DNA Methylation Dynamics in Health and Disease
(Closed)
Share This Topical Collection
Editors
 Prof. Dr. Lorenzo Chiariotti
Prof. Dr. Lorenzo Chiariotti
Prof. Dr. Lorenzo Chiariotti
E-Mail
Website
Collection Editor
Department of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples “Federico II”, 80131 Naples, Italy
Interests: tumor epigenetics; brain tumors; molecular biology
Special Issues, Collections and Topics in MDPI journals
 Dr. Mariella Cuomo
Dr. Mariella Cuomo
Dr. Mariella Cuomo
E-Mail
Collection Editor
Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università di Napoli "Federico II", via S. Pansini, 5, 80131 Naples, Italy
Topical Collection Information
Dear Colleagues,
Epigenetic processes are the main mechanisms that cells use to regulate spatio-temporal expression patterns during development. Among epigenetic machineries, DNA methylation has attracted a great deal of interest in the scientific community over the years. Despite its high stability in the genome, it has been widely demonstrated that the addition and removal of a methyl group to a CG doublet follows specific trajectories that lead the cells to acquire, maintain, and read their genome messages in a plastic manner. In this scenario, DNA methylation assumes a key role in the underlying developmental or pathological processes. During mammalian development, methylation and demethylation events occur, and specific DNA methylation profiles are sculpted for each somatic cell. In this way, DNA methylation decides how and where genes turn on or off in each cell type. Thus, an alteration in the DNA methylation picture during development may lead to several disorders, even later in life. Therefore, many efforts have been made to disentangle the active variations of the DNA methylation landscape and to correlate them to physiological and pathological processes. Moreover, the recent and extraordinary advances in single-cell technologies and ultra-deep sequencing have made it possible to depict the DNA methylation panorama at an unprecedented resolution and with extremely high accuracy. New insight in cell-to-cell DNA methylation heterogeneity, together with recent findings on different kinds of cytosine modifications (hydroxymethylation, etc.) and the emerging dynamic nature of DNA methylation and demethylation events, open novel unexpected roads for DNA methylation studies—especially in discerning stochastic from well-orchestrated epigenetic events.
This Collection will, therefore, focus on the dynamics of DNA methylation in biological processes, such as imprinting, X-inactivation, and DNA methylation changes during development and stem cell differentiation. It will also discuss the correlation between gene-specific and genome-wide DNA methylation changes in diseases such as cancer, genetic diseases, and neuropsychiatric diseases. Finally, it will give attention to the genomic distribution of DNA methylation at high resolution in various organisms, cell types, and diseases.
Prof. Dr. Lorenzo Chiariotti
Dr. Mariella Cuomo
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- DNA methylation
- cytosine hydroxymethylation in heath and disease
- non-CpG methylation
- epigenetics
- DNMTs
- demethylation
- DNA methylation dynamics
- epialleles
- epipolymorphisms
- epimutations
- TETs enzymes
- cell-to-cell methylation heterogeneity
Published Papers (8 papers)
Open AccessArticle
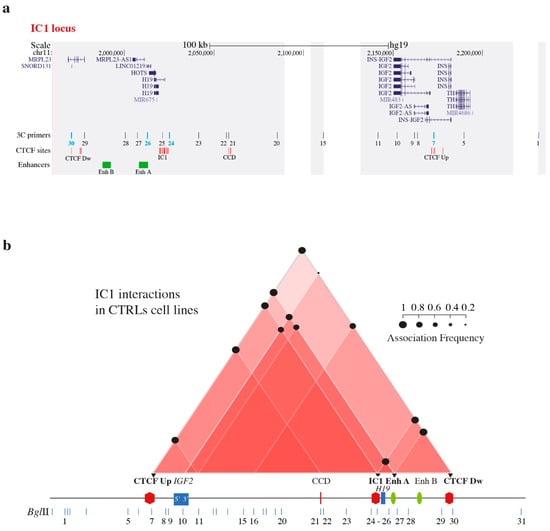
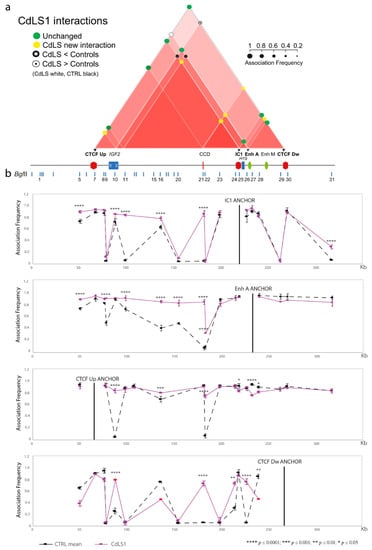
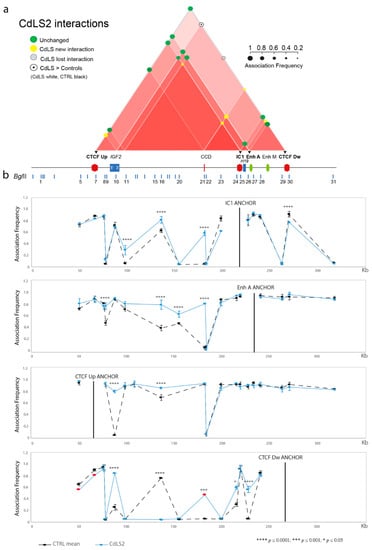
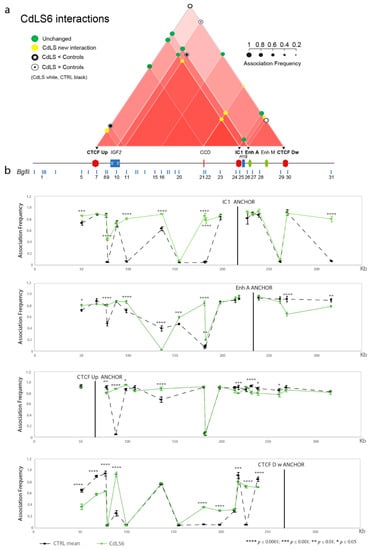
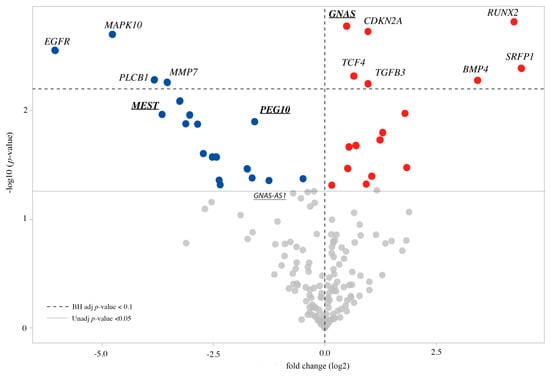
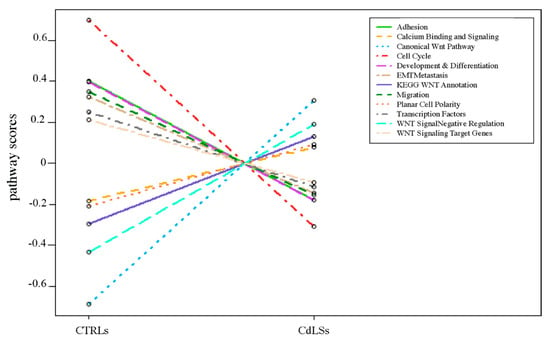
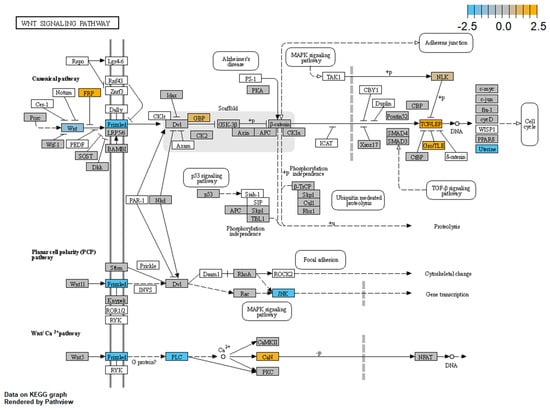
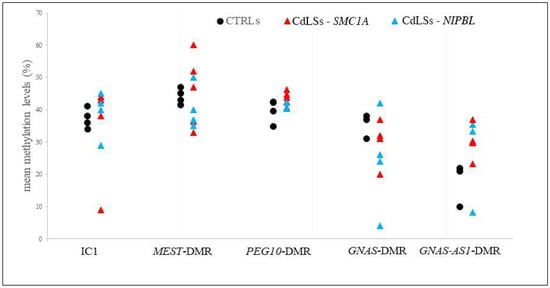
Cohesin Mutations Induce Chromatin Conformation Perturbation of the H19/IGF2 Imprinted Region and Gene Expression Dysregulation in Cornelia de Lange Syndrome Cell Lines
by
Silvana Pileggi, Marta La Vecchia, Elisa Adele Colombo, Laura Fontana, Patrizia Colapietro, Davide Rovina, Annamaria Morotti, Silvia Tabano, Giovanni Porta, Myriam Alcalay, Cristina Gervasini, Monica Miozzo and Silvia Maria Sirchia
Cited by 2 | Viewed by 1895
Abstract
Traditionally, Cornelia de Lange Syndrome (CdLS) is considered a cohesinopathy caused by constitutive mutations in cohesin complex genes. Cohesin is a major regulator of chromatin architecture, including the formation of chromatin loops at the imprinted
IGF2/
H19 domain. We used 3C analysis
[...] Read more.
Traditionally, Cornelia de Lange Syndrome (CdLS) is considered a cohesinopathy caused by constitutive mutations in cohesin complex genes. Cohesin is a major regulator of chromatin architecture, including the formation of chromatin loops at the imprinted
IGF2/
H19 domain. We used 3C analysis on lymphoblastoid cells from CdLS patients carrying mutations in
NIPBL and
SMC1A genes to explore 3D chromatin structure of the
IGF2/
H19 locus and evaluate the influence of cohesin alterations in chromatin architecture. We also assessed quantitative expression of imprinted
loci and WNT pathway genes, together with DMR methylation status of the imprinted genes. A general impairment of chromatin architecture and the emergence of new interactions were found. Moreover, imprinting alterations also involved the expression and methylation levels of imprinted genes, suggesting an association among cohesin genetic defects, chromatin architecture impairment, and imprinting network alteration. The WNT pathway resulted dysregulated: canonical WNT, cell cycle, and WNT signal negative regulation were the most significantly affected subpathways. Among the deregulated pathway nodes, the key node of the frizzled receptors was repressed. Our study provides new evidence that mutations in genes of the cohesin complex have effects on the chromatin architecture and epigenetic stability of genes commonly regulated by high order chromatin structure.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
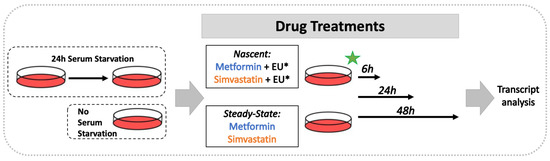
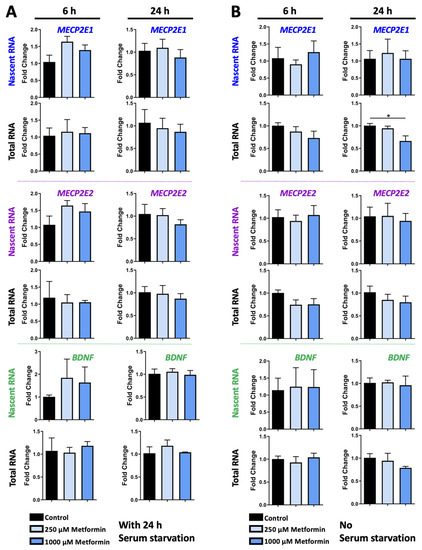
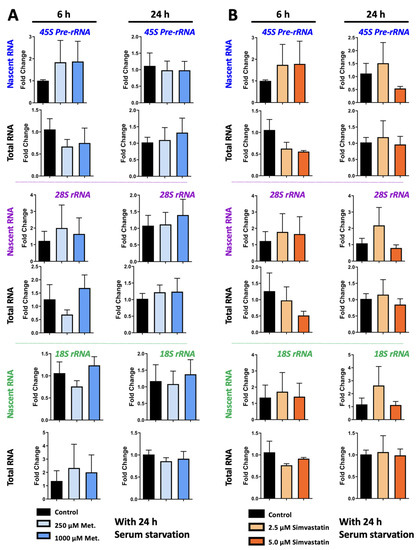
Transcriptional Regulation of MECP2E1-E2 Isoforms and BDNF by Metformin and Simvastatin through Analyzing Nascent RNA Synthesis in a Human Brain Cell Line
by
Marjorie Buist, David Fuss and Mojgan Rastegar
Cited by 11 | Viewed by 3107
Abstract
Methyl CpG binding protein 2 (MeCP2) is the main DNA methyl-binding protein in the brain that binds to 5-methylcytosine and 5-hydroxymethyl cytosine.
MECP2 gene mutations are the main origin of Rett Syndrome (RTT), a neurodevelopmental disorder in young females. The disease has no
[...] Read more.
Methyl CpG binding protein 2 (MeCP2) is the main DNA methyl-binding protein in the brain that binds to 5-methylcytosine and 5-hydroxymethyl cytosine.
MECP2 gene mutations are the main origin of Rett Syndrome (RTT), a neurodevelopmental disorder in young females. The disease has no existing cure, however, metabolic drugs such as metformin and statins have recently emerged as potential therapeutic candidates. In addition, induced
MECP2-BDNF homeostasis regulation has been suggested as a therapy avenue. Here, we analyzed nascent RNA synthesis versus steady state total cellular RNA to study the transcriptional effects of metformin (an anti-diabetic drug) on
MECP2 isoforms (
E1 and
E2) and
BNDF in a human brain cell line. Additionally, we investigated the impact of simvastatin (a cholesterol lowering drug) on transcriptional regulation of
MECP2E1/E2-BDNF. Metformin was capable of post-transcriptionally inducing
BDNF and/or
MECP2E1, while transcriptionally inhibiting
MECP2E2. In contrast simvastatin significantly inhibited
BDNF transcription without significantly impacting
MECP2E2 transcripts. Further analysis of ribosomal
RNA transcripts confirmed that the drug neither individually nor in combination affected these fundamentally important transcripts. Experimental analysis was completed in conditions of the presence or absence of serum starvation that showed minimal impact for serum deprival, although significant inhibition of steady state
MECP2E1 by simvastatin was only detected in non-serum starved cells. Taken together, our results suggest that metformin controls
MECP2E1/E2-BDNF transcriptionally and/or post-transcriptionally, and that simvastatin is a potent transcriptional inhibitor of
BDNF. The transcriptional effect of these drugs on
MECP2E1/E2-BDNF were not additive under these tested conditions, however, either drug may have potential application for related disorders.
Full article
►▼
Show Figures
Open AccessFeature PaperEditor’s ChoiceReview
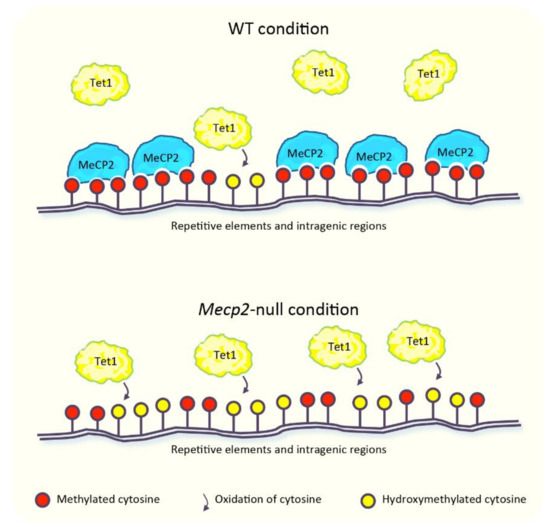
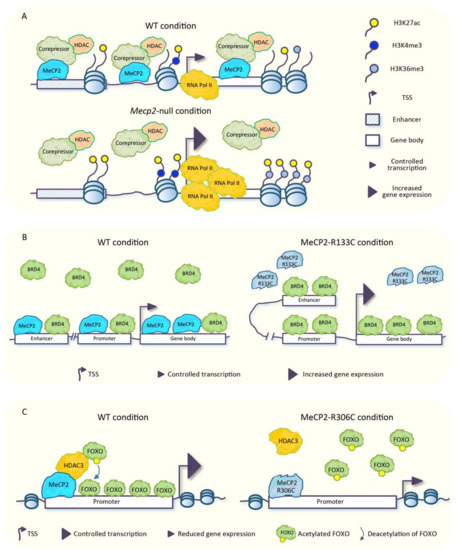
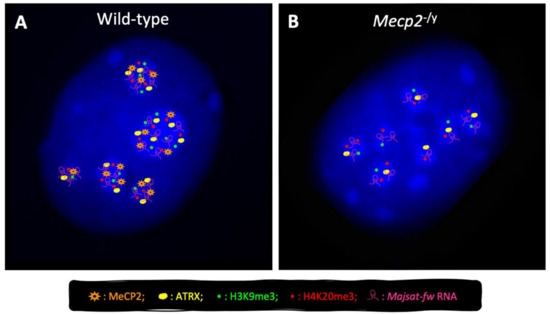
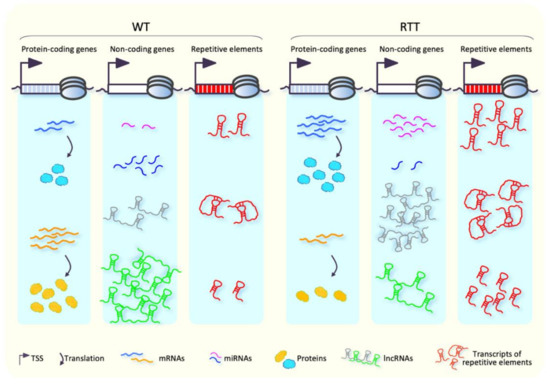
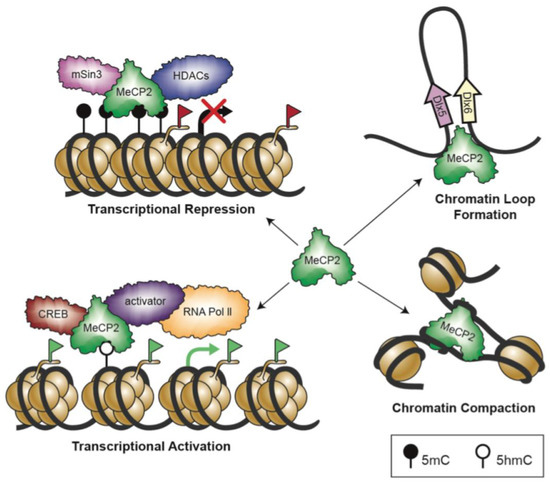
Transcriptomic and Epigenomic Landscape in Rett Syndrome
by
Domenico Marano, Salvatore Fioriniello, Maurizio D’Esposito and Floriana Della Ragione
Cited by 9 | Viewed by 4573
Abstract
Rett syndrome (RTT) is an extremely invalidating, cureless, developmental disorder, and it is considered one of the leading causes of intellectual disability in female individuals. The vast majority of RTT cases are caused by de novo mutations in the X-linked Methyl-CpG binding protein
[...] Read more.
Rett syndrome (RTT) is an extremely invalidating, cureless, developmental disorder, and it is considered one of the leading causes of intellectual disability in female individuals. The vast majority of RTT cases are caused by de novo mutations in the X-linked Methyl-CpG binding protein 2 (
MECP2) gene, which encodes a multifunctional reader of methylated DNA. MeCP2 is a master epigenetic modulator of gene expression, with a role in the organization of global chromatin architecture. Based on its interaction with multiple molecular partners and the diverse epigenetic scenario, MeCP2 triggers several downstream mechanisms, also influencing the epigenetic context, and thus leading to transcriptional activation or repression. In this frame, it is conceivable that defects in such a multifaceted factor as MeCP2 lead to large-scale alterations of the epigenome, ranging from an unbalanced deposition of epigenetic modifications to a transcriptional alteration of both protein-coding and non-coding genes, with critical consequences on multiple downstream biological processes. In this review, we provide an overview of the current knowledge concerning the transcriptomic and epigenomic alterations found in RTT patients and animal models.
Full article
►▼
Show Figures
Open AccessReview
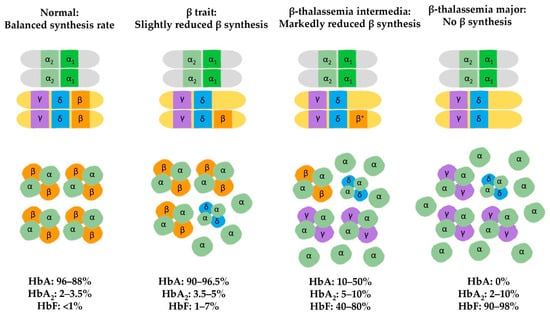
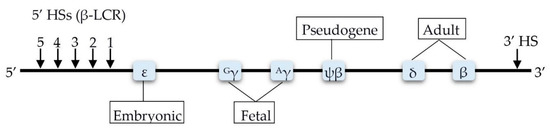
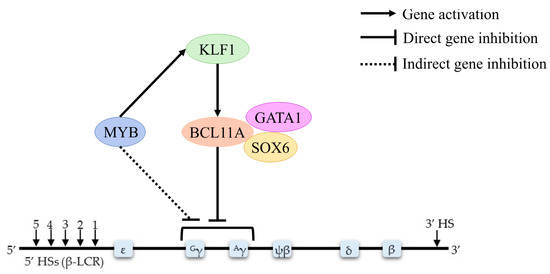
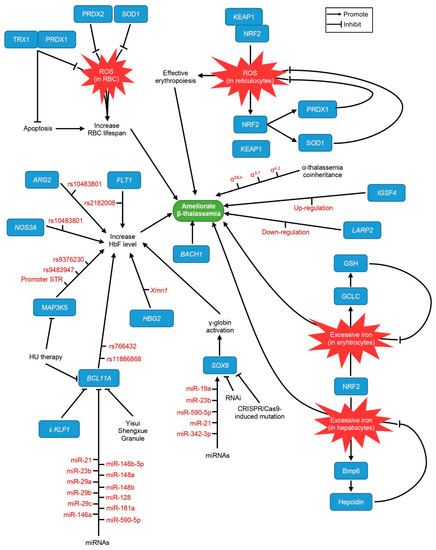
Epigenetic Insights and Potential Modifiers as Therapeutic Targets in β–Thalassemia
by
Nur Atikah Zakaria, Md Asiful Islam, Wan Zaidah Abdullah, Rosnah Bahar, Abdul Aziz Mohamed Yusoff, Ridhwan Abdul Wahab, Shaharum Shamsuddin and Muhammad Farid Johan
Cited by 13 | Viewed by 7213
Abstract
Thalassemia, an inherited quantitative globin disorder, consists of two types, α– and
β–thalassemia.
β–thalassemia is a heterogeneous disease that can be asymptomatic, mild, or even severe. Considerable research has focused on investigating its underlying etiology. These studies found that DNA hypomethylation
[...] Read more.
Thalassemia, an inherited quantitative globin disorder, consists of two types, α– and
β–thalassemia.
β–thalassemia is a heterogeneous disease that can be asymptomatic, mild, or even severe. Considerable research has focused on investigating its underlying etiology. These studies found that DNA hypomethylation in the β–globin gene cluster is significantly related to fetal hemoglobin (HbF) elevation. Histone modification reactivates γ-globin gene expression in adults and increases β–globin expression. Down-regulation of γ–globin suppressor genes, i.e.,
BCL11A,
KLF1,
HBG-XMN1,
HBS1L-MYB, and
SOX6, elevates the HbF level.
β–thalassemia severity is predictable through
FLT1,
ARG2,
NOS2A, and
MAP3K5 gene expression.
NOS2A and
MAP3K5 may predict the
β–thalassemia patient’s response to hydroxyurea, a HbF-inducing drug. The transcription factors NRF2 and
BACH1 work with antioxidant enzymes, i.e.,
PRDX1,
PRDX2,
TRX1, and
SOD1, to protect erythrocytes from oxidative damage, thus increasing their lifespan. A single
β–thalassemia-causing mutation can result in different phenotypes, and these are predictable by
IGSF4 and
LARP2 methylation as well as long non-coding RNA expression levels. Finally, the coinheritance of
β–thalassemia with α–thalassemia ameliorates the
β–thalassemia clinical presentation. In conclusion, the management of
β–thalassemia is currently limited to genetic and epigenetic approaches, and numerous factors should be further explored in the future.
Full article
►▼
Show Figures
Open AccessReview
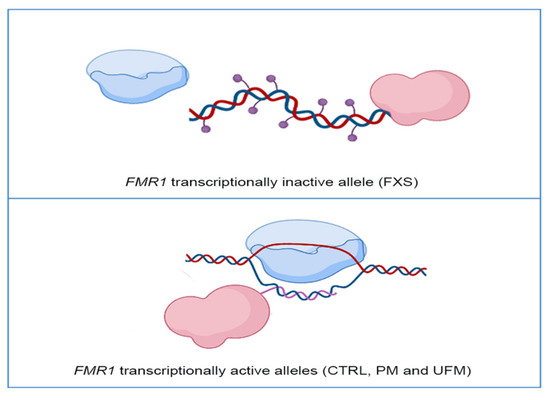
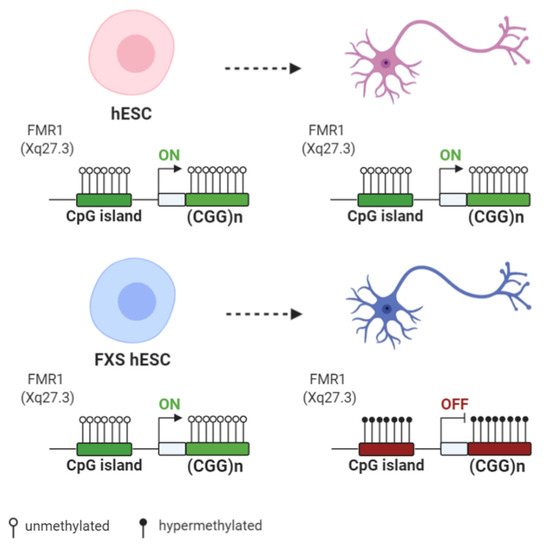
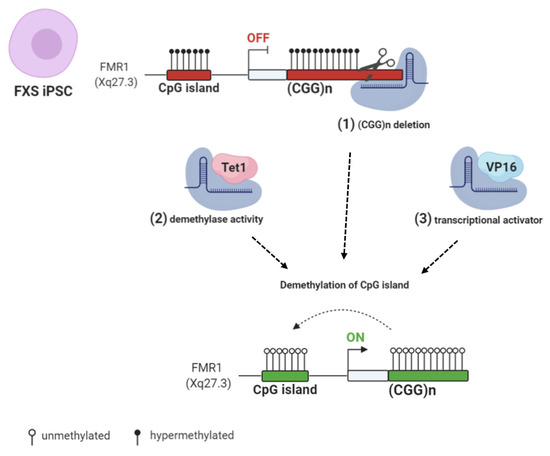
DNA Methylation, Mechanisms of FMR1 Inactivation and Therapeutic Perspectives for Fragile X Syndrome
by
Veronica Nobile, Cecilia Pucci, Pietro Chiurazzi, Giovanni Neri and Elisabetta Tabolacci
Cited by 17 | Viewed by 4409
Abstract
Among the inherited causes of intellectual disability and autism, Fragile X syndrome (FXS) is the most frequent form, for which there is currently no cure. In most FXS patients, the
FMR1 gene is epigenetically inactivated following the expansion over 200 triplets of a
[...] Read more.
Among the inherited causes of intellectual disability and autism, Fragile X syndrome (FXS) is the most frequent form, for which there is currently no cure. In most FXS patients, the
FMR1 gene is epigenetically inactivated following the expansion over 200 triplets of a CGG repeat (FM: full mutation).
FMR1 encodes the Fragile X Mental Retardation Protein (FMRP), which binds several mRNAs, mainly in the brain. When the FM becomes methylated at 10–12 weeks of gestation, the
FMR1 gene is transcriptionally silent. The molecular mechanisms involved in the epigenetic silencing are not fully elucidated. Among FXS families, there is a rare occurrence of males carrying a FM, which remains active because it is not methylated, thus ensuring enough FMRPs to allow for an intellectual development within normal range. Which mechanisms are responsible for sparing these individuals from being affected by FXS? In order to answer this critical question, which may have possible implications for FXS therapy, several potential epigenetic mechanisms have been described. Here, we focus on current knowledge about the role of DNA methylation and other epigenetic modifications in
FMR1 gene silencing.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
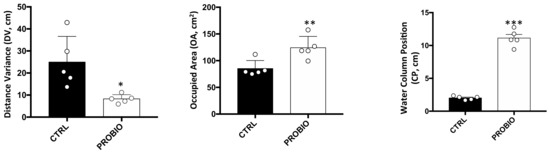
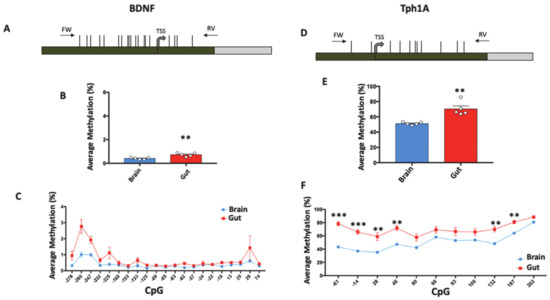
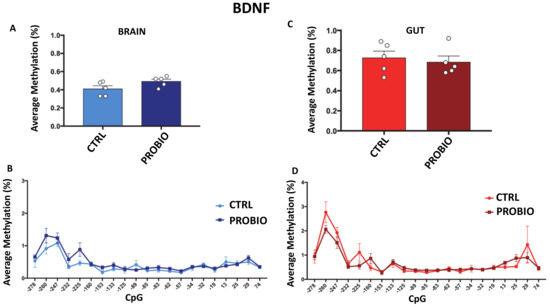
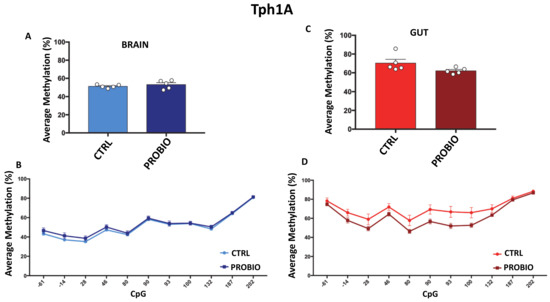
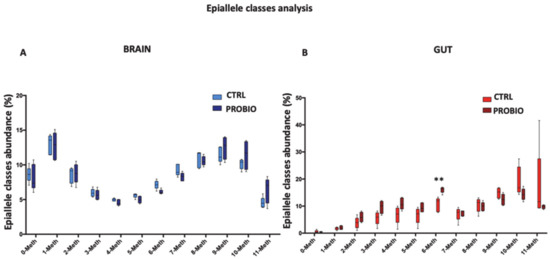
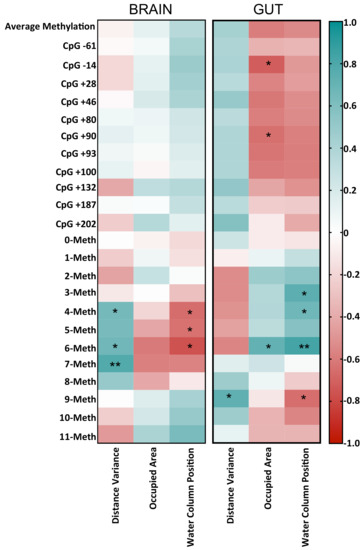
DNA Methylation Profiles of Tph1A and BDNF in Gut and Brain of L. Rhamnosus-Treated Zebrafish
by
Mariella Cuomo, Luca Borrelli, Rosa Della Monica, Lorena Coretti, Giulia De Riso, Luna D’Angelo Lancellotti di Durazzo, Alessandro Fioretti, Francesca Lembo, Timothy G. Dinan, John F. Cryan, Sergio Cocozza and Lorenzo Chiariotti
Cited by 19 | Viewed by 3174
Abstract
The bidirectional microbiota–gut–brain axis has raised increasing interest over the past years in the context of health and disease, but there is a lack of information on molecular mechanisms underlying this connection. We hypothesized that change in microbiota composition may affect brain epigenetics
[...] Read more.
The bidirectional microbiota–gut–brain axis has raised increasing interest over the past years in the context of health and disease, but there is a lack of information on molecular mechanisms underlying this connection. We hypothesized that change in microbiota composition may affect brain epigenetics leading to long-lasting effects on specific brain gene regulation. To test this hypothesis, we used
Zebrafish (Danio Rerio) as a model system. As previously shown, treatment with high doses of probiotics can modulate behavior in
Zebrafish, causing significant changes in the expression of some brain-relevant genes, such as
BDNF and
Tph1A. Using an ultra-deep targeted analysis, we investigated the methylation state of the
BDNF and
Tph1A promoter region in the brain and gut of probiotic-treated and untreated
Zebrafishes. Thanks to the high resolution power of our analysis, we evaluated cell-to-cell methylation differences. At this resolution level, we found slight DNA methylation changes in probiotic-treated samples, likely related to a subgroup of brain and gut cells, and that specific DNA methylation signatures significantly correlated with specific behavioral scores.
Full article
►▼
Show Figures
Open AccessReview
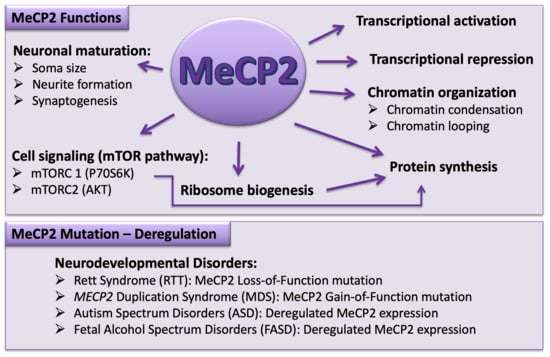
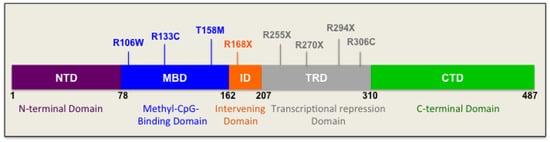
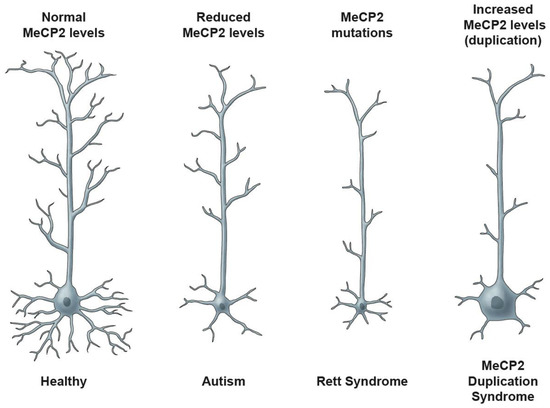
Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease
by
Shervin Pejhan and Mojgan Rastegar
Cited by 28 | Viewed by 12097
Abstract
Rett Syndrome (RTT) is a severe, rare, and progressive developmental disorder with patients displaying neurological regression and autism spectrum features. The affected individuals are primarily young females, and more than 95% of patients carry
de novo mutation(s) in the Methyl-CpG-Binding Protein 2 (
[...] Read more.
Rett Syndrome (RTT) is a severe, rare, and progressive developmental disorder with patients displaying neurological regression and autism spectrum features. The affected individuals are primarily young females, and more than 95% of patients carry
de novo mutation(s) in the Methyl-CpG-Binding Protein 2 (
MECP2) gene. While the majority of RTT patients have
MECP2 mutations (classical RTT), a small fraction of the patients (atypical RTT) may carry genetic mutations in other genes such as the cyclin-dependent kinase-like 5 (
CDKL5) and
FOXG1. Due to the neurological basis of RTT symptoms, MeCP2 function was originally studied in nerve cells (neurons). However, later research highlighted its importance in other cell types of the brain including glia. In this regard, scientists benefitted from modeling the disease using many different cellular systems and transgenic mice with loss- or gain-of-function mutations. Additionally, limited research in human postmortem brain tissues provided invaluable findings in RTT pathobiology and disease mechanism. MeCP2 expression in the brain is tightly regulated, and its altered expression leads to abnormal brain function, implicating MeCP2 in some cases of autism spectrum disorders. In certain disease conditions, MeCP2 homeostasis control is impaired, the regulation of which in rodents involves a regulatory microRNA (
miR132) and brain-derived neurotrophic factor (BDNF). Here, we will provide an overview of recent advances in understanding the underlying mechanism of disease in RTT and the associated genetic mutations in the
MECP2 gene along with the pathobiology of the disease, the role of the two most studied protein variants (MeCP2E1 and MeCP2E2 isoforms), and the regulatory mechanisms that control MeCP2 homeostasis network in the brain, including BDNF and
miR132.
Full article
►▼
Show Figures
Open AccessArticle
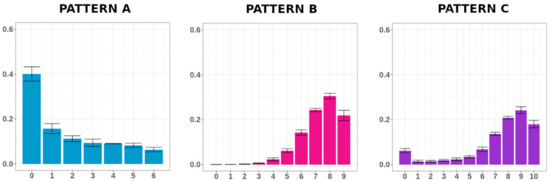
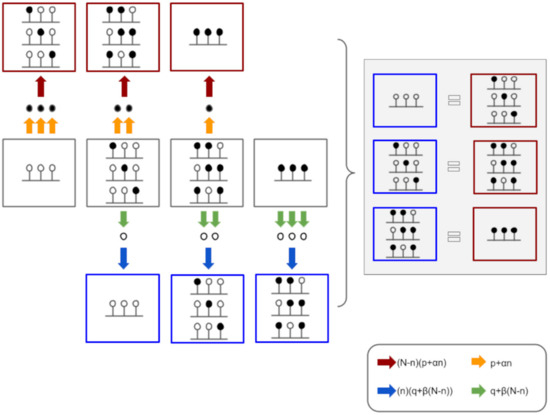
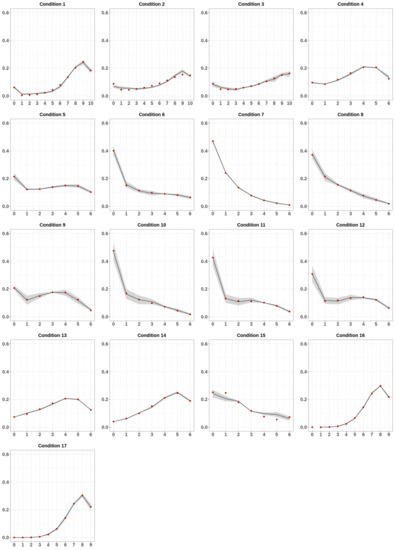
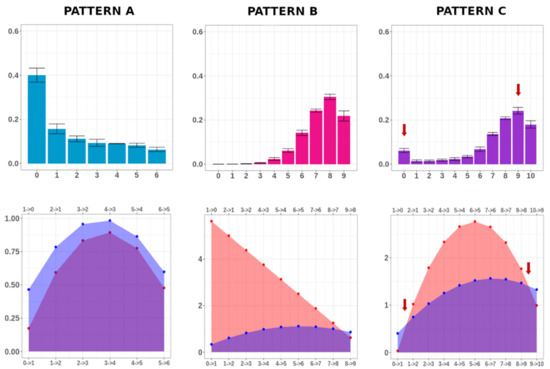
Modeling DNA Methylation Profiles through a Dynamic Equilibrium between Methylation and Demethylation
by
Giulia De Riso, Damiano Francesco Giuseppe Fiorillo, Annalisa Fierro, Mariella Cuomo, Lorenzo Chiariotti, Gennaro Miele and Sergio Cocozza
Cited by 7 | Viewed by 2135
Abstract
DNA methylation is a heritable epigenetic mark that plays a key role in regulating gene expression. Mathematical modeling has been extensively applied to unravel the regulatory mechanisms of this process. In this study, we aimed to investigate DNA methylation by performing a high-depth
[...] Read more.
DNA methylation is a heritable epigenetic mark that plays a key role in regulating gene expression. Mathematical modeling has been extensively applied to unravel the regulatory mechanisms of this process. In this study, we aimed to investigate DNA methylation by performing a high-depth analysis of particular loci, and by subsequent modeling of the experimental results. In particular, we performed an in-deep DNA methylation profiling of two genomic loci surrounding the transcription start site of the D-Aspartate Oxidase and the D-Serine Oxidase genes in different samples (n = 51). We found evidence of cell-to-cell differences in DNA methylation status. However, these cell differences were maintained between different individuals, which indeed showed very similar DNA methylation profiles. Therefore, we hypothesized that the observed pattern of DNA methylation was the result of a dynamic balance between DNA methylation and demethylation, and that this balance was identical between individuals. We hence developed a simple mathematical model to test this hypothesis. Our model reliably captured the characteristics of the experimental data, suggesting that DNA methylation and demethylation work together in determining the methylation state of a locus. Furthermore, our model suggested that the methylation status of neighboring cytosines plays an important role in this balance.
Full article
►▼
Show Figures
Planned Papers
The below list represents only planned manuscripts. Some of these
manuscripts have not been received by the Editorial Office yet. Papers
submitted to MDPI journals are subject to peer-review.
Title: Chromatin conformation and gene expression alterations of the H19-IGF2 imprinted region in cell lines derived from patients with Cornelia de Lange syndrome
Authors: Monica Miozzo; et al.
Affiliation: Department of Health Sciences, Medical Genetics, Università degli Studi di Milano, 20142 Milano, Italy
Title: To be determined
Authors: Mojgan Rastegar; et al.
Affiliation: University of Manitoba, Winnipeg, MB, Canada
Title: To be determined
Authors: Hongmei Zhang; et al.
Affiliation: Division of Epidemiology, Biostatistics and Environmental Health, School of Public Health, University of Memphis, Memphis, TN 38111, USA



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}