evol. 2018, 9, 373–379. [Google Scholar] [CrossRef]
- Ward, D.F. Modelling the potential geographic distribution of invasive ant species in New Zealand. Biol. Invas. 2007, 9, 723–735. [Google Scholar] [CrossRef]
- Brown, J.H. Macroecology; University of Chicago Press: Chicago, IL, USA, 1995. [Google Scholar]
- Kolokotrones, T.; Savage, V.; Deeds, E.J.; Fontana, W. Curvature in metabolic scaling. Nature 2010, 464, 753. [Google Scholar] [CrossRef]
- McGill, B. Towards a unification of unified theories of biodiversity. Ecol. Lett. 2010, 13, 627–642. [Google Scholar] [CrossRef]
- Harte, J.; Zillio, T.; Conlisk, E.; Smith, A. Maximum entropy and the state-variable approach to macroecology. Ecology 2008, 89, 2700–2711. [Google Scholar] [CrossRef]
- Harte, J.; Smith, A.; Storch, D. Biodiversity scales from plots to biomes with a universal species–area curve. Ecol. Lett. 2009, 12, 789–797. [Google Scholar] [CrossRef]
- Favretti, M. Remarks on the Maximum Entropy principle with Application to the Maximum Entropy Theory of Ecology. Entropy 2018, 20, 11. [Google Scholar] [CrossRef]
- Harte, J. Maximum Entropy and Theory Construction: A Reply to Favretti. Entropy 2018, 20, 285. [Google Scholar] [CrossRef]
- Favretti, M. Maximum Entropy Theory of Ecology: A Reply to Harte. Entropy 2018, 20, 308. [Google Scholar] [CrossRef]
- Brummer, A.B.; Newman, E.A. Derivations of the Core Functions of the Maximum Entropy Theory of Ecology. Entropy 2019, 21, 712. [Google Scholar] [CrossRef]
- Bertram, J.; Newman, E.; Dewar, R. Comparison of two maximum entropy models highlights the metabolic structure of metacommunities as a key determinant of local community assembly. Ecol. Model. 2019, 407. [Google Scholar] [CrossRef]
- Xiao, X.; McGlinn, D.J.; White, E.P. A strong test of the maximum entropy theory of ecology. Am. Nat. 2015, 185, E70–E80. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.; Harte, M.; Lowell, N.; Wilber, M.; Harte, J. Empirical tests of within-and across-species energetics in a diverse plant community. Ecology 2014, 95, 2815–2825. [Google Scholar] [CrossRef]
- Rominger, A.; Merow, C. meteR: An R package for testing the Maximum Entropy Theory of Ecology. Methods Ecol. Evol. 2017, 8, 241–247. [Google Scholar] [CrossRef]
- Gruner, D. Geological age ecosystem development and local resource constraints on arthropod community structure in the Hawaiian Islands. Biol. J. Linn. Soc. 2007, 90, 551–570. [Google Scholar] [CrossRef]
- Silverberg, J.; Bierbaum, M.; Sethna, J.; Cohen, I. Collective Motion of Humans in Mosh and Circle Pits at Heavy Metal Concerts. Phys. Rev. Lett. 2013, 110. [Google Scholar] [CrossRef]
- Duh, A.; Rupnik, M.S.; Korošak, D. Collective behavior of social bots is encoded in their temporal twitter activity. Big Data 2018, 6. [Google Scholar] [CrossRef]
- Lee, E.D.; Broedersz, C.P.; Bialek, W. Statistical Mechanics of the US Supreme Court. J. Stat. Phys. 2015, 160, 275–301. [Google Scholar] [CrossRef] [Green Version]
- Kadirvelu, B.; Hayashi, Y.; Nasuto, S. Inferring structural connectivity using Ising couplings in models of neuronal networks. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Bressler, S.L.; Seth, A.K. Wiener–Granger causality: A well established methodology. Neuroimage 2011, 58, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Mora, T.; Bialek, W. Are biological systems poised at criticality? J. Stat. Phys. 2011, 144, 268–302. [Google Scholar] [CrossRef]
- Bialek, W. Biophysics Searching for Principles; Princeton University Press: Princeton, NJ, USA, 2012. [Google Scholar]
- Tkačik, G.; Marre, O.; Mora, T.; Amodei, D.; Berry, M.J.; Bialek, W. The simplest maximum entropy model for collective behavior in a neural network. J. Stat. Mech. Theory Exp. 2013, 2013, P03011. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenario | State Space | Observables and Average values |
|---|---|---|
| Amino acids in proteins | Average amino acid occurrence on a given site | |
| and average co-occurrences of amino acids on site-pairs | ||
| Retinal ganglion cells | Firing rates and pairwise correlations | |
| Whole brain networks | Activation rates and pairwise correlations | |
| Plant communities | Average value of traits | |
| Macroecologic biodiversity | Average abundance per species and average over species | |
| of the total metabolic rate of the individuals within the species. | ||
| US Supreme Court | Pairwise correlations |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cofré, R.; Herzog, R.; Corcoran, D.; Rosas, F.E. A Comparison of the Maximum Entropy Principle Across Biological Spatial Scales. Entropy 2019, 21, 1009. https://doi.org/10.3390/e21101009
Cofré R, Herzog R, Corcoran D, Rosas FE. A Comparison of the Maximum Entropy Principle Across Biological Spatial Scales. Entropy. 2019; 21(10):1009. https://doi.org/10.3390/e21101009
Chicago/Turabian StyleCofré, Rodrigo, Rubén Herzog, Derek Corcoran, and Fernando E. Rosas. 2019. "A Comparison of the Maximum Entropy Principle Across Biological Spatial Scales" Entropy 21, no. 10: 1009. https://doi.org/10.3390/e21101009