Molecular Docking and Structure-Based Virtual Screenings as Efficient Tools in Bioorganic and Medicinal Chemistry
Share This Topical Collection
Editor
 Dr. Laurent Soulère
Dr. Laurent Soulère
Dr. Laurent Soulère
E-Mail
Website
Collection Editor
Institut de Chimie et Biochimie Moléculaire et Supramoléculaire, 69100 Villeurbanne, France
Interests: bioorganic chemistry; quorum sensing modulation; FtsZ inhibitors; molecular docking; virtual screening
Topical Collection Information
Dear Colleagues,
Molecular docking has become a classical tool to gain insight into structure–activity relationships by analyzing the binding modes of drug candidates to molecular targets. This technique also permits the in-silico design of active biomolecules. Structure-based virtual screenings have also changed traditional screenings by decreasing the number of tested compounds using in silico filters. For docking and docking-based virtual screening, the availability of compound databases and structures of protein targets obtained by biophysical techniques or homology modeling is essential to develop new strategies for discovering new bioactive molecules.
This Topical Collection aims at reporting new achievements in molecular docking and virtual screening studies in the field of bioorganic and medicinal chemistry, in terms of strategies, methodologies, and case studies.
Dr. Laurent Soulère
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- Molecular docking
- Virtual screening
- In silico design
- Compound databases
- Binding mode studies
- Proteins targets
- Bioactive molecules
Published Papers (9 papers)
Open AccessArticle
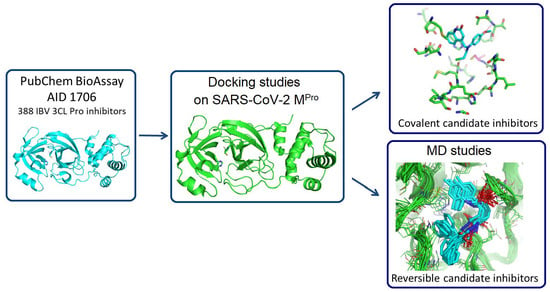
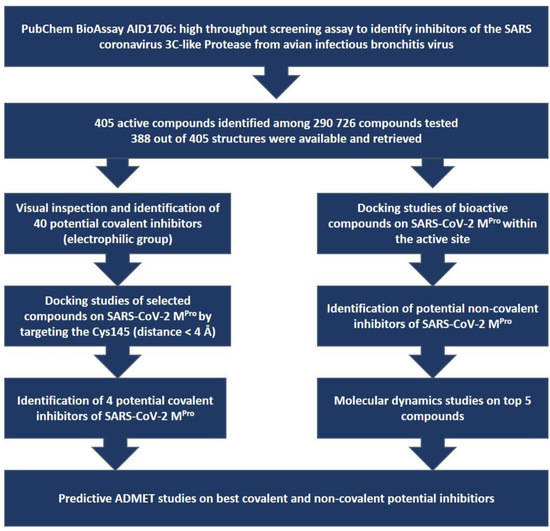
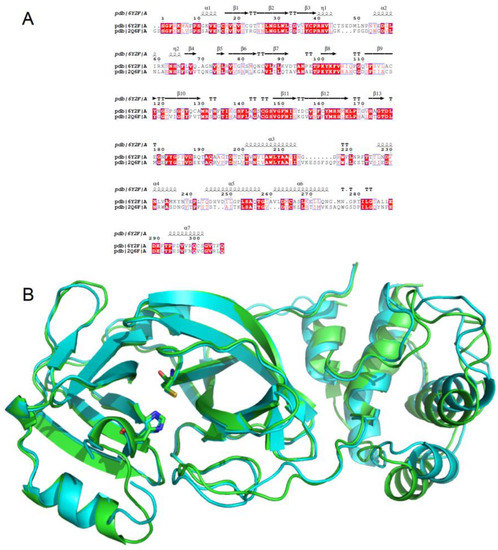
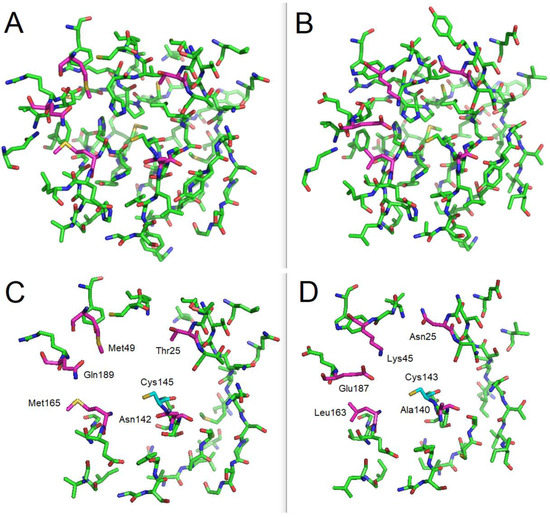
In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Main Protease among a PubChem Database of Avian Infectious Bronchitis Virus 3CLPro Inhibitors
by
Laurent Soulère, Thibaut Barbier and Yves Queneau
Viewed by 1231
Abstract
Remarkable structural homologies between the main proteases of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the avian infectious bronchitis virus (IBV) were revealed by comparative amino-acid sequence and 3D structural alignment. Assessing whether reported IBV 3CLPro inhibitors could also interact with
[...] Read more.
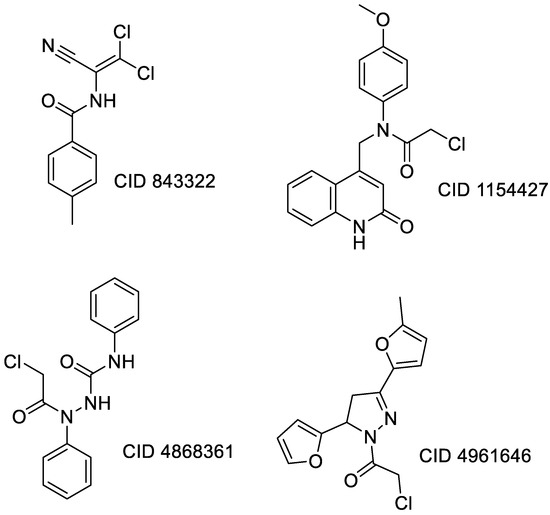
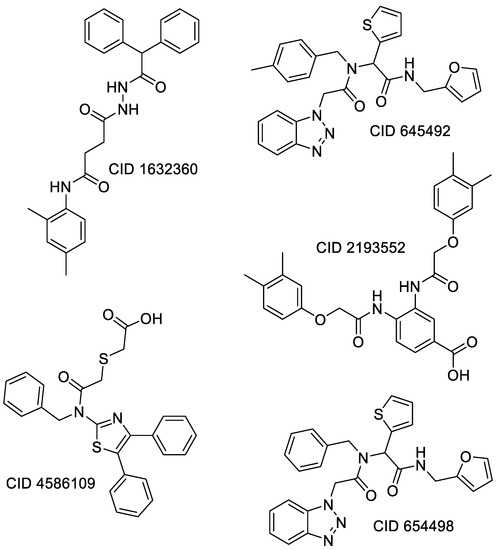
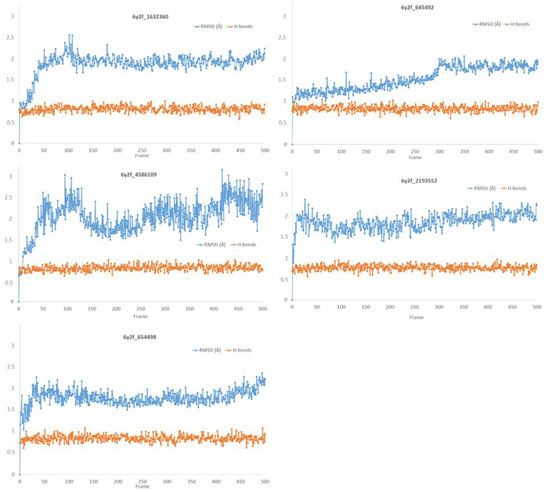
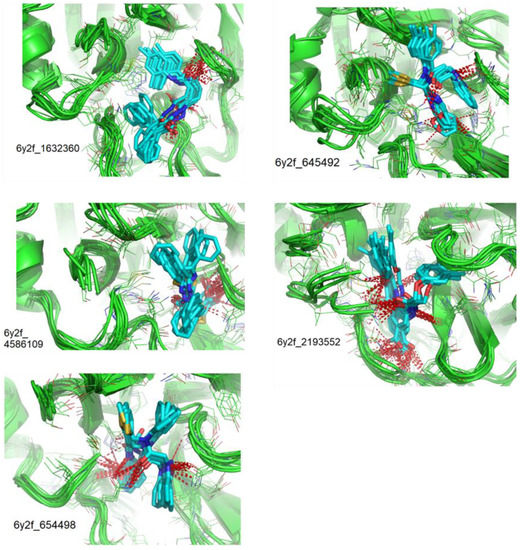
Remarkable structural homologies between the main proteases of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the avian infectious bronchitis virus (IBV) were revealed by comparative amino-acid sequence and 3D structural alignment. Assessing whether reported IBV 3CLPro inhibitors could also interact with SARS-CoV-2 has been undertaken in silico using a PubChem BioAssay database of 388 compounds active on the avian infectious bronchitis virus 3C-like protease. Docking studies of this database on the SARS-CoV-2 protease resulted in the identification of four covalent inhibitors targeting the catalytic cysteine residue and five non-covalent inhibitors for which the binding was further investigated by molecular dynamics (MD) simulations. Predictive ADMET calculations on the nine compounds suggest promising pharmacokinetic properties.
Full article
►▼
Show Figures
Open AccessArticle
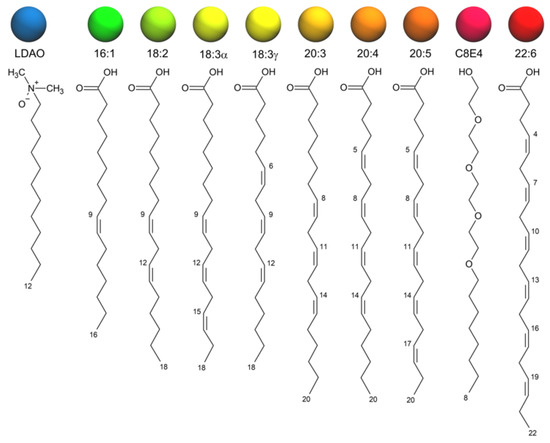
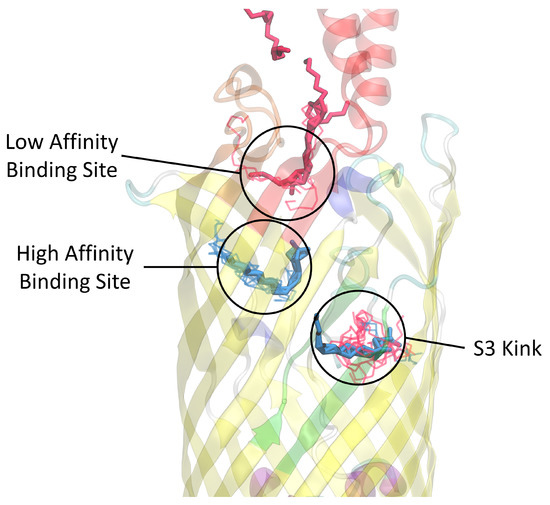
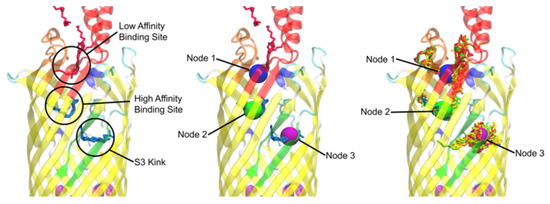
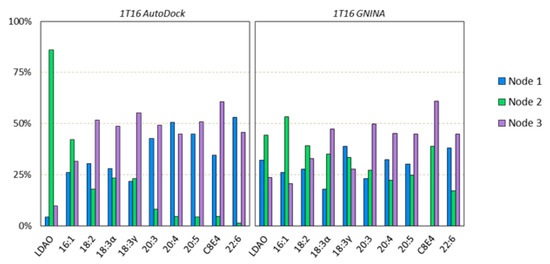
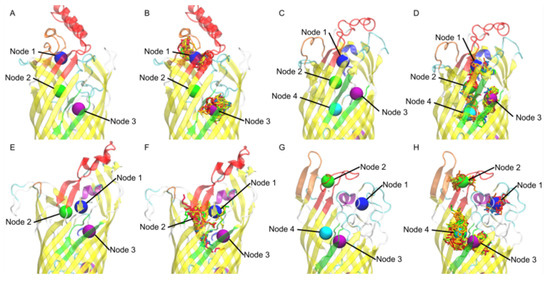
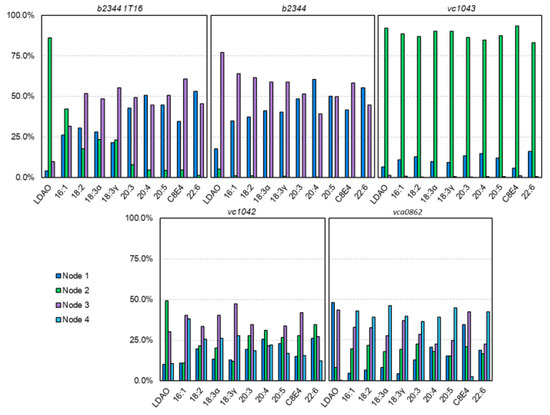
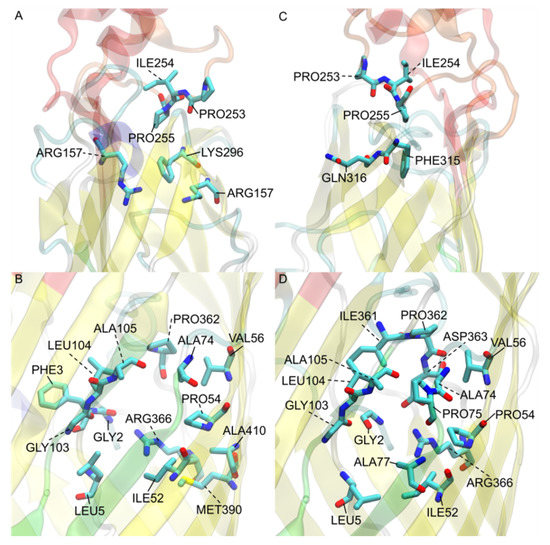
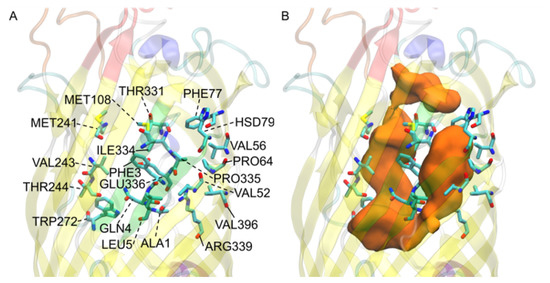
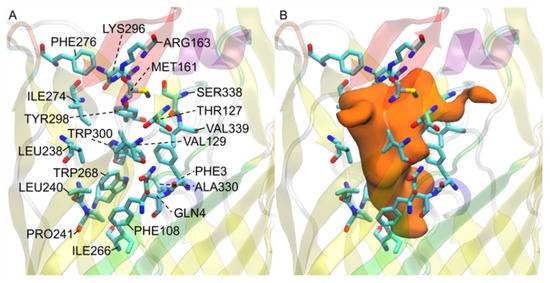
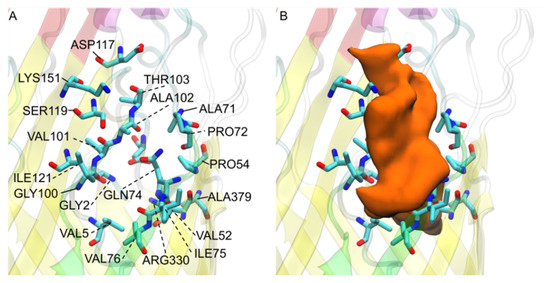
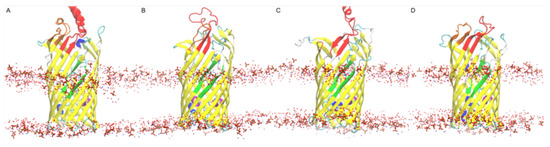
Simulated Docking Predicts Putative Channels for the Transport of Long-Chain Fatty Acids in Vibrio cholerae
by
Andrew Turgeson, Lucas Morley, David Giles and Bradley Harris
Cited by 4 | Viewed by 1384
Abstract
Fatty acids (FA) play an important role in biological functions, such as membrane homeostasis, metabolism, and as signaling molecules. FadL is the only known protein that uptakes long-chain fatty acids in Gram-negative bacteria, and this uptake has traditionally been thought to be limited
[...] Read more.
Fatty acids (FA) play an important role in biological functions, such as membrane homeostasis, metabolism, and as signaling molecules. FadL is the only known protein that uptakes long-chain fatty acids in Gram-negative bacteria, and this uptake has traditionally been thought to be limited to fatty acids up to 18 carbon atoms in length. Recently however, it was found
Vibrio cholerae has the ability to uptake fatty acids greater than 18 carbon atoms and this uptake corresponds to bacterial survivability. Using
E. coli’s FadL as a template,
V. cholerae FadL homologs
vc1042,
vc1043, and
vca0862 have been computationally folded, simulated on an atomistic level using Molecular Dynamics, and docked
in silico to analyze the FadL transport channels. For the
vc1042 and
vc1043 homologs, these transport channels have more structural accommodations for the many rigid unsaturated bonds of long-chain polyunsaturated fatty acids, while the
vca0862 homolog was found to lack transport channels within the signature beta barrel of FadL proteins.
Full article
►▼
Show Figures
Open AccessArticle
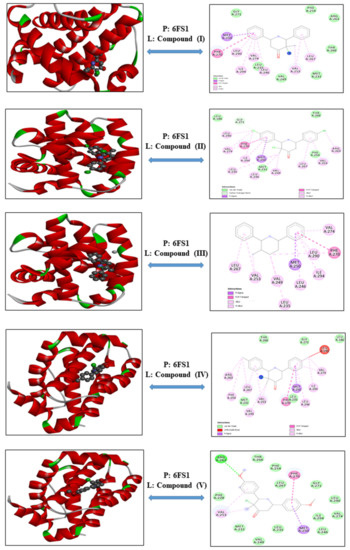
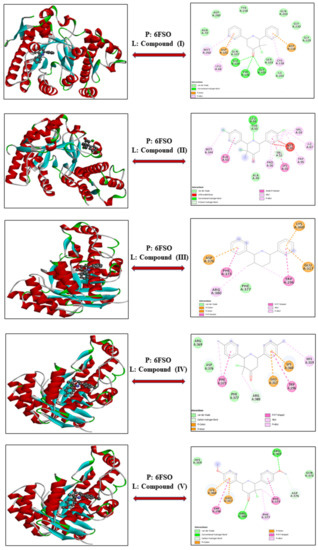
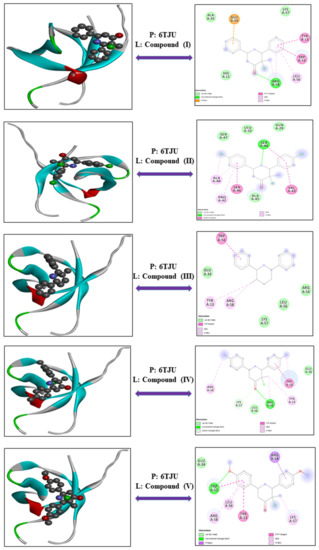
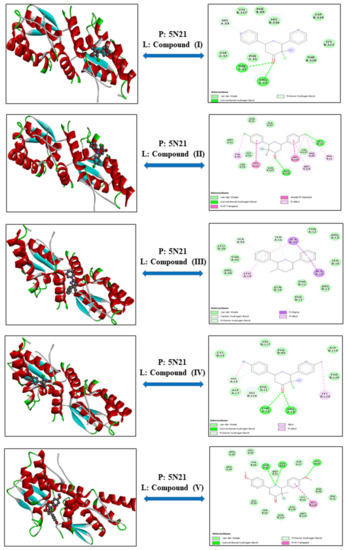
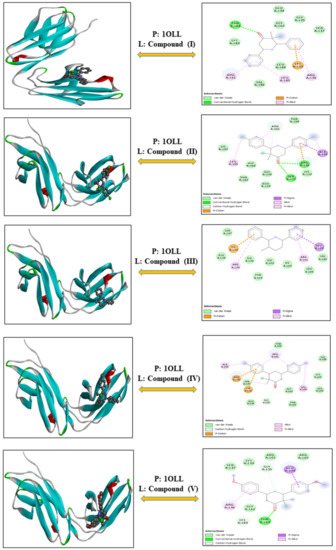
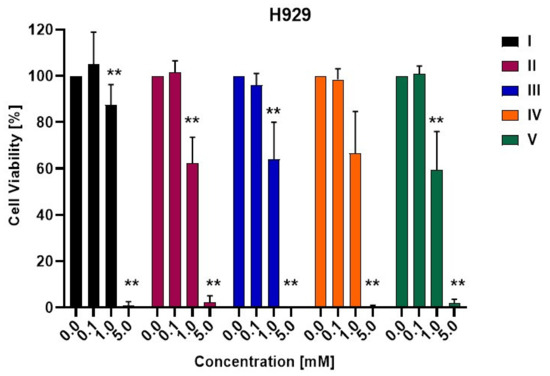
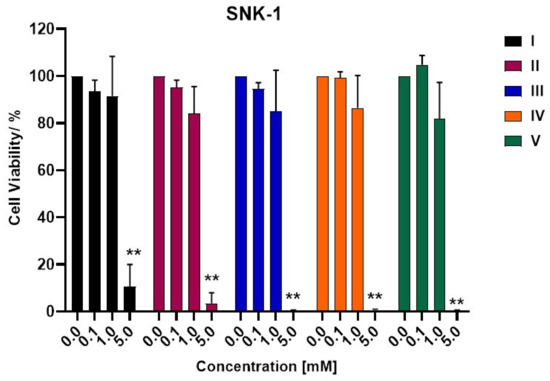
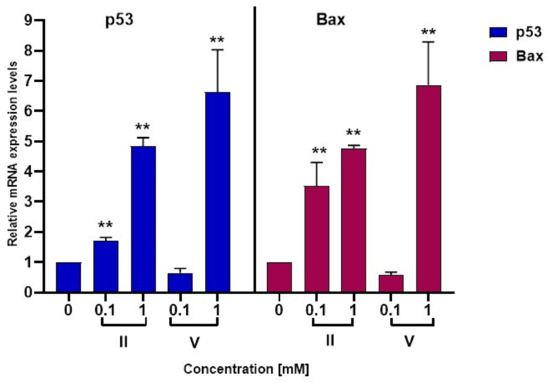
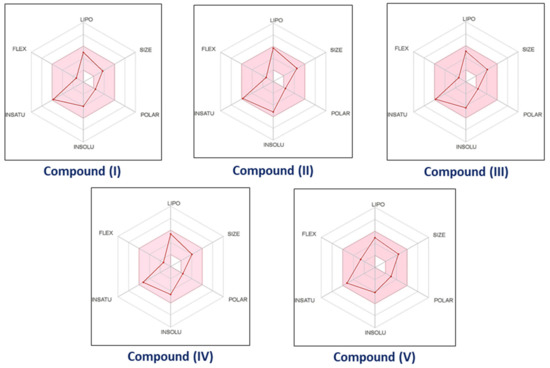
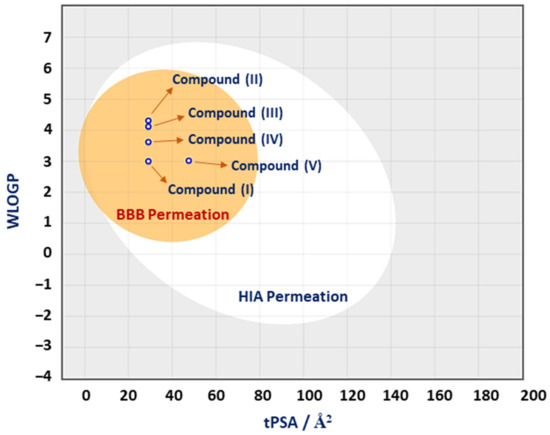
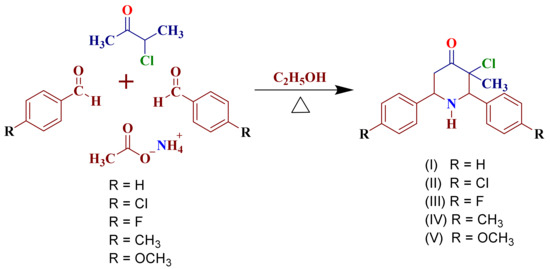
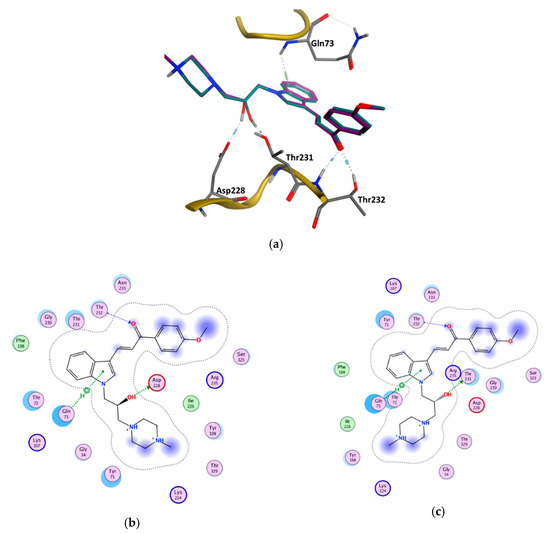
3-Chloro-3-methyl-2,6-diarylpiperidin-4-ones as Anti-Cancer Agents: Synthesis, Biological Evaluation, Molecular Docking, and In Silico ADMET Prediction
by
Arulraj Ramalingam, Nurulhuda Mustafa, Wee Joo Chng, Mouna Medimagh, Sivakumar Sambandam and Noureddine Issaoui
Cited by 12 | Viewed by 2120
Abstract
Piperidine pharmacophore-containing compounds have demonstrated therapeutic efficacy against a range of diseases and are now being investigated in cancer. A series of 3-chloro-3-methyl-2,6-diarylpiperidin-4-ones, compounds (
I–
V) were designed and synthesized for their evaluation as a potential anti-cancer agent. Compounds
II
[...] Read more.
Piperidine pharmacophore-containing compounds have demonstrated therapeutic efficacy against a range of diseases and are now being investigated in cancer. A series of 3-chloro-3-methyl-2,6-diarylpiperidin-4-ones, compounds (
I–
V) were designed and synthesized for their evaluation as a potential anti-cancer agent. Compounds
II and
IV reduced the growth of numerous hematological cancer cell lines while simultaneously increasing the mRNA expression of apoptosis-promoting genes, p53 and Bax. Molecular docking analyses confirmed that compounds can bind to 6FS1, 6FSO (myeloma), 6TJU (leukemia), 5N21, and 1OLL (NKTL). Computational ADMET research confirmed the essential physicochemical, pharmacokinetic, and drug-like characteristics of compounds (
I–
V). The results revealed that these compounds interact efficiently with active site residues and that compounds (
II) and (
V) can be further evaluated as potential therapeutic candidates.
Full article
►▼
Show Figures
Open AccessArticle
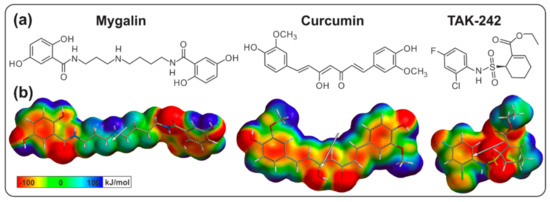
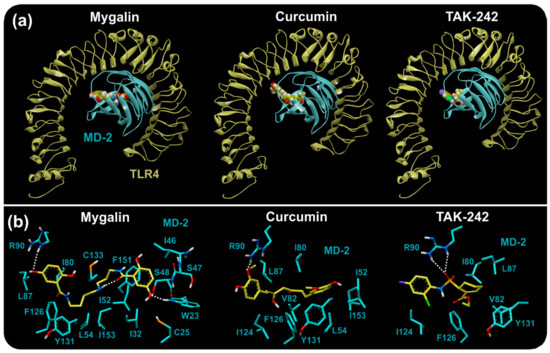
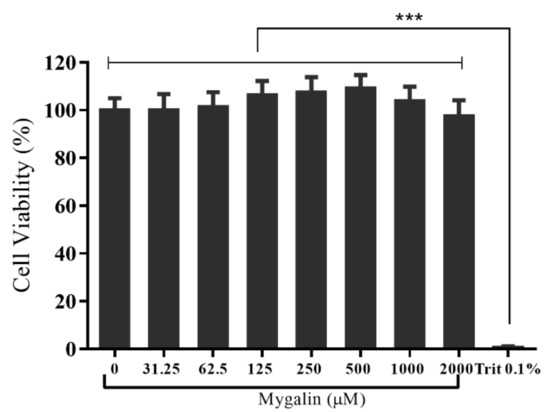
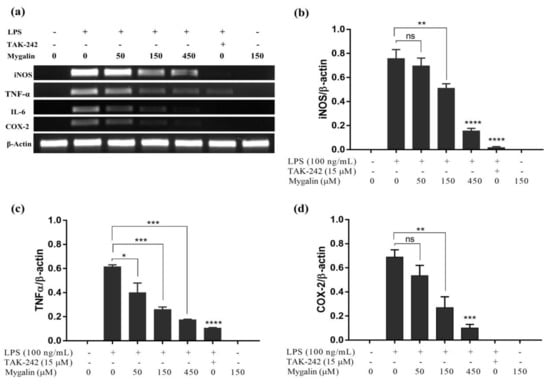
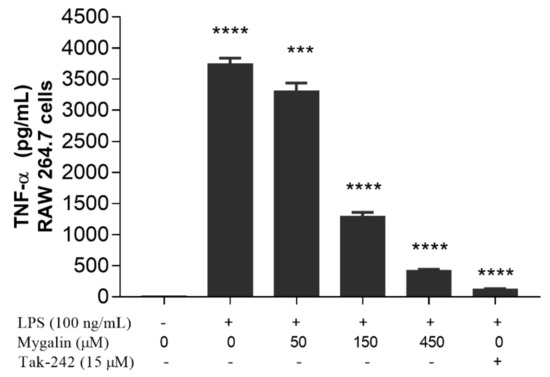
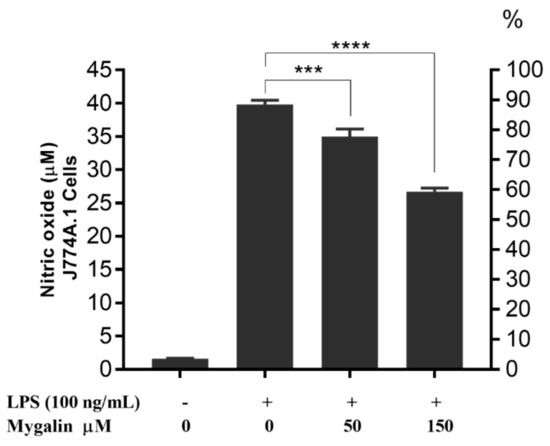
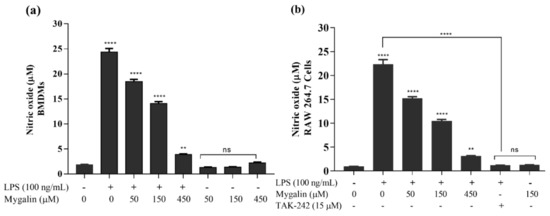
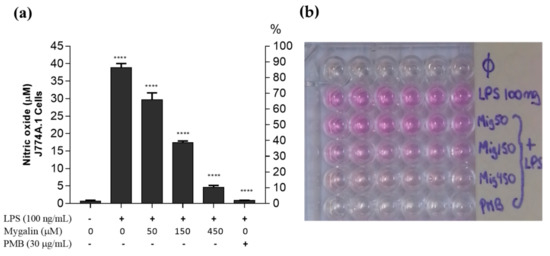
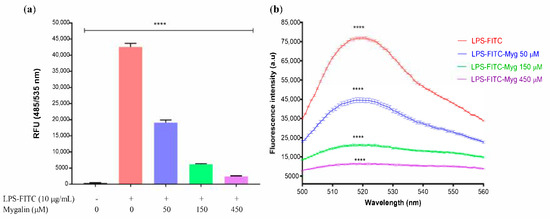
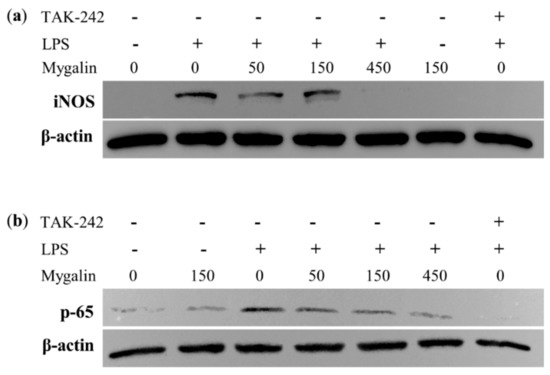
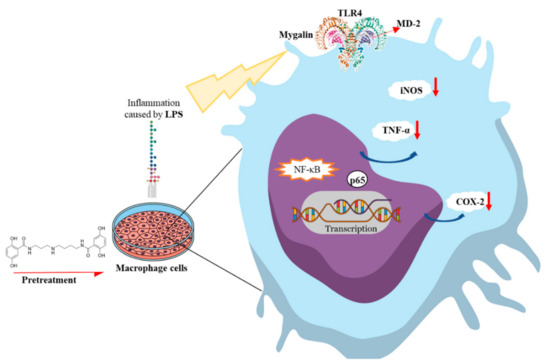
Acylpolyamine Mygalin as a TLR4 Antagonist Based on Molecular Docking and In Vitro Analyses
by
Abraham Espinoza-Culupú, Ricardo Vázquez-Ramírez, Mariella Farfán-López, Elizabeth Mendes, Maria Notomi Sato, Pedro Ismael da Silva Junior and Monamaris Marques Borges
Cited by 7 | Viewed by 3676
Abstract
Toll-like receptors (TLRs) are transmembrane proteins that are key regulators of innate and adaptive immune responses, particularly TLR4, and they have been identified as potential drug targets for the treatment of disease. Several low-molecular-weight compounds are being considered as new drug targets for
[...] Read more.
Toll-like receptors (TLRs) are transmembrane proteins that are key regulators of innate and adaptive immune responses, particularly TLR4, and they have been identified as potential drug targets for the treatment of disease. Several low-molecular-weight compounds are being considered as new drug targets for various applications, including as immune modulators. Mygalin, a 417 Da synthetic bis-acylpolyamine, is an analog of spermidine that has microbicidal activity. In this study, we investigated the effect of mygalin on the innate immune response based on a virtual screening (VS) and molecular docking analysis. Bone marrow-derived macrophages and the cell lines J774A.1 and RAW 264.7 stimulated with lipopolysaccharide (LPS) were used to confirm the data obtained in silico. Virtual screening and molecular docking suggested that mygalin binds to TLR4 via the protein myeloid differentiation factor 2 (MD-2) and LPS. Macrophages stimulated by mygalin plus LPS showed suppressed gene expression of tumor necrosis factor (TNF-α), interleukine 6 (IL-6), cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), as well as inhibition of signaling protein p65 of the nuclear factor κB (NF-κB), resulting in decreased production of nitric oxide (NO) and TNF-α. These results indicate that mygalin has anti-inflammatory potential, being an attractive option to be explored. In addition, we reinforce the importance of virtual screening analysis to assist in the discovery of new drugs.
Full article
►▼
Show Figures
Open AccessArticle
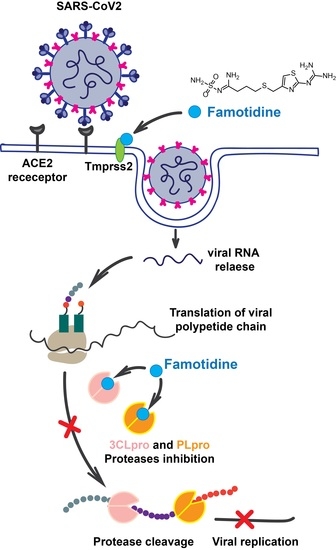
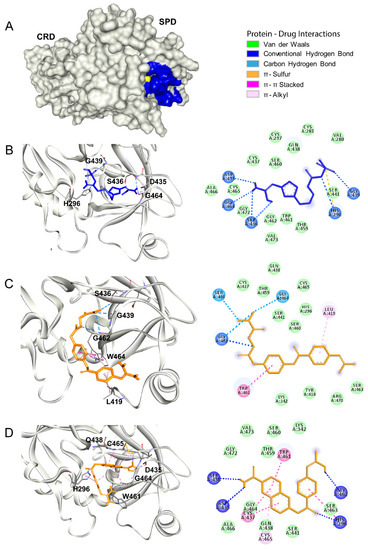
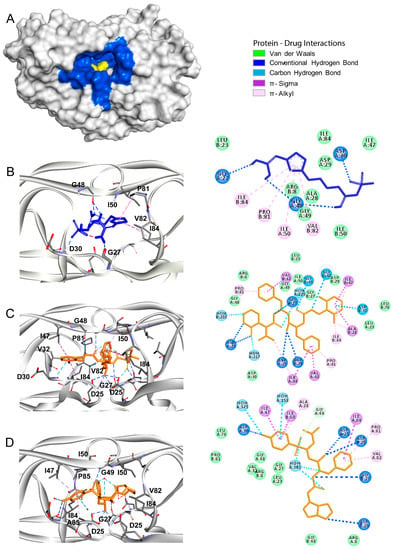
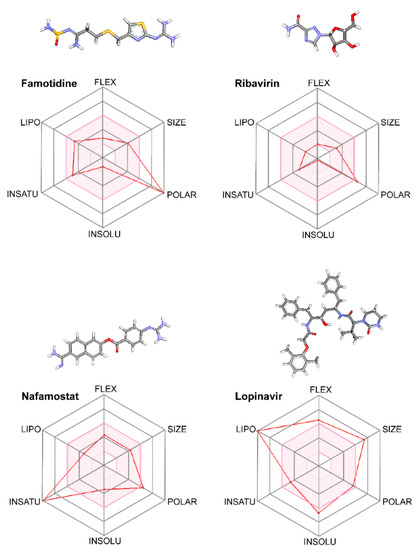
Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis
by
Joseph T. Ortega, Maria Luisa Serrano and Beata Jastrzebska
Cited by 40 | Viewed by 9975
Abstract
The pandemic associated with Severe Acute Respiratory Syndrome Coronavirus type 2 (SARS-CoV2) and its disease named COVID-19 challenged the scientific community to discover effective therapeutic solutions in a short period. Repurposing existing drugs is one viable approach that emphasizes speed during these urgent
[...] Read more.
The pandemic associated with Severe Acute Respiratory Syndrome Coronavirus type 2 (SARS-CoV2) and its disease named COVID-19 challenged the scientific community to discover effective therapeutic solutions in a short period. Repurposing existing drugs is one viable approach that emphasizes speed during these urgent times. Famotidine, a class A G protein-coupled receptor antagonist used for the treatment of gastroesophageal reflux was recently identified in an in silico screening. Additionally, a recent retrospective clinical report showed that the treatment with famotidine provided a good outcome in patients infected with SARS-CoV2. A clinical trial testing effectiveness of famotidine in combination with hydroxychloroquine is currently ongoing in the United States (US). In the 1990s, famotidine was described as an antiviral agent against human immunodeficiency virus (HIV). Interestingly, some HIV protease inhibitors are presently being used against SARS-CoV2. However, it is not clear if famotidine could be effective against SARS-CoV2. Thus, by using a computational analysis, we aimed to examine if the antiviral effect of famotidine could be related to the inhibition of proteases involved in the virus replication. Our results showed that famotidine could interact within the catalytic site of the three proteases associated with SARS-CoV2 replication. However, weak binding affinity of famotidine to these proteases suggests that a successful famotidine therapy could likely be achieved only in combination with other antiviral drugs. Finally, analysis of famotidine’s pharmacokinetic parameters indicated that its effect against SARS-CoV2 infection could be reached only upon intravenous administration. This work will contribute to the pharmacological knowledge of famotidine as an antiviral agent against SARS-CoV2.
Full article
►▼
Show Figures
Open AccessArticle
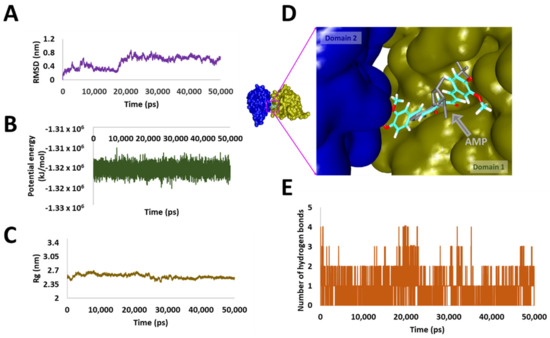
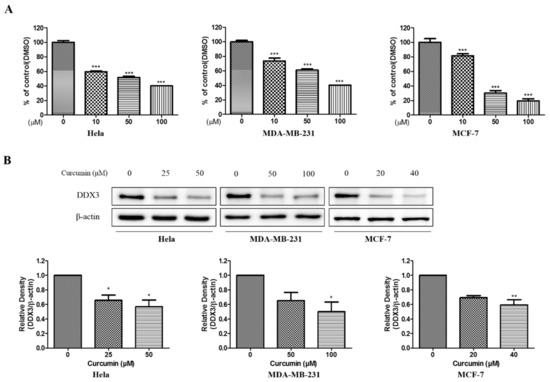
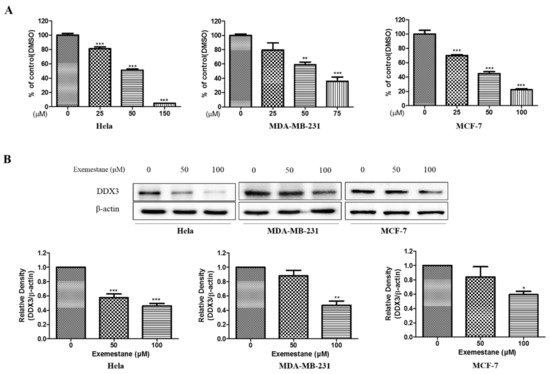
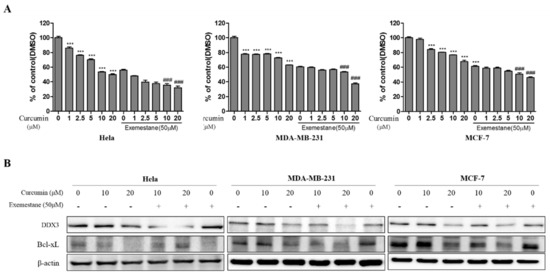
A Computational Approach with Biological Evaluation: Combinatorial Treatment of Curcumin and Exemestane Synergistically Regulates DDX3 Expression in Cancer Cell Lines
by
Shailima Rampogu, Seong Min Kim, Minky Son, Ayoung Baek, Chanin Park, Gihwan Lee, Yumi Kim, Gon Sup Kim, Ju Hyun Kim and Keun Woo Lee
Cited by 13 | Viewed by 3129
Abstract
DDX3 belongs to RNA helicase family that demonstrates oncogenic properties and has gained wider attention due to its role in cancer progression, proliferation and transformation. Mounting reports have evidenced the role of DDX3 in cancers making it a promising target to abrogate DDX3
[...] Read more.
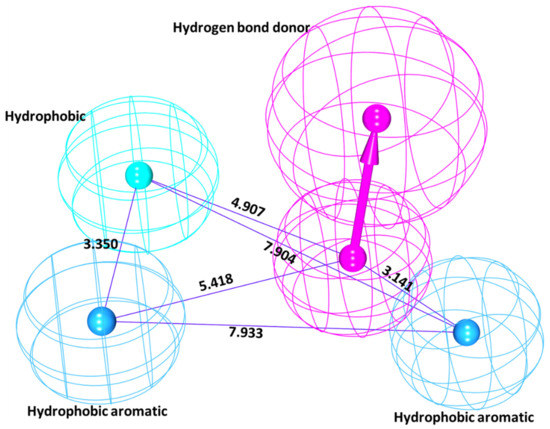
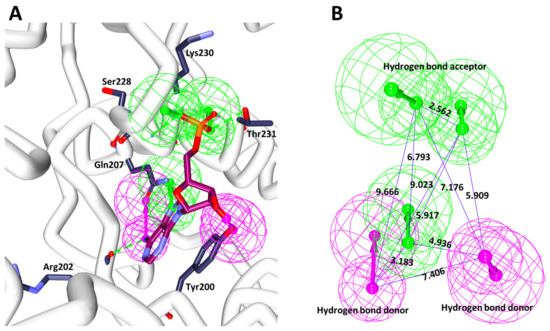
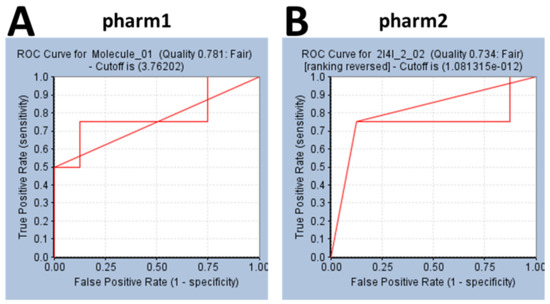
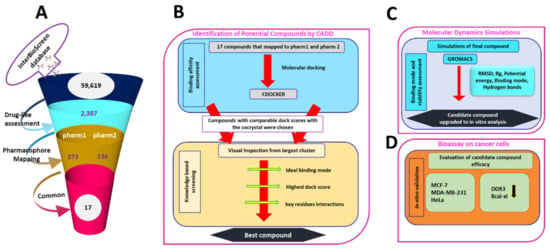
DDX3 belongs to RNA helicase family that demonstrates oncogenic properties and has gained wider attention due to its role in cancer progression, proliferation and transformation. Mounting reports have evidenced the role of DDX3 in cancers making it a promising target to abrogate DDX3 triggered cancers. Dual pharmacophore models were generated and were subsequently validated. They were used as 3D queries to screen the InterBioScreen database, resulting in the selection of curcumin that was escalated to molecular dynamics simulation studies. In vitro anti-cancer analysis was conducted on three cell lines such as MCF-7, MDA-MB-231 and HeLa, which were evaluated along with exemestane. Curcumin was docked into the active site of the protein target (PDB code 2I4I) to estimate the binding affinity. The compound has interacted with two key residues and has displayed stable molecular dynamics simulation results. In vitro analysis has demonstrated that both the candidate compounds have reduced the expression of DDX3 in three cell lines. However, upon combinatorial treatment of curcumin (10 and 20 μM) and exemestane (50 μM) a synergism was exhibited, strikingly downregulating the DDX3 expression and has enhanced apoptosis in three cell lines. The obtained results illuminate the use of curcumin as an alternative DDX3 inhibitor and can serve as a chemical scaffold to design new small molecules.
Full article
►▼
Show Figures
Open AccessArticle
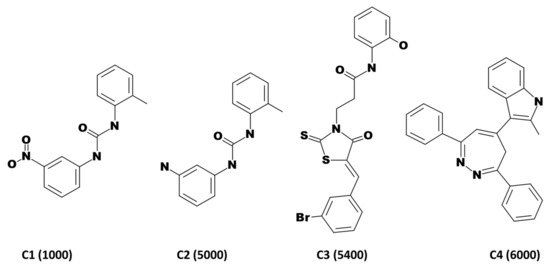
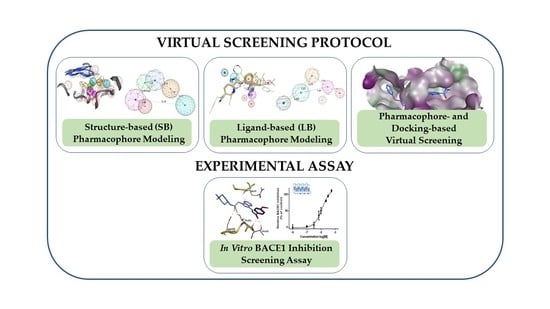
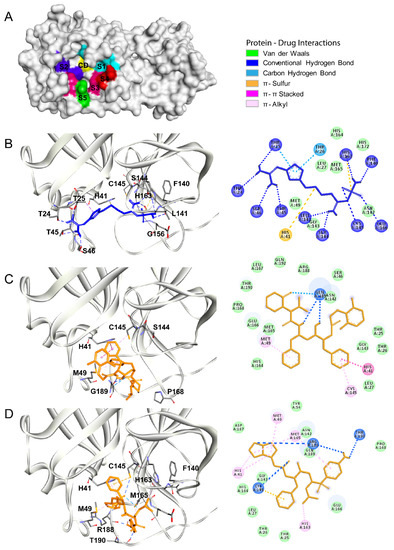
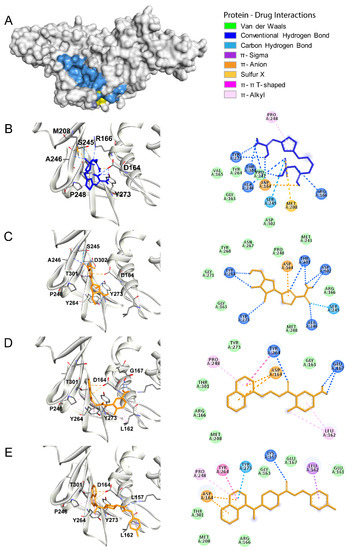
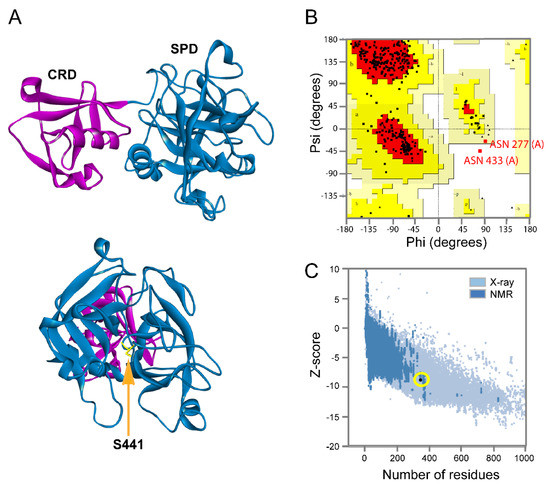
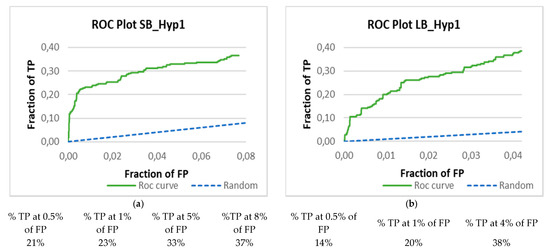
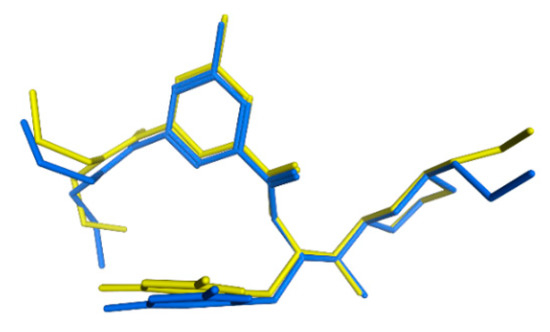
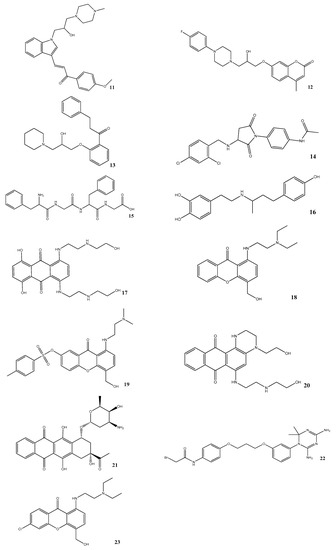
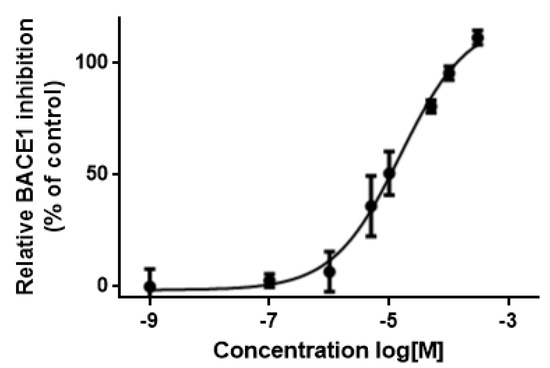
Combining Virtual Screening Protocol and In Vitro Evaluation towards the Discovery of BACE1 Inhibitors
by
Judite R. M. Coimbra, Salete J. Baptista, Teresa C. P. Dinis, Maria M. C. Silva, Paula I. Moreira, Armanda E. Santos and Jorge A. R. Salvador
Cited by 15 | Viewed by 4305
Abstract
The treatment options for a patient diagnosed with Alzheimer’s disease (AD) are currently limited. The cerebral accumulation of amyloid-β (Aβ) is a critical molecular event in the pathogenesis of AD. When the amyloidogenic β-secretase (BACE1) is inhibited, the production of Aβ peptide is
[...] Read more.
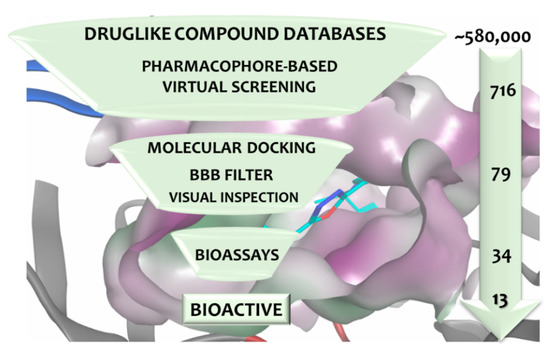
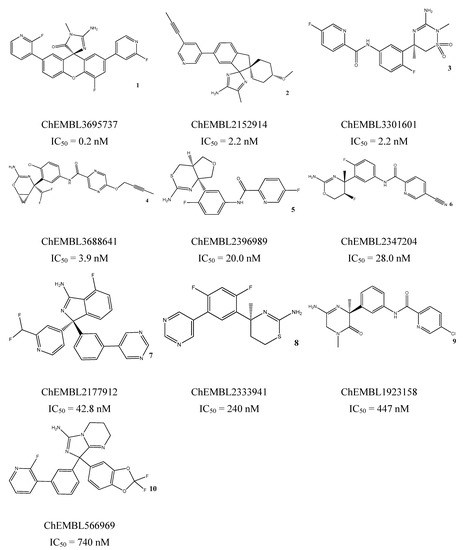
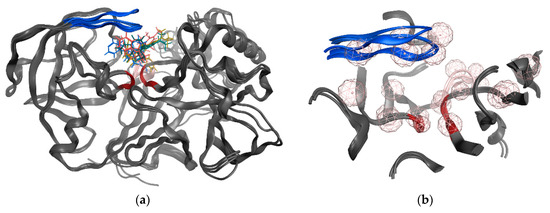
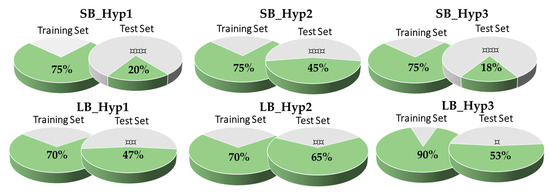
The treatment options for a patient diagnosed with Alzheimer’s disease (AD) are currently limited. The cerebral accumulation of amyloid-β (Aβ) is a critical molecular event in the pathogenesis of AD. When the amyloidogenic β-secretase (BACE1) is inhibited, the production of Aβ peptide is reduced. Henceforth, the main goal of this study is the discovery of new small bioactive molecules that potentially reach the brain and inhibit BACE1. The work was conducted by a customized molecular modelling protocol, including pharmacophore-based and molecular docking-based virtual screening (VS). Structure-based (SB) and ligand-based (LB) pharmacophore models were designed to accurately screen several drug-like compound databases. The retrieved hits were subjected to molecular docking and in silico filtered to predict their ability to cross the blood–brain barrier (BBB). Additionally, 34 high-scoring compounds structurally distinct from known BACE1 inhibitors were selected for in vitro screening assay, which resulted in 13 novel hit-compounds for this relevant therapeutic target. This study disclosed new BACE1 inhibitors, proving the utility of combining computational and in vitro approaches for effectively predicting anti-BACE1 agents in the early drug discovery process.
Full article
►▼
Show Figures
Open AccessArticle
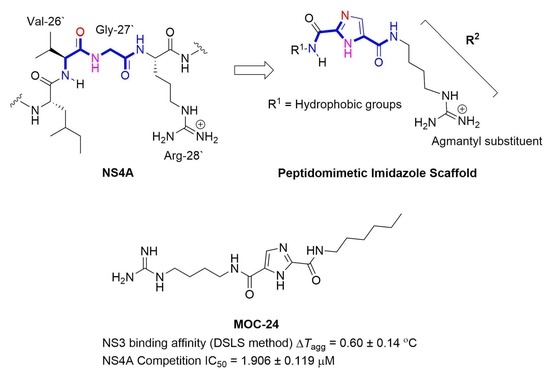
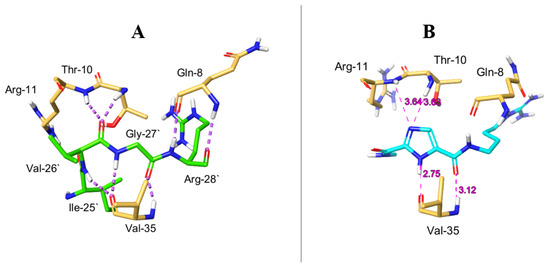
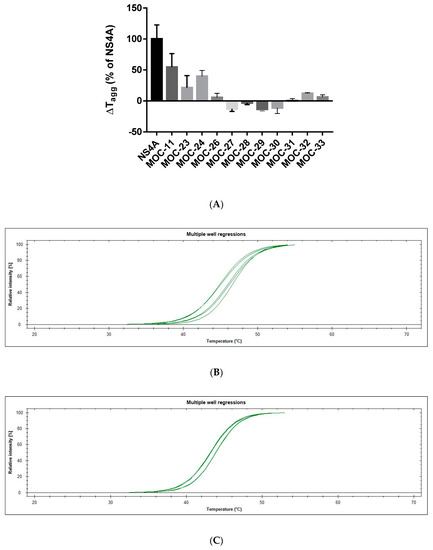
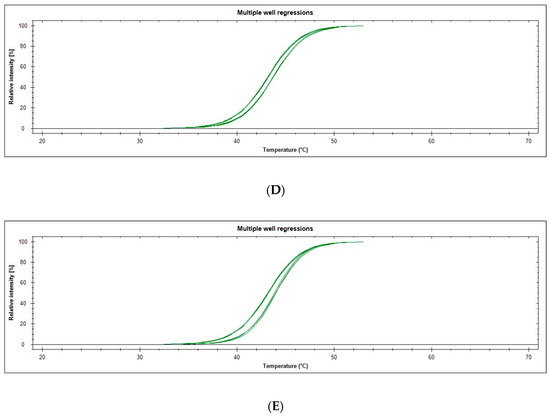
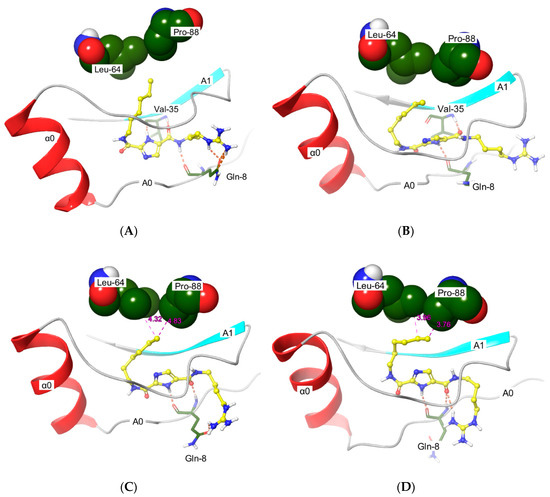
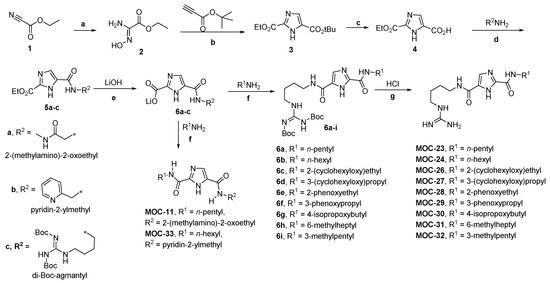
1H-Imidazole-2,5-Dicarboxamides as NS4A Peptidomimetics: Identification of a New Approach to Inhibit HCV-NS3 Protease
by
Abdelsattar M. Omar, Mahmoud A. Elfaky, Stefan T. Arold, Sameh H. Soror, Maan T. Khayat, Hani Z. Asfour, Faida H. Bamane and Moustafa E. El-Araby
Cited by 5 | Viewed by 3973
Abstract
The nonstructural (NS) protein NS3/4A protease is a critical factor for hepatitis C virus (HCV) maturation that requires activation by NS4A. Synthetic peptide mutants of NS4A were found to inhibit NS3 function. The bridging from peptide inhibitors to heterocyclic peptidomimetics of NS4A has
[...] Read more.
The nonstructural (NS) protein NS3/4A protease is a critical factor for hepatitis C virus (HCV) maturation that requires activation by NS4A. Synthetic peptide mutants of NS4A were found to inhibit NS3 function. The bridging from peptide inhibitors to heterocyclic peptidomimetics of NS4A has not been considered in the literature and, therefore, we decided to explore this strategy for developing a new class of NS3 inhibitors. In this report, a structure-based design approach was used to convert the bound form of NS4A into 1
H-imidazole-2,5-dicarboxamide derivatives as first generation peptidomimetics. This scaffold mimics the buried amino acid sequence Ile-25` to Arg-28` at the core of NS4A
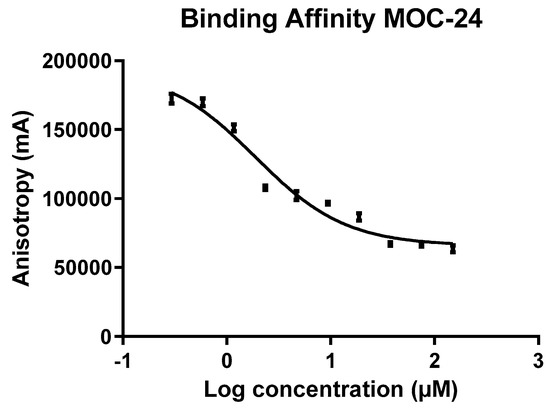
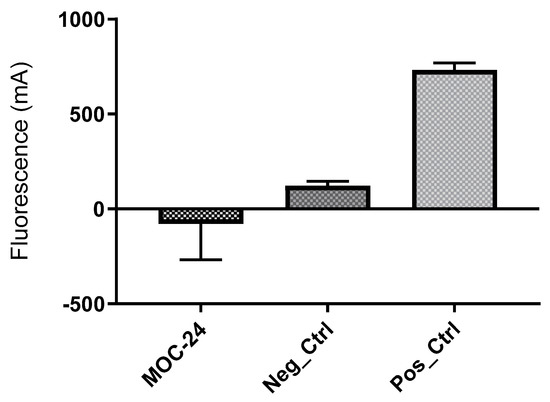
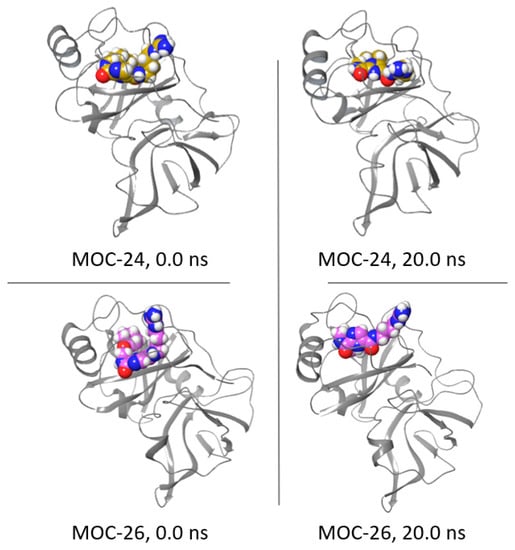
21`–33` needed to activate the NS3 protease. Some of the synthesized compounds (Coded MOC) were able to compete with and displace NS4A
21`–33` for binding to NS3. For instance,
N5-(4-guanidinobutyl)-
N2-(
n-hexyl)-1
H-imidazole-2,5-dicarboxamide (MOC-24) inhibited the binding of NS4A
21`–33` with a competition half maximal inhibitory concentration (IC
50) of 1.9 ± 0.12 µM in a fluorescence anisotropy assay and stabilized the denaturation of NS3 by increasing the aggregation temperature (40% compared to NS4A
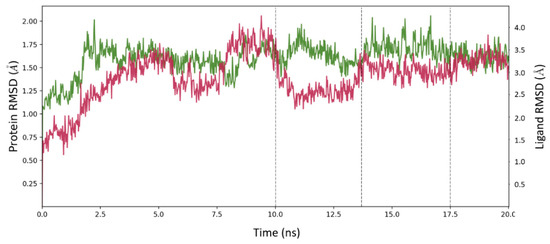
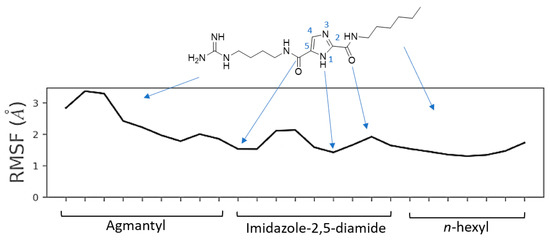
21`–33`). MOC-24 also inhibited NS3 protease activity in a fluorometric assay. Molecular dynamics simulations were conducted to rationalize the differences in structure–activity relationship (SAR) between the active MOC-24 and the inactive MOC-26. Our data show that MOC compounds are possibly the first examples of NS4A peptidomimetics that have demonstrated promising activities against NS3 proteins.
Full article
►▼
Show Figures
Open AccessArticle
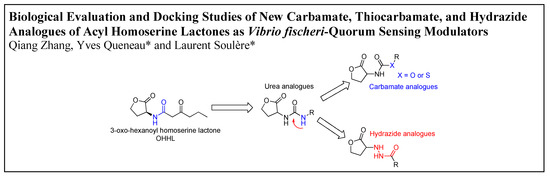
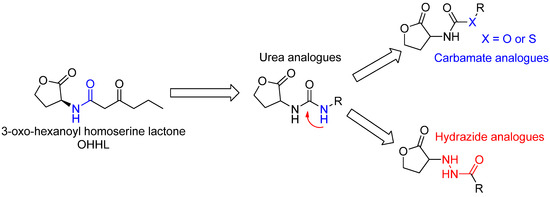
Biological Evaluation and Docking Studies of New Carbamate, Thiocarbamate, and Hydrazide Analogues of Acyl Homoserine Lactones as Vibrio fischeri-Quorum Sensing Modulators
by
Qiang Zhang, Yves Queneau and Laurent Soulère
Cited by 3 | Viewed by 2848
Abstract
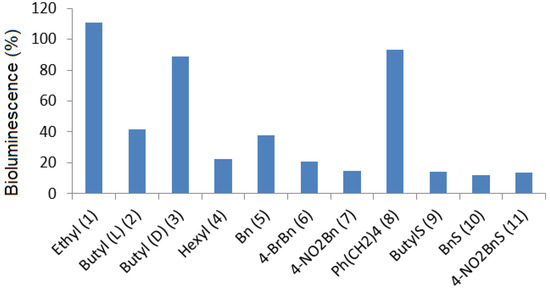
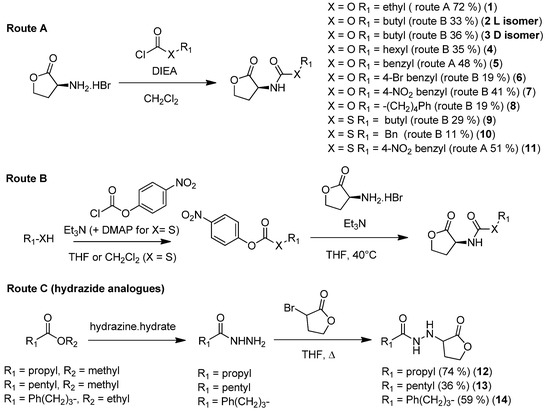
A series of carbamate, thiocarbamate, and hydrazide analogues of acylhomoserine lactones (AHLs) were synthesized and their ability to modulate
Vibrio fischeri-quorum sensing was evaluated. The compounds in the series exhibit variable side chain length and the possible presence of a diversely substituted
[...] Read more.
A series of carbamate, thiocarbamate, and hydrazide analogues of acylhomoserine lactones (AHLs) were synthesized and their ability to modulate
Vibrio fischeri-quorum sensing was evaluated. The compounds in the series exhibit variable side chain length and the possible presence of a diversely substituted phenyl substituent. Biological evaluation on the
Vibrio fischeri quorum sensing system revealed that the ethyl substituted carbamate (
1) display a weak agonistic activity whereas compounds with longer chain length or benzyl substituents display significant antagonistic activity. The most active compounds in the series were the 4-nitrobenzyl carbamate and thiocarbamate
7 and
11 which exhibited an IC
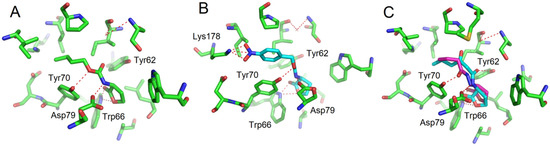
50 value of about 20 µM. These activities are in the range of other reported of AHL-structurally related quorum sensing (QS) inhibitors. Docking experiments conducted on the LuxR model showed that, compared to the natural ligand OHHL, the additional heteroatom of the carbamate group induces a new hydrogen bond with Tyr70 leading to a different global hydrogen-bond network. Tyr70 is an important residue in the binding site and is strictly conserved in the LuxR family. For the 4-nitrobenzyl carbamate and thiocarbamate analogues, the docking results highlight an additional hydrogen bond between the nitro group and Lys178. For hydrazide analogues, which are deprived of any activity, docking shows that the orientation of the carbonyl group is opposite as compared with the natural ligand, leading to the absence of a H-bond between the C=O with Tyr62. This suggests that, either this later interaction, or the influence of the C=O orientation on the overall ligand conformation, are essential for the biological activity.
Full article
►▼
Show Figures



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}