
The Neurokinin-1 Receptor: A Promising Antitumor Target




Abstract
:1. Introduction
2. Neurokinin-1 Receptor and Cancer
2.1. Neurokinin-1 Receptor: General Findings
2.2. Substance P and Neurokinin-1 Receptor Antagonists
2.3. Neurokinin-1 Receptor Isoforms and Cancer
2.4. The Substance P/Neurokinin-1 Receptor System and Cancer: Key Points
2.5. The Substance P/Neurokinin-1 Receptor System as a Cancer Predictive Factor
2.6. The Neurokinin-1 Receptor Is Crucial for the Viability of Cancer Cells
2.7. Neurokinin-1 Receptor and EGFR, Akt, and HER-2
2.8. Neurokinin-1 Receptor and the Warburg Effect
2.9. Neurokinin-1 Receptor Isoforms Balance
2.10. Neurokinin-1 Receptor and Inflammation
2.11. Neurokinin-1 Receptor, Metastasis, and Angiogenesis
2.12. Neurokinin-1 Receptor and Substance P Located in the Nucleus of Cancer Cells
2.13. Neurokinin-1 Receptor and Nerve Terminals
2.14. Substance P: Contradictory Findings
3. Antitumor Strategies Targeting the Neurokinin-1 Receptor
4. Neurokinin-1 Receptor Antagonists: Limitations and Challenges
5. Conclusions and Future Directions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Robinson, P.; Coveñas, R.; Muñoz, M. Combination therapy of chemotherapy or radiotherapy and the neurokinin-1 receptor antagonist aprepitant: A new antitumor strategy? Curr. Med. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Ilmer, M.; Kappler, R.; Berger, M. Therapeutic innovations for targeting hepatoblastoma. Anticancer Res. 2016, 36, 5577–5592. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Ward, R.; Barton, M. The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin. Oncol. 2004, 16, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B. The war on cancer. Lancet 1996, 347, 1377–1381. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Coveñas, R. The galaninergic system: A target for cancer treatment. Cancers 2022, 14, 3755. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Coveñas, R. The neurotensinergic system: A target for cancer treatment. Curr. Med. Chem. 2022, 29, 3221–3260. [Google Scholar] [CrossRef]
- Kastin, A.J. Handbook of Biologically Active Peptides, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Colucci, R.; Blandizzi, C.; Ghisu, N.; Florio, T.; Del Tacca, M. Somatostatin inhibits colon cancer cell growth through cyclooxygenase-2 downregulation. Br. J. Pharmacol. 2008, 155, 198–209. [Google Scholar] [CrossRef]
- Coveñas, R.; Muñoz, M. Involvement of the substance P/neurokinin-1 receptor system in cancer. Cancers 2022, 14, 3539. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. The neurokinin-1 receptor antagonist aprepitant, a new drug for the treatment of hematological malignancies: Focus on acute myeloid leukemia. J. Clin. Med. 2020, 9, 1659. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Mirzavi, F.; Aghaee-Bakhtiari, S.H.; Hashemy, S.I. SP/NK1R system regulates carcinogenesis in prostate cancer: Shedding light on the antitumoral function of aprepitant. Biochim. Biophys. Acta Mol. Cell. Res. 2022, 1869, 119221. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Neurokinin receptor antagonism: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Kolorz, J.; Demir, S.; Gottschlich, A.; Beirith, I.; Ilmer, M.; Lüthy, D.; Walz, C.; Dorostkar, M.M.; Magg, T.; Hauck, F.; et al. The neurokinin-1 receptor is a target in pediatric rhabdoid tumors. Curr. Oncol. 2021, 29, 94–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-W.; Li, L.; Hu, W.-Q.; Hu, M.-N.; Tao, Y.; Hu, H.; Miao, X.-K.; Yang, W.-L.; Zhu, Q.; Mou, L.-Y. Neurokinin-1 receptor promotes non-small cell lung cancer progression through transactivation of EGFR. Cell Death Dis. 2022, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.F.; Argüelles, S.; Rosso, M.; Medina, R.; Coveñas, R.; Ayala, A.; Muñoz, M. The neurokinin-1 receptor is essential for the viability of human glioma cells: A possible target for treating glioblastoma. Biomed. Res. Int. 2022, 2022, 6291504. [Google Scholar] [CrossRef] [PubMed]
- González-Moles, M.A.; Ramos-García, P.; Esteban, F. Significance of the overexpression of substance P and its receptor NK-1R in head and neck carcinogenesis: A systematic review and meta-analysis. Cancers 2021, 13, 1349. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Coveñas, R. Neurokinin-1 receptor. In Encyclopedia of Signaling Molecules, 2nd ed.; Choi, S., Ed.; Springer: Cham, Switzerland, 2018; pp. 3437–3445. [Google Scholar]
- Muñoz, M.; Coveñas, R.; Substance, P. Encyclopedia of Endocrine Diseases, 2nd ed.; Huhtaniemi, I., Martini, L., Eds.; Academic Press: Oxford, UK, 2019; Volume 1, pp. 571–578. [Google Scholar]
- dSNP (Single Nucleotide Polymorphism Database). Single Nucleotide Polymorphisms. 2020. Available online: https://www-ncbi-nlm-nih-gov.ezproxy.usal.es/snp/ (accessed on 20 September 2022).
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef]
- Xue, L.; Sun, Q.; Zhao, H.; Rovira, X.; Gai, S.; He, Q.; Pin, J.-P.; Liu, J.; Rondard, P. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 2019, 10, 2765. [Google Scholar] [CrossRef] [Green Version]
- Meyer, B.H.; Segura, J.M.; Martinez, K.L.; Hovius, R.; George, N.; Johnsson, K.; Vogel, H. FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2138–2143. [Google Scholar] [CrossRef] [Green Version]
- Terpager, M.; Scholl, D.J.; Kubale, V.; Martini, L.; Elling, C.E.; Schwartz, T.W. Construction of covalently coupled, concatameric dimers of 7TM receptors. J. Recept. Signal Transduct. Res. 2009, 29, 235–245. [Google Scholar] [CrossRef]
- GPCR Database. 2022. Available online: https://gpcrdb.org/protein/nk1r_human (accessed on 20 September 2022).
- Muñoz, M.; Coveñas, R. Neurokinin/Tachykin receptors. In Encyclopedia of Molecular Pharmacology, 3rd ed.; Offermanns, S., Rosenthal, W., Eds.; Springer: Cham, Switzerland, 2021; pp. 1093–1103. [Google Scholar]
- Roush, E.D.; Kwatra, M.M. Human substance P receptor expressed in Chinese hamster ovary cells directly activates g (alpha q/11), g (alpha s), g (alpha o). FEBS Lett. 1998, 428, 291–294. [Google Scholar] [CrossRef]
- Mitsuhashi, M.; Ohashi, Y.; Shichijo, S.; Christian, C.; Sudduth-Klinger, J.; Harrowe, G.; Payan, D.G. Multiple intracellular signaling pathways of the neuropeptide substance P receptor. J. Neurosci. Res. 1992, 32, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Sagan, S.; Chassaing, G.; Pradier, L.; Lavielle, S. Tachykinin peptides affect differently the second messenger pathways after binding to CHO-expressed human NK-1 receptors. J. Pharmacol. Exp. Ther. 1996, 276, 1039–1048. [Google Scholar] [PubMed]
- Takeda, Y.; Blount, P.; Sachais, B.S.; Hershey, A.D.; Raddatz, R.; Krause, J.E. Ligand binding kinetics of substance P and neurokinin A receptors stably expressed in Chinese hamster ovary cells and evidence for differential stimulation of inositol 1, 4, 5 trisphosphate and cyclic amp second messenger responses. J. Neurochem. 1992, 59, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Ramkissoon, S.H.; Patel, H.J.; Taborga, M.; Rameshwar, P. G protein-coupled receptors in haematopoietic disruption. Expert Opin. Biol. Ther. 2006, 6, 109–120. [Google Scholar] [CrossRef]
- Nakajima, Y.; Tsuchida, K.; Negishi, M.; Ito, S.; Nakanishi, S. Direct linkage of three tachykinin receptors to stimulation of both phosphatidylinositol hydrolysis and cyclic AMP cascades in transfected Chinese hamster ovary cells. J. Biol. Chem. 1992, 267, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Hastrup, H.; Raffetseder, U.; Martini, L.; Schwartz, T.W. Two active molecular phenotypes of the tachykinin NK1 receptor revealed by G-protein fusions and mutagenesis. J. Biol. Chem. 2001, 276, 19793–19799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khawaja, A.M.; Rogers, D.F. Tachykinins: Receptor to effector. Int. J. Biochem. Cell Biol. 1996, 28, 721–738. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Coveñas, R. Neurokinin-1 receptor antagonists against hepatoblastoma. Cancers 2019, 11, 1258. [Google Scholar] [CrossRef] [Green Version]
- Harris, G.C.; Aston-Jones, G. Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome. Nature 1994, 371, 155–157. [Google Scholar] [CrossRef]
- PDB. Protein Data Bank (PDB). Available online: https://pdb101.rcsb.org (accessed on 20 September 2022).
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: A modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Li, J.; Zeng, Q.; Zhang, Y.; Li, X.; Hu, H.; Miao, X.; Yang, W.; Zhang, W.; Song, X.; Mou, L.; et al. Neurokinin-1 receptor mediated breast cancer cell migration by increased expression of MMP-2 and MMP-14. Eur. J. Cell Biol. 2016, 95, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Li, J.; Hu, H.; Miao, X.; Song, X.; Yang, W.; Zeng, Q.; Mou, L.; Wang, R. Human hemokinin-1 promotes migration of melanoma cells and increases MMP-2 and MT1-MMP expression by activating tumor cell NK1 receptors. Peptides 2016, 83, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Kang, Y.; Zhou, Y.; Zeng, Q.; Song, H.; Wang, R. Neurokinin-1 receptor directly mediates glioma cell migration by up-regulation of matrix metalloproteinase-2 (MMP-2) and membrane type 1-matrix metalloproteinase (MT1-MMP). J. Biol. Chem. 2013, 288, 306–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.; Paige, C.J. Hemokinin-1 has substance P-like function in U-251 MG astrocytoma cells. A pharmacological and functional study. J. Neuroimmunol. 2005, 164, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014, 46, 1727–1750. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Martínez-Armesto, J.; Coveñas, R. NK-1 receptor antagonists as antitumor drugs: A survey of the literature from 2000 to 2011. Expert Opin. Ther. Pat. 2012, 22, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Rost, K.; Fleischer, F.; Nieber, K. Neurokinin-1 receptor antagonists: Between hope and disappointment. Med. Monatsschr. Pharm. 2006, 29, 200–205. [Google Scholar]
- Kramer, M.S.; Cutler, N.; Feighner, J.; Shrivastava, R.; Carman, J.; Sramek, J.J.; Reines, S.A.; Liu, G.; Snavely, D.; Wyatt-Knowles, E.; et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 1998, 281, 1640–1645. [Google Scholar] [CrossRef]
- Keller, M.; Montgomery, S.; Ball, W.; Morrison, M.; Snavely, D.; Liu, G.; Hargreaves, R.; Hietala, J.; Lines, C.; Beebe, K.; et al. Lack of efficacy of the substance P (neurokinin 1 receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol. Psychiatry 2006, 59, 216–223. [Google Scholar] [CrossRef]
- Deng, X.T.; Tang, S.M.; Wu, P.Y.; Li, Q.P.; Ge, X.X.; Xu, B.M.; Wang, H.-S.; Miao, L. SP/NK-1R promotes gallbladder cancer cell proliferation and migration. J. Cell. Mol. Med. 2019, 23, 7961–7973. [Google Scholar] [CrossRef] [Green Version]
- Friess, H.; Zhu, Z.; Liard, V.; Shi, X.; Shrikhande, S.V.; Wang, L.; Lieb, K.; Korc, M.; Palma, C.; Zimmermann, A.; et al. Neurokinin-1 receptor expression and its potential effects on tumor growth in human pancreatic cancer. Lab. Investig. 2003, 83, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Nizam, E.; Erin, N. Differential consequences of neurokinin receptor 1 and 2 antagonists in metastatic breast carcinoma cells; effects independent of substance P. Biomed. Pharmacother. 2018, 108, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.M. The role of human neutrophil elastase in the development of human emphysema. Zhonghua Jie He He Hu Xi Za Zhi 1989, 12, 347–349. [Google Scholar] [PubMed]
- Mohammadi, F.; Javid, H.; Afshari, A.R.; Mashkani, B.; Hashemy, S.I. Substance P accelerates the progression of human esophageal squamous cell carcinoma via MMP-2, MMP-9, VEGF-A, and VEGFR1 overexpression. Mol. Biol. Rep. 2020, 47, 4263–4272. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.; Javid, H.; Alaei, A.; Hashemy, S.I. New insight into the role of substance P/neurokinin-1 receptor system in breast cancer progression and its crosstalk with microRNAs. Clin. Genet. 2020, 98, 322–330. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Dong, D.; Wang, Z.; Ji, W.; Yu, M.; Zhang, F.; Niu, R.; Zhou, Y. MiR-34b/c-5p and the neurokinin-1 receptor regulate breast cancer cell proliferation and apoptosis. Cell Prolif. 2019, 52, e12527. [Google Scholar] [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar] [PubMed]
- Zhou, Y.; Zhao, L.; Xiong, T.; Chen, X.; Zhang, Y.; Yu, M.; Yang, J.; Yao, Z. Roles of full-length and truncated neurokinin-1 receptors on tumor progression and distant metastasis in human breast cancer. Breast Cancer Res. Treat. 2013, 140, 49–61. [Google Scholar] [CrossRef]
- Lai, J.P.; Lai, S.; Tuluc, F.; Tansky, M.F.; Kilpatrick, L.E.; Leeman, S.E.; Douglas, S.D. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 12605–12610. [Google Scholar] [CrossRef] [Green Version]
- Simeonidis, S.; Castagliuolo, I.; Pan, A.; Liu, J.; Wang, C.C.; Mykoniatis, A.; Pasha, A.; Valenick, L.; Sougioultzis, S.; Zhao, D.; et al. Regulation of the NK-1 receptor gene expression in human macrophage cells via an NF-kappa B site on its promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 2957–2962. [Google Scholar] [CrossRef] [Green Version]
- Goode, T.; O’Connor, T.; Hopkins, A.; Moriarty, D.; O’Sullivan, G.C.; Collins, J.K.; O’Donoghue, D.; Baird, A.W.; O’Connell, J.; Shanahan, F. Neurokinin-1 receptor (NK-1R) expression is induced in human colonic epithelial cells by pro-inflammatory cytokines and mediates proliferation in response to substance P. J. Cell. Physiol. 2003, 197, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.; Muñoz, M.; Berger, M. The role of the neurokinin-1 receptor in the microenvironment of inflammation and cancer. Sci. World J. 2012, 2012, 381434. [Google Scholar] [CrossRef] [Green Version]
- Spitsin, S.; Pappa, V.; Douglas, S.D. Truncation of neurokinin-1 receptor—Negative regulation of substance P signaling. J. Leukoc. Biol. 2018, 103, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Neth, O.; Ilmer, M.; Garnier, A.; Salinas-Martín, M.V.; de Agustin Asencio, J.C.; von Schweinitz, D.; Kappler, R.; Muñoz, M. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J. Hepatol. 2014, 60, 985–994. [Google Scholar] [CrossRef]
- Singh, D.; Joshi, D.D.; Hameed, M.; Qian, J.; Gascón, P.; Maloof, P.B.; Mosenthal, A.; Rameshwar, P. Increased expression of preprotachykinin-I and neurokinin receptors in human breast cancer cells: Implications for bone marrow metastasis. Proc. Natl. Acad. Sci. USA 2000, 97, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Mozafari, M.; Ebrahimi, S.; Darban, R.A.; Hashemy, S.I. Potential in vitro therapeutic effects of targeting SP/NK1R system in cervical cancer. Mol. Biol. Rep. 2021, 49, 1067–1076. [Google Scholar] [CrossRef]
- Douglas, S.D.; Leeman, S.E. Neurokinin-1 receptor: Functional significance in the immune system in reference to selected infections and inflammation. Ann. N. Y. Acad. Sci. 2011, 1217, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.G.; Odersky, A.; Szymansky, A.; Seiler, M.; Althoff, K.; Beckers, A.; Speleman, F.; Schäfers, S.; De Preter, K.; Astrahanseff, K.; et al. Targeting tachykinin receptors in neuroblastoma. Oncotarget 2017, 8, 430–443. [Google Scholar] [CrossRef] [Green Version]
- Pohl, A.; Kappler, R.; Muhlig, J.; von Schweinitz, D.; Berger, M. Expression of truncated neurokinin-1 receptor in childhood neuroblastoma is independent of tumor biology and stage. Anticancer Res. 2017, 37, 6079–6085. [Google Scholar] [CrossRef]
- Harford-Wright, E.; Lewis, K.M.; Vink, R.; Ghabriel, M.N. Evaluating the role of substance P in the growth of brain tumors. Neuroscience 2014, 261, 85–94. [Google Scholar] [CrossRef]
- Golestaneh, M.; Firoozrai, M.; Javid, H.; Hashemy, S.I. The substance P/neurokinin-1 receptor signaling pathway mediates metastasis in human colorectal SW480 cancer cells. Mol. Biol. Rep. 2022, 49, 4893–4900. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Wang, N.; Zhang, R.; Dong, D.; Liu, R.; Zhang, L.; Ji, W.; Yu, M.; Zhang, F.; et al. TGFβ regulates NK1R-Tr to affect the proliferation and apoptosis of breast cancer cells. Life Sci. 2020, 256, 117674. [Google Scholar] [CrossRef]
- Beirith, I.; Renz, B.W.; Mudusetti, S.; Ring, N.S.; Kolorz, J.; Koch, D.; Bazhin, A.V.; Berger, M.; Wang, J.; Angele, M.K.; et al. Identification of the neurokinin-1 receptor as targetable stratification factor for drug repurposing in pancreatic cancer. Cancers 2021, 13, 2703. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F.D.; Coveñas, R. The neurokinin-1 receptor: Structure dynamics and signaling. Receptors 2022, 1, 54–71. [Google Scholar] [CrossRef]
- Wang, F.; Liu, S.; Liu, J.; Feng, F.; Guo, Y.; Zhang, W.; Zheng, G.; Wang, Q.; Cai, L.; Guo, M.; et al. SP promotes cell proliferation in esophageal squamous cell carcinoma through the NK1R/Hes1 axis. Biochem. Biophys. Res. Commun. 2019, 514, 1210–1216. [Google Scholar] [CrossRef]
- Davoodian, M.; Boroumand, N.; Mehrabi Bahar, M.; Jafarian, A.H.; Asadi, M.; Hashemy, S.I. Evaluation of serum level of substance P and tissue distribution of NK-1 receptor in breast cancer. Mol. Biol. Rep. 2019, 46, 1285–1293. [Google Scholar] [CrossRef]
- Hennig, I.M.; Laissue, J.A.; Horisberger, U.; Reubi, J.C. Substance-P receptors in human primary neoplasms: Tumoral and vascular localization. Int. J. Cancer 1995, 61, 786–792. [Google Scholar] [CrossRef]
- Gharaee, N.; Pourali, L.; Jafarian, A.H.; Hashemy, S.I. Evaluation of serum level of substance P and tissue distribution of NK-1 receptor in endometrial cancer. Mol. Biol. Rep. 2018, 45, 2257–2262. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L.; Pacini, M.; Geppetti, P.; Alessandri, G.; Maggi, C.A. Substance P stimulates neovascularization in vivo and proliferation of cultured endothelial cells. Microvasc. Res. 1990, 40, 264–278. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. The neurokinin-1 receptor antagonist aprepitant: An intelligent bullet against cancer? Cancers 2020, 12, 2682. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.W.; Zhao, D.; Zhan, Y.; Moyer, M.P.; Pothoulakis, C. Substance P mediates antiapoptotic responses in human colonocytes by Akt activation. Proc. Natl. Acad. Sci. USA 2007, 104, 2013–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulenwider, H.D.; Smith, B.M.; Nichenko, A.S.; Carpenter, J.M.; Nennig, S.E.; Cheng, K.; Rice, K.C.; Schank, J.R. Cellular and behavioral effects of lipopolysaccharide treatment are dependent upon neurokinin-1 receptor activation. J. Neuroinflammation 2018, 15, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak-Drzewiecka, A.; Ratajewski, M.; Wagner, W.; Dastych, J. HIF-1alpha is up-regulated in activated mast cells by a process that involves calcineurin and NFAT. J. Immunol. 2008, 181, 1665–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, M.; Rosso, M. The NK-1 receptor antagonist aprepitant as a broad-spectrum antitumor drug. Investig. New Drugs 2010, 28, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Rosso, M.; Robles-Frías, M.J.; Salinas-Martín, M.V.; Coveñas, R. Immunolocalization of the neurokinin-1 receptor: A new target in the treatment of human malignant melanoma. Lab. Investig. 2010, 90, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Coveñas, R. Safety of neurokinin-1 receptor antagonists. Expert Opin. Drug Saf. 2013, 12, 673–685. [Google Scholar] [CrossRef]
- Mehboob, R.; Kurdi, M.; Baeesa, S.; Fawzy Halawa, T.; Tanvir, I.; Maghrabi, Y.; Hakamy, S.; Saeedi, R.; Moshref, R.; Nasief, H.; et al. Immunolocalization of neurokinin 1 receptor in WHO grade 4 astrocytomas, oral squamous cell, and urothelial carcinoma. Folia Neuropathol. 2022, 60, 165–176. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Coveñas, R. A new frontier in the treatment of cancer: NK-1 receptor antagonists. Curr. Med. Chem. 2010, 17, 504–516. [Google Scholar] [CrossRef]
- Li, Y.; Douglas, S.D.; Ho, W. Human stem cells express substance P gene and its receptor. J. Hematother. Stem Cell Res. 2000, 9, 445–452. [Google Scholar] [CrossRef]
- Dubon, M.J.; Park, K.S. Substance P enhances the proliferation and migration potential of murine bone marrow-derived mesenchymal stem cell-like cell lines. Exp. Ther. Med. 2015, 9, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Kim, H.J.; Cho, D.C.; Bae, J.S.; Park, S.W. Substance P stimulates proliferation of spinal neural stem cells in spinal cord injury via the mitogen-activated protein kinase signaling pathway. Spine J. 2015, 15, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Vykoukal, J.; Hubertus, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M.; Ilmer, M. Targeting the neurokinin-1 receptor inhibits growth of human colon cancer cells. Int. J. Oncol. 2015, 47, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, M.; Coveñas, R. Neurokinin-1 receptor antagonists as anticancer drugs. Lett. Drug Des. Discov. 2019, 16, 1110–1129. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, M.; Tong, Y.; Liu, X.; Zhang, L.; Dong, D.; Shao, J.; Zhou, Y. miR-206 promotes cancer progression by targeting full-length neurokinin-1 receptor in breast cancer. Technol. Cancer Res. Treat. 2019, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Castro, T.A.; Cohen, M.C.; Rameshwar, P. The expression of neurokinin-1 and preprotachykinin-1 in breast cancer cells depends on the relative degree of invasive and metastatic potential. Clin. Exp. Metastasis 2005, 22, 621–628. [Google Scholar] [CrossRef]
- Chen, X.Y.; Ru, G.Q.; Ma, Y.Y.; Xie, J.; Chen, W.Y.; Wang, H.J.; Wang, S.B.; Li, L.; Jin, K.T.; He, X.L.; et al. High expression of substance P and its receptor neurokinin-1 receptor in colorectal cancer is associated with tumor progression and prognosis. OncoTargets Ther. 2016, 9, 3595–3602. [Google Scholar] [CrossRef] [Green Version]
- Garnier, A.; Ilmer, M.; Becker, K.; Häberle, B.; von Schweinitz, D.; Kappler, R.; Berger, M. Truncated neurokinin-1 receptor is an ubiquitous antitumor target in hepatoblastoma, and its expression is independent of tumor biology and stage. Oncol. Lett. 2016, 11, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Feng, F.; Xu, G.; Zhang, H.; Hong, L.; Yang, J. Elevated SP/NK-1R in esophageal carcinoma promotes esophageal carcinoma cell proliferation and migration. Gene 2015, 560, 205–210. [Google Scholar] [CrossRef]
- Feng, F.; Yang, J.; Tong, L.; Yuan, S.; Tian, Y.; Hong, L.; Wang, W.; Zhang, H. Substance P immunoreactive nerve fibres are related to gastric cancer differentiation status and could promote proliferation and migration of gastric cancer cells. Cell Biol. Int. 2011, 35, 623–629. [Google Scholar] [CrossRef]
- González-Moles, M.A.; Brener, S.; Ruiz-Avila, I.; Gil-Montoya, J.A.; Tostes, D.; Bravo, M.; Esteban, F. Substance P and NK-1R expression in oral precancerous epithelium. Oncol. Rep. 2009, 22, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Al-Keilani, M.S.; Elstaty, R.I.; Alqudah, M.A.; Alkhateeb, A.M. Immunohistochemical expression of substance P in breast cancer and its association with prognostic parameters and Ki-67 index. PLoS ONE 2021, 16, e0252616. [Google Scholar] [CrossRef]
- Al-Keilani, M.S.; Elstaty, R.I.; Alqudah, M.A. The prognostic potential of neurokinin 1 receptor in breast cancer and its relationship with Ki-67 index. Int. J. Breast Cancer 2022, 2022, 4987912. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Glioma, and neurokinin-1 receptor antagonists: A new therapeutic approach. Anticancer Agents Med. Chem. 2020, 19, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R.; Esteban, F.; Redondo, M. The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J. Biosci. 2015, 40, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, S.; Boada, M.D. Neuropeptide-induced modulation of carcinogenesis in a metastatic breast cancer cell line (MDA-MB-231LUC+). Cancer Cell Int. 2018, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- García-Recio, S.; Fuster, G.; Fernández-Nogueira, P.; Pastor-Arroyo, E.M.; Park, S.Y.; Mayordomo, C.; Ametller, E.; Mancino, M.; González-Farré, X.; Russnes, H. Substance P autocrine signaling contributes to persistent HER2 activation that drives malignant progression and drug resistance in breast cancer. Cancer Res. 2013, 73, 6424–6434. [Google Scholar] [CrossRef] [Green Version]
- Mayordomo, C.; García-Recio, S.; Ametller, E.; Fernández-Nogueira, P.; Pastor-Arroyo, E.M.; Vinyals, L.; Casas, I.; Gascón, P.; Almendro, V. Targeting of substance P induces cancer cell death and decreases the steady state of EGFR and Her2. J. Cell. Physiol. 2012, 227, 1358–1366. [Google Scholar] [CrossRef]
- Castagliuolo, I.; Valenick, L.; Liu, J.; Pothoulakis, C. Epidermal growth factor receptor transactivation mediates substance P-induced mitogenic responses in U-373 MG cells. J. Biol. Chem. 2000, 275, 26545–26550. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E. Glioblastoma: Synergy of growth promotion between CCL5 and NK-1R can be thwarted by blocking CCL5 with miraviroc, an FDA-approved anti-HIV drug, and blocking NK-1R with aprepitant, an FDA-approved anti-nausea drug. J. Clin. Pharm. Ther. 2010, 35, 657–663. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Li, X.F.; Yuan, G.Q.; Hu, H.; Song, X.Y.; Li, J.Y.; Miao, X.K.; Zhou, T.X.; Yang, W.L.; Zhang, X.W.; et al. β-Arrestin 1 has an essential role in neurokinin-1 receptor-mediated glioblastoma cell proliferation and G2/M phase transition. J. Biol. Chem. 2017, 292, 8933–8947. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; García-Recio, S.; Gascón, P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr. Drug Targets 2010, 11, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Richardson, M.D.; Bigner, D.D.; Kwatra, M.M. Signal transduction through substance P receptor in human glioblastoma cells: Roles for Src and PKCδ. Cancer Chem. Pharmacol. 2005, 56, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Esteban, F.; Ramos-García, P.; Muñoz, M.; González-Moles, M.A. Substance P and neurokinin 1 receptor in chronic inflammation and cancer of the head and neck: A review of the literature. Int. J. Environ. Res. Public Health 2021, 19, 375. [Google Scholar] [CrossRef] [PubMed]
- De Fea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Déry, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmann, S.; Seep, J.; Jonas, L. Delay of neutrophil apoptosis by the neuropeptide substance P: Involvement of caspase cascade. Peptides 2001, 22, 661–670. [Google Scholar] [CrossRef]
- Hartmann, W.; Küchler, J.; Koch, A.; Friedrichs, N.; Waha, A.; Endl, E.; Czerwitzki, J.; Metzger, D.; Steiner, S.; Wurst, P.; et al. Activation of phosphatidylinositol-30-kinase/AKT signaling is essential in hepatoblastoma survival. Clin. Cancer Res. 2009, 15, 4538–4545. [Google Scholar] [CrossRef] [Green Version]
- Breuleux, M.; Klopfenstein, M.; Stephan, C.; Doughty, C.A.; Barys, L.; Maira, S.M.; Kwiatkowski, D.; Lane, H.A. Increased AKT s473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther. 2009, 8, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/AKT signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, T.; Kwatra, S.G.; Goldsmith, L.E.; Richardson, M.D.; Cox, E.A.; Sampson, J.H.; Kwatra, M.M. A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J. Neurochem. 2009, 109, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Medrano, S.; Gruenstein, E.; Dimlich, R.V.W. Substance P receptors on human astrocytoma cells are linked to glycogen breakdown. Neurosci. Lett. 1994, 167, 14–18. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Fernández-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Liu, D.; Qiu, Z.; Huang, Y.; Chen, B.; Wang, L.; Xu, H.; Huang, N.; Liu, L.; Li, W. GSK3β overexpression indicates poor prognosis, and its inhibition reduces cell proliferation and survival of non-small cell lung cancer cells. PLoS ONE 2014, 9, e91231. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I. The transcription factor FOXM1c is activated by protein kinase CK2, protein kinase A (PKA), c-Src, and Raf-1. Biochem. Biophys. Res. Commun. 2011, 413, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Ogo, H.; Kuroyanagi, N.; Inoue, A.; Nishio, H.; Hirai, Y.; Akiyama, M.; DiMaggio, D.A.; Krause, J.E.; Nakata, Y. Human astrocytoma cells (U-87 MG) exhibit a specific substance P binding site with the characteristics of an NK-1 receptor. J. Neurochem. 1996, 67, 1813–1820. [Google Scholar] [CrossRef]
- Singh, S.; Kumaravel, S.; Dhole, S.; Roy, S.; Pavan, V.; Chakraborty, S. Neuropeptide substance P enhances inflammation-mediated tumor signaling pathways and migration and proliferation of head and neck cancers. Indian J. Surg. Oncol. 2021, 12, 93–102. [Google Scholar] [CrossRef]
- Ho, W.Z.; Kaufman, D.; Uvaydova, M.; Douglas, S.D. Substance P augments interleukin-10 and tumor necrosis factor-alpha release by human cord blood monocytes and macrophages. J. Neuroimmunol. 1996, 71, 73–80. [Google Scholar] [CrossRef]
- Weller, M.; Stevens, A.; Sommer, N.; Melms, A.; Dichgans, J.; Wiethölter, H. Comparative analysis of cytokine patterns in immunological, infectious, and oncological neurological disorders. J. Neurol. Sci. 1991, 104, 215–221. [Google Scholar] [CrossRef]
- Frei, K.; Piani, D.; Malipiero, U.V.; Van Meir, E.; de Tribolet, N.; Fontana, A. Granulocyte-macrophage colony-stimulating factor GM-CSF production by glioblastoma cells. Despite the presence of inducing signals, GM-CSF is not expressed in vivo. J. Immunol. 1992, 148, 3140–3146. [Google Scholar] [PubMed]
- Lotz, M.; Vaughan, J.H.; Carson, D.A. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science 1988, 241, 1218–1221. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Targeting NK-1 receptors to prevent and treat pancreatic cancer: A new therapeutic approach. Cancers 2015, 7, 1215–1232. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, E.; Leeman, S.E.; Watts, L.A.; Coukos, J.A.; O’Brien, M.J.; Cerda, S.R.; Farraye, F.A.; Stucchi, A.F.; Becker, J.M. Truncated neurokinin-1 receptor is increased in colonic epithelial cells from patients with colitis-associated cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 17420–17425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinos-Quintana, A.; Trujillo-Hacha, P.; Piruat, J.I.; Bejarano-García, J.A.; García-Guerrero, E.; Pérez-Simón, J.A.; Muñoz, M. Human acute myeloid leukemia cells express neurokinin-1 receptor, which is involved in the antileukemiceffect of neurokinin-1 receptor antagonists. Investig. New Drugs 2019, 37, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Downie, J.; Bayliss, J.; Stephenson, A.; Bluebond-Langner, M. Long-term daily administration of aprepitant for the management of intractable nausea and vomiting in children with life-limiting conditions: A case series. J. Pain Symptom Manag. 2021, 62, e225–e231. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Parrilla, J.; Rosso, M.; Coveñas, R. Antipruritic vs. antitumour action of aprepitant: A question of dose. Acta Derm. Venereol. 2019, 99, 620–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashash, D.; Safaroghli-Azar, A.; Bayati, S.; Razani, E.; Pourbagheri-Sigaroodi, A.; Gharehbaghian, A.; Momeny, M.; Sanjadi, M.; Rezaie-Tavirani, M.; Ghaffari, S.H. Neurokinin-1 receptor (NK1R) inhibition sensitizes APL cells to anti-tumor effect of arsenic trioxide via restriction of NF-κB axis: Shedding new light on resistance to aprepitant. Int. J. Biochem. Cell Biol. 2018, 103, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance, and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, E.; Pei, G.; Zhao, Z.; Kim, S.T.; German, A.; Robinson, P. Substance P antagonism as a novel therapeutic option to enhance efficacy of cisplatin in triple-negative breast cancer and protect PC12 cells against cisplatin-induced oxidative stress and apoptosis. Cancers 2021, 13, 5178. [Google Scholar] [CrossRef]
- Lee, Y.; Gude, N.A.; Thistlethwaite, P.A.; Sussman, M.A.; Gottlieb, R.A.; Gustafsson, Å.B. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation 2010, 121, 675–683. [Google Scholar] [CrossRef]
- Ilmer, M.; Garnier, A.; Vykoukal, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M. Targeting the neurokinin-1 receptor compromises canonical Wnt signaling in hepatoblastoma. Mol. Cancer Ther. 2015, 14, 2712. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Yuan, S.; Cheng, J.; Kang, S.; Zhao, W.; Zhang, J. Substance P promotes the progression of endometrial adenocarcinoma. Int. J. Gynecol. Cancer 2016, 26, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Drell, T.L., IV; Lindecke, A.; Niggemann, B.; Kaltschmidt, C.; Zaenker, K.S.; Entschladen, F. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int. J. Cancer 2004, 112, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, G.; Ma, Q.; Li, W.; Liu, J.; Han, L.; Duan, W.; Xu, Q.; Liu, H.; Wang, Z.; et al. Neurotransmitter substance P mediates pancreatic cancer perineural invasion via NK-1R in cancer cells. Mol. Cancer Res. 2013, 11, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, P.L.; Jiang, S.; Fu, Y.; Avraham, S.; Avraham, H.K. The proinflammatory peptide substance P promotes blood-brain barrier breaching by breast cancer cells through changes in microvascular endothelial cell tight junctions. Int. J. Cancer 2014, 134, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Rosso, M.; Coveñas, R. Triple-negative breast cancer: How neurokinin-1 receptor antagonists could be used as a new therapeutic approach. Mini Rev. Med. Chem. 2020, 20, 408–417. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Li, Y.; Wang, X.; Lu, Q.; Wang, D.; Qi, C.; Li, C.; Li, Z.; Lian, B.; et al. Combined anti-hepatocellular carcinoma therapy inhibit drug-resistance and metastasis via targeting “substance P-hepatic stellate cells-hepatocellular carcinoma” axis. Biomaterials 2021, 276, 121003. [Google Scholar] [CrossRef]
- Razmgah, M.M.; Ghahremanloo, A.; Javid, H.; AlAlikhan, A.; Afshari, A.-R.; Hashemy, S.I. The effect of substance P and its specific antagonist (aprepitant) on the expression of MMP-2, MMP-9, VEGF, and VEGFR in ovarian cancer cells. Mol. Biol. Rep. 2022, 49, 9307–9314. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Watnick, R.S. The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Bielenberg, D.R.; Lifshits, E.; Heymach, J.V.; Klagsbrun, M. Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc. Res. 2008, 76, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Sharif, T.R.; Sharif, M. Substance P-induced mitogenesis in human astrocytoma cells correlates with activation of the mitogen-activated protein kinase signaling pathway. Cancer Res. 1996, 1, 4983–4991. [Google Scholar] [PubMed]
- Ji, T.; Ma, K.; Wu, H.; Cao, T. A substance P (SP)/neurokinin-1 receptor axis promotes perineural invasion of pancreatic cancer and is affected by lncRNA LOC389641. J. Immunol. Res. 2022, 2022, 5582811. [Google Scholar] [CrossRef]
- Nagakawa, O.; Ogasawara, M.; Fujii, H.; Murakami, K.; Murata, J.; Fuse, H.; Saiki, I. Effect of prostatic neuropeptides on invasion and migration of PC-3 prostate cancer cells. Cancer Lett. 1998, 133, 27–33. [Google Scholar] [CrossRef]
- Tebas, P.; Spitsin, S.; Barrett, J.S.; Tuluc, F.; Elci, O.; Korelitz, J.J.; Wagner, W.; Winters, A.; Kim, D.; Catalano, R.; et al. Reduction of soluble CD163, substance P, programmed death 1 and inflammatory markers: Phase 1B trial of aprepitant in HIV-1-infected adults. AIDS 2015, 29, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Mehner, C.; Hockla, A.; Miller, E.; Ran, S.; Radisky, D.C.; Radisky, E.S. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple-negative breast cancer. Oncotarget 2014, 5, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Javid, H.; Asadi, J.; Zahedi Avval, F.; Afshari, A.R.; Hashemy, S.I. The role of substance P/neurokinin 1 receptor in the pathogenesis of esophageal squamous cell carcinoma through constitutively active PI3K/Akt/NF-κB signal transduction pathways. Mol. Biol. Rep. 2020, 47, 2253–2263. [Google Scholar] [CrossRef]
- Saito, H.; Yoshizawa, H.; Yoshimori, K.; Katakami, N.; Katsumata, N.; Kawahara, M.; Eguchi, K. Efficacy and safety of single-dose fosaprepitant in the prevention of chemotherapy-induced nausea and vomiting in patients receiving high-dose cisplatin: A multicentre, randomized, double-blind, placebo-controlled phase 3 trial. Ann. Oncol. 2013, 24, 1067–1073. [Google Scholar] [CrossRef]
- García-Recio, S.; Gascón, P. Biological and pharmacological aspects of the NK-1receptor. BioMed. Res. Int. 2015, 2015, 495704. [Google Scholar] [CrossRef] [Green Version]
- Girish, C.; Manikandan, S. Aprepitant: A substance P antagonist for chemotherapy-induced nausea and vomiting. Indian J. Cancer 2007, 44, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Crespo, J.C.; Crespo, J.P.; Coveñas, R. Neurokinin-1 receptor antagonist aprepitant, and radiotherapy, successful combination therapy in a patient with lung cancer: A case report. Mol. Clin. Oncol. 2019, 11, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tattersall, F.D.; Rycroft, W.; Cumberbatch, M.; Mason, G.; Tye, S.; Williamson, D.J.; Hale, J.J.; Mills, S.G.; Finke, P.E.; MacCoss, M.; et al. The novel NK-1 receptor antagonist MK-0869 (L-754,030) and its water-soluble phosphoryl prodrug, L-758,298, inhibit acute and delayed cisplatin-induced emesis in ferrets. Neuropharmacology 2000, 39, 652–663. [Google Scholar] [CrossRef]
- Majkowska-Pilip, A.; Halik, P.K.; Gniazdowska, E. The significance of NK1 receptor ligands and their application in targeted radionuclide tumor therapy. Pharmaceutics 2019, 11, 443. [Google Scholar] [CrossRef] [Green Version]
- Halik, P.K.; Lipiński, P.F.J.; Matalińska, J.; Koźmiński, P.; Misicka, A.; Gniazdowska, E. Radiochemical synthesis, and evaluation of novel radio conjugates of neurokinin 1 receptor antagonist aprepitant dedicated for NK-1R-positive tumors. Molecules 2020, 25, 3756. [Google Scholar] [CrossRef]
- Sooho, Y.; Jieun, A.; Changhee, P.; Dohyun, K.; Jaehwi, L. Design, and characterization of phosphatidylcholine-based solid dispersions of aprepitant for enhanced solubility and dissolution. Pharmaceutics 2020, 12, 407. [Google Scholar] [CrossRef]
- Olver, I.; Shelukar, S.; Thompson, K.C. Nanomedicines in the treatment of emesis during chemotherapy: Focus on aprepitant. Int. J. Nanomed. 2007, 2, 13–18. [Google Scholar] [CrossRef]
- Muñoz, M.; Berger, M.; Rosso, M.; González-Ortega, A.; Carranza, A.; Coveñas, R. Antitumor activity of neurokinin-1 receptor antagonists in MG-63 human osteosarcoma xenografts. Int. J. Oncol. 2014, 44, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Legi, A.; Rodriguez, E.; Eckols, T.K.; Mistry, C.; Robinson, P. Substance P antagonism prevents chemotherapy-induced cardiotoxicity. Cancers 2021, 13, 1732. [Google Scholar] [CrossRef]
- García-Aranda, M.; Téllez, T.; McKenna, L.; Redondo, M. Neurokinin-1 receptor (NK-1R) antagonists as a new strategy to overcome cancer resistance. Cancers 2022, 14, 2255. [Google Scholar] [CrossRef]
- Castro-Obregón, S.; Rao, R.V.; del Rio, G.; Chen, S.F.; Poksay, K.S.; Rabizadeh, S.; Vesce, S.; Zhang, X.; Swanson, R.A.; Bredesen, D.E. Alternative, nonapoptotic programmed cell death: Mediation by arrestin 2, ERK2, and Nur77. J. Biol. Chem. 2004, 279, 17543–17553. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.E.; Chung, E.; Son, Y. A neuropeptide, Substance-P, directly induces tissue-repairing M2-like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNγ. Sci. Rep. 2017, 7, 9417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Wang, X.; Meng, Y.; Ma, J.; Zhang, Q.; Wang, G.S.L.; Cheng, X.; Hong, X.; Wang, Y.; Yan, Z.; et al. A novel mechanism of endoplasmic reticulum stress- and c-myc-degradation-mediated therapeutic benefits of antineurokinin-1 receptor drugs in colorectal cancer. Adv. Sci. 2021, 8, e2101936. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Huang, H.; Huang, F.; Yang, T.; Zhang, T.; Wu, H.; Zhou, H.; Chen, Q.; Shi, Y.; Sun, Y.; et al. Neurokinin-1 receptor is an effective oxidative stress through mitochondrial calcium overload. Proc. Natl. Acad. Sci. USA 2019, 116, 19635–19645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javid, H.; Mohammadi, F.; Zahiri, E.; Hashemy, S.I. The emerging role of substance P/neurokinin-1 receptor signaling pathways in growth and development of tumor cells. J. Physiol. Biochem. 2019, 75, 415–421. [Google Scholar] [CrossRef]
- Bayati, S.; Razani, E.; Bashash, D.; Safaroghli-Azar, A.; Safa, M.; Ghaffari, S.H. Antileukemic effects of neurokinin-1 receptor inhibition on hematologic malignant cells: A novel therapeutic potential for aprepitant. Anticancer Drugs 2018, 29, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.S.; Kim, S.E.; Jeon, S.H.; Lee, J.S.; Choi, K.Y. Both ERK and Wnt/beta-catenin pathways are involved in Wnt3a-induced proliferation. J. Cell Sci. 2005, 118, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Javid, H.; Afshari, A.-R.; Zahedi Avva, F.; Asadi, J.; Hashemy, S.I. Aprepitant promotes caspase-dependent apoptotic cell death and G2/M arrest through PI3K/Akt/NF- κ B axis in cancer stem-like esophageal squamous cell carcinoma spheres. Biomed. Res. Int. 2021, 2021, 8808214. [Google Scholar] [CrossRef]
- Fu, S.; Jin, D.; Liu, S.; Wang, L.; Wang, Z.; Mei, G.; Zou, Z.L.; Wu, J.Q.; Xu, Z.Y. Protective effect of neuropeptide substance P on bone marrow mesenchymal stem cells against apoptosis induced by serum deprivation. Stem Cells Int. 2015, 2015, 270328. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ramena, G.; Elble, R.C. The role of cancer stem cells in relapse of solid tumors. Front. Biosci. 2012, 4, 1528–1541. [Google Scholar] [CrossRef]
- Korfi, F.; Javid, H.; Darban, R.A.; Hashemy, S.I. The effect of SP/NK1R on the expression and activity of catalase and superoxide dismutase in glioblastoma cancer cells. Biochem. Res. Int. 2021, 2021, 6620708. [Google Scholar] [CrossRef] [PubMed]
- Ghahremani, F.; Sabbaghzadeh, R.; Ebrahimi, S.; Javid, H.; Ghahremani, J.; Hashemy, S.I. Pathogenic role of the SP/NK1R system in GBM cells through inhibiting the thioredoxin system. Iran J. Basic Med. Sci. 2021, 24, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.; Darban, R.A.; Javid, H.; Hashemy, S.I. The therapeutic potential of aprepitant in glioblastoma cancer cells through redox modification. BioMed. Res. Int. 2022, 2022, 8540403. [Google Scholar] [CrossRef]
- Muñoz, M.; González-Ortega, A.; Coveñas, R. The NK-1 receptor is expressed in human leukemia and is involved in the antitumor action of aprepitant and other NK-1 receptor antagonists on acute lymphoblastic leukemia cell lines. Investig. New Drugs 2012, 30, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Karayama, M.; Inui, N.; Kuroishi, S.; Nakano, H.; Nakamura, Y.; Yokomura, K.; Toyoshima, M.; Shirai, T.; Masuda, M.; et al. Efficacy and safety of high dose aprepitant treatment in patients with advanced non-small cell lung cancer. Lung Cancer 2014, 84, 259–264. [Google Scholar] [CrossRef]
- Robinson, P.; Kasembeli, M.; Bharadwaj, U.; Engineer, N.; Eckols, K.T.; Tweardy, D.J. Substance P receptor signaling mediates doxorubicin-induced cardiomyocyte apoptosis and triple-negative breast cancer chemoresistance. BioMed. Res. Int. 2016, 2016, 1959270. [Google Scholar] [CrossRef] [Green Version]
- Un, H.; Ugan, R.A.; Kose, D.; Bayir, Y.; Cadirci, E.; Selli, J.; Halici, Z. A novel effect of aprepitant: Protection for cisplatin-induced nephrotoxicity and hepatotoxicity. Eur. J. Pharmacol. 2020, 880, 173168. [Google Scholar] [CrossRef]
- Kitchens, C.A.; McDonald, P.R.; Pollack, I.F.; Wipf, P.; Lazo, J.S. Synergy between microtubule destabilizing agents and neurokinin 1 receptor antagonists identified by an siRNA synthetic lethal screen. FASEB J. 2009, 23, 756.13. [Google Scholar] [CrossRef]
- Alfieri, A.B.; Cubeddu, L.X. Efectos de los antagonistas de los receptores NK1 y de la dexametasona sobre la inflamación neurogénica inducida por ciclofosfamida y por radiación X, en la rata. Arch. Venez. Farmacol. Ter. 2004, 23, 61–66. [Google Scholar]
- Alfieri, A.B.; Cubeddu, L.X. Role of NK1 receptors on cisplatin-induced nephrotoxicity in the rat. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 361, 334–338. [Google Scholar] [CrossRef]
- Muñoz, M.; González-Ortega, A.; Salinas-Martín, M.V.; Carranza, A.; García-Recio, S.; Almendro, V.; Coveñas, R. The neurokinin-1 receptor antagonist aprepitant is a promising candidate for the treatment of breast cancer. Int. J. Oncol. 2014, 45, 1658–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, R.E.; Ramiro, S.; Lladó, S.; Toro, S.; Coveñas, R.; Muñoz, M. Antitumor action of temozolomide, ritonavir, and aprepitant against human glioma cells. J. Neurooncol. 2016, 126, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Oncoprevent Gmbh. Neurokinin-1 Receptor Antagonists for Use in a Method of Prevention of Cancer; WO2015101596; Oncoprevent Gmbh: Berlin, Germany, 2015. [Google Scholar]
- NK 1 IP Ltd. Use of Non-Peptidic NK1 Receptor Antagonists for the Production of Apoptosis in Tumor Cells; US2020054620; NK 1 IP Ltd.: Sevilla, Spain, 2020. [Google Scholar]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multi-drug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Lange, R.A.; Parsons, H.; Andrews, T.; Aune, G.J. The tell-tale heart: Molecular and cellular responses to childhood anthracycline exposure. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1379–H1389. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, A.; Kleinerman, E.S. Anthracycline-induced cardiotoxicity: Causes, mechanisms, and prevention. Adv. Exp. Med. Biol. 2020, 1257, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.Y.; Jung, S.Y.; Jennings, N.B.; Rodriguez-Aguayo, C.; Peng, G.; Lee, S.R.; Kim, S.B.; Kim, K.; Leem, S.H.; Lin, S.Y.; et al. FOXM1 mediates Dox resistance in breast cancer by enhancing DNA repair. Carcinogenesis 2012, 33, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consejo Superior Investigación. Antagonists of NK1 Receptors Derived from Carbohydrates, Production Methods, and Medical Use; US2019092802; Consejo Superior Investigación: Sevilla, Spain, 2019. [Google Scholar]
- Rodriguez, F.D.; Coveñas, R. Involvement of the substance P/neurokinin-1 receptor system in alcohol use disorders. In Advances in Medicine and Biology; Berhardt, L.V., Ed.; Nova Science: New York, NY, USA, 2022; Volume 194, pp. 151–172. [Google Scholar]
- Snider, R.M.; Constantine, J.W.; Lowe, J.A., 3rd; Longo, K.P.; Lebel, W.S.; Woody, H.A.; Drozda, S.E.; Desai, M.C.; Vinick, F.J.; Spencer, R.W. A potent non-peptide antagonist of the substance P (NK1) receptor. Science 1991, 251, 435–437. [Google Scholar] [CrossRef]
- Hale, J.J.; Mills, S.G.; MacCoss, M.; Finke, P.E.; Cascieri, M.A.; Sadowski, S.; Ber, E.; Chicchi, G.G.; Kurtz, M.; Metzger, J.; et al. Structural optimization affording 2-(R)-(1-(R)-3, 5-bis(trifluoromethyl)phenylethoxy)-3-(S)-(4-fluoro)phenyl-4- (3-oxo-1,2,4-triazole-5-yl)methyl morpholine, a potent, orally active, long-acting morpholine acetal human NK-1 receptor antagonist. J. Med. Chem. 1998, 41, 4607–4614. [Google Scholar] [CrossRef]
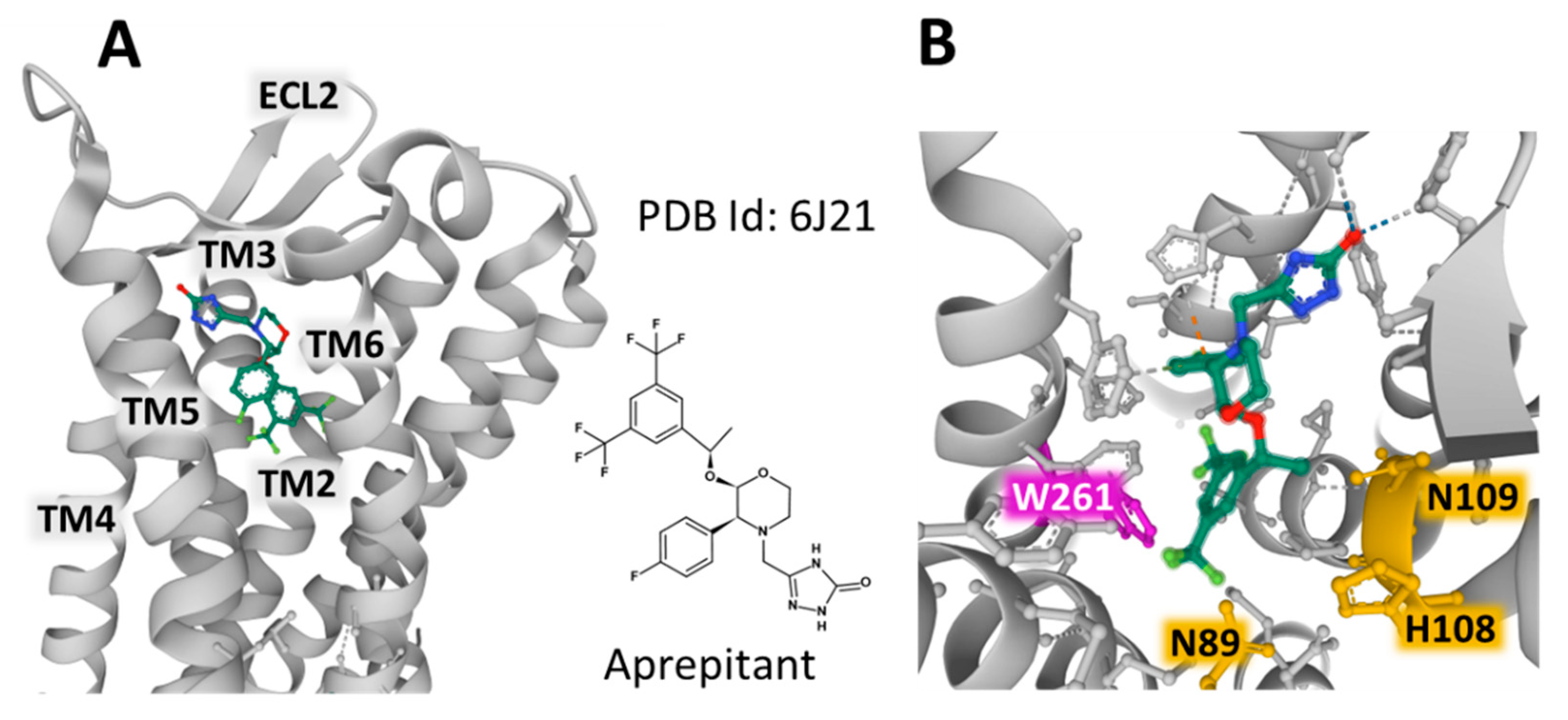
- Chen, S.; Lu, M.; Liu, D.; Yang, L.; Yi, C.; Ma, L.; Zhang, H.; Liu, Q.; Frinurer, T.M.; Wang, M.W.; et al. Human substance P receptor binding mode of the antagonist drug aprepitant by NMR and crystallography. Nat. Commun. 2019, 10, 638. [Google Scholar] [CrossRef]
- KingDraw Free Software. Available online: http://www.kingdraw.cn/en/index.html (accessed on 20 September 2022).
- Recio, R.; Vengut-Climent, E.; Mouillac, B.; Orcel, H.; López-Lázaro, M.; Calderón-Montaño, J.M.; Alvarez, E.; Khiar, N.; Fernández, I. Design, synthesis, and biological studies of a library of NK1-receptor ligands based on a 5-arylthiosubstituted 2-amino-4,6-diaryl-3-cyano-4H-pyran core: Switch from antagonist to agonist effect by chemical modification. Eur. J. Med. Chem. 2017, 138, 644–660. [Google Scholar] [CrossRef]
- Muñoz, M.; Muñoz, M.E.; Morell, F.; Coveñas, R. Why use aprepitant as a cough suppressant in lung cancer when at higher doses it could also exert an antitumor action? Arch. Bronconeumol. 2022, 58, 727–728. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
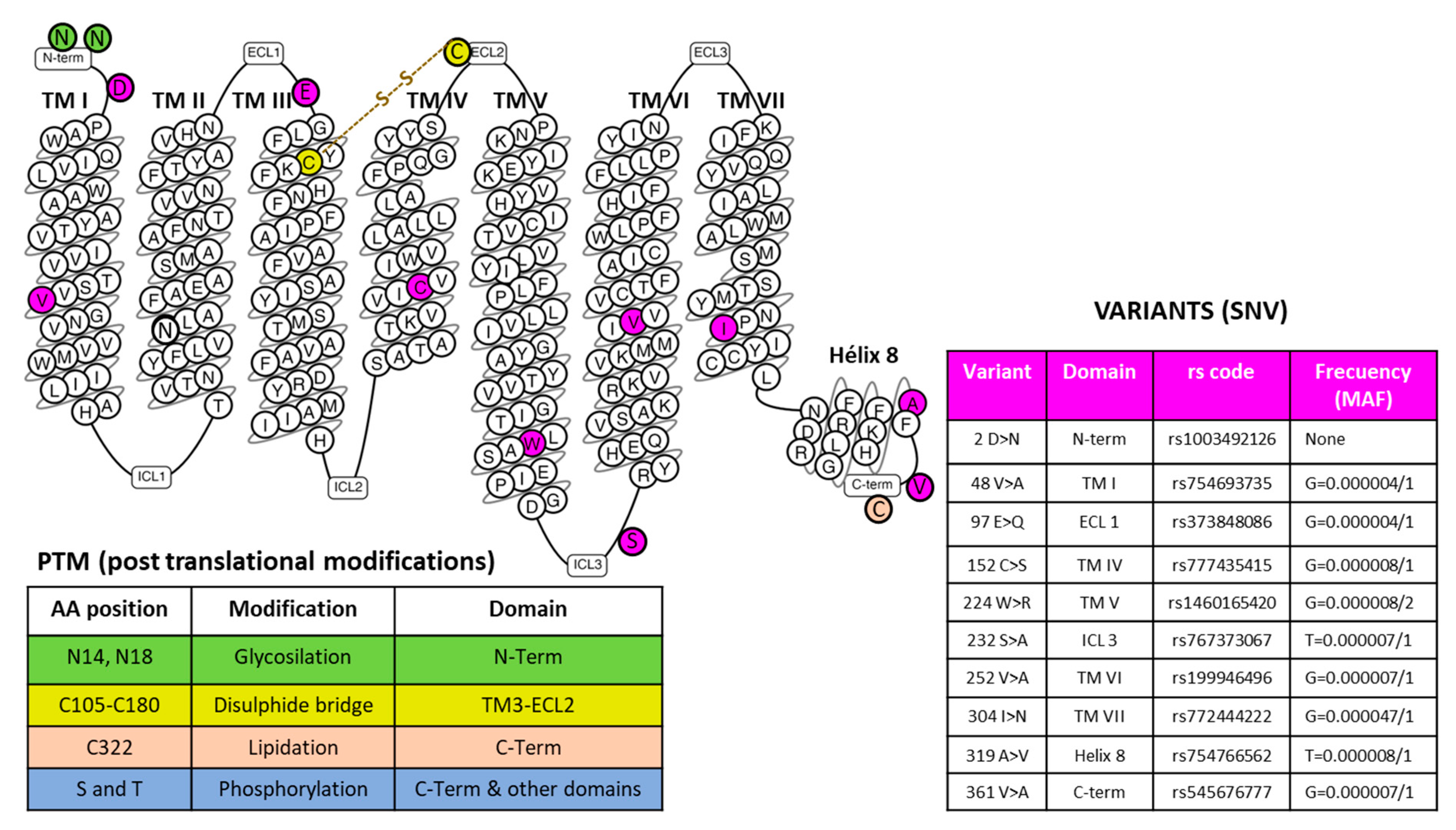
| NK-1R: preferential affinity for SP/hemokinin-1 [17] |
| NK-1R: SP binds to extracellular loops; non-peptide NK-1R antagonists between III and VI transmembrane segments [25] |
| Gln165, His197, and 265 NK-1R residues: regulate the binding of non-peptide NK-1R antagonists [25] |
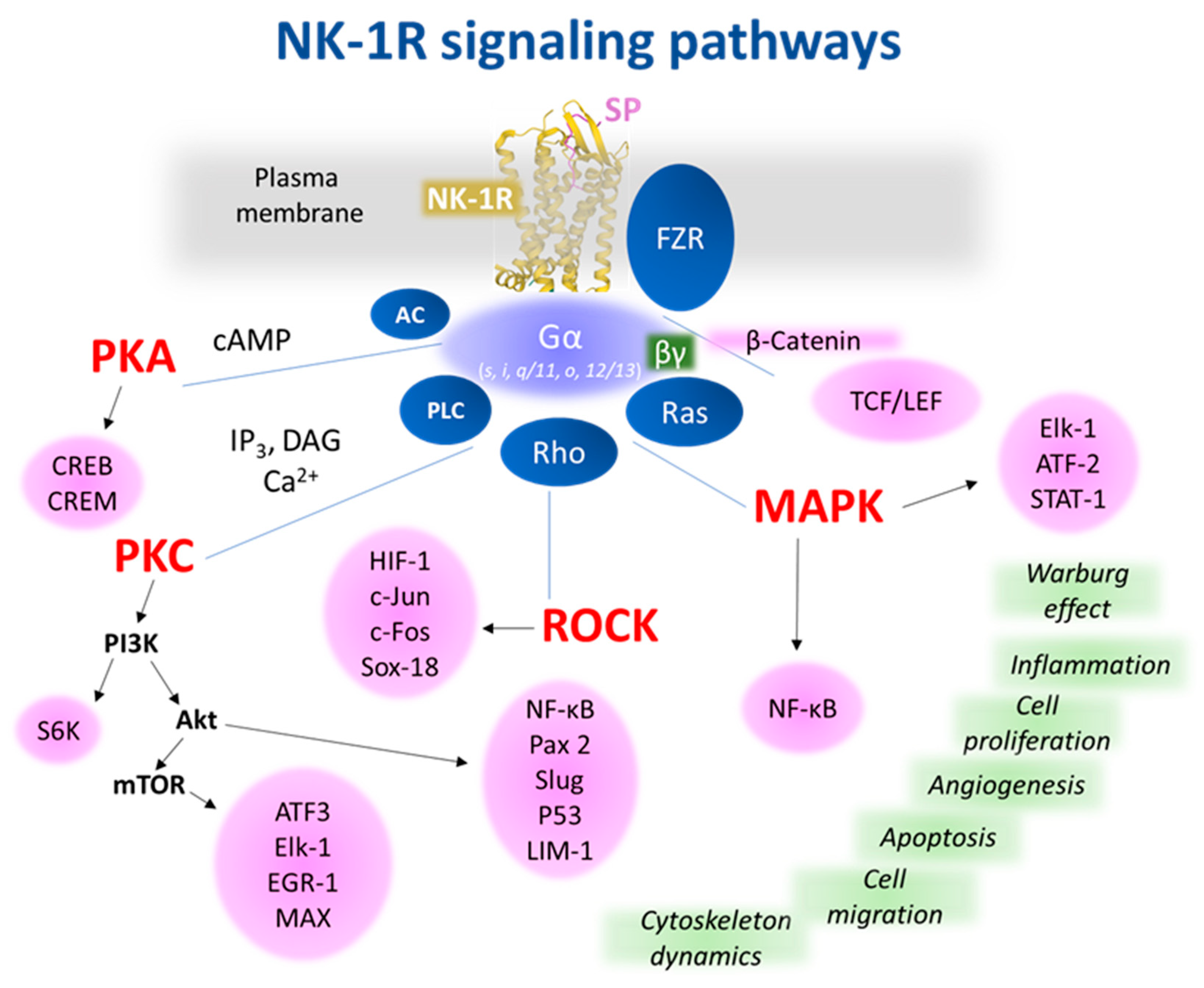
| NK-1R: coupled to Gαq, Gαs, Gαi, Gαo, and Gα12/13 proteins [25,26,27,28,29] |
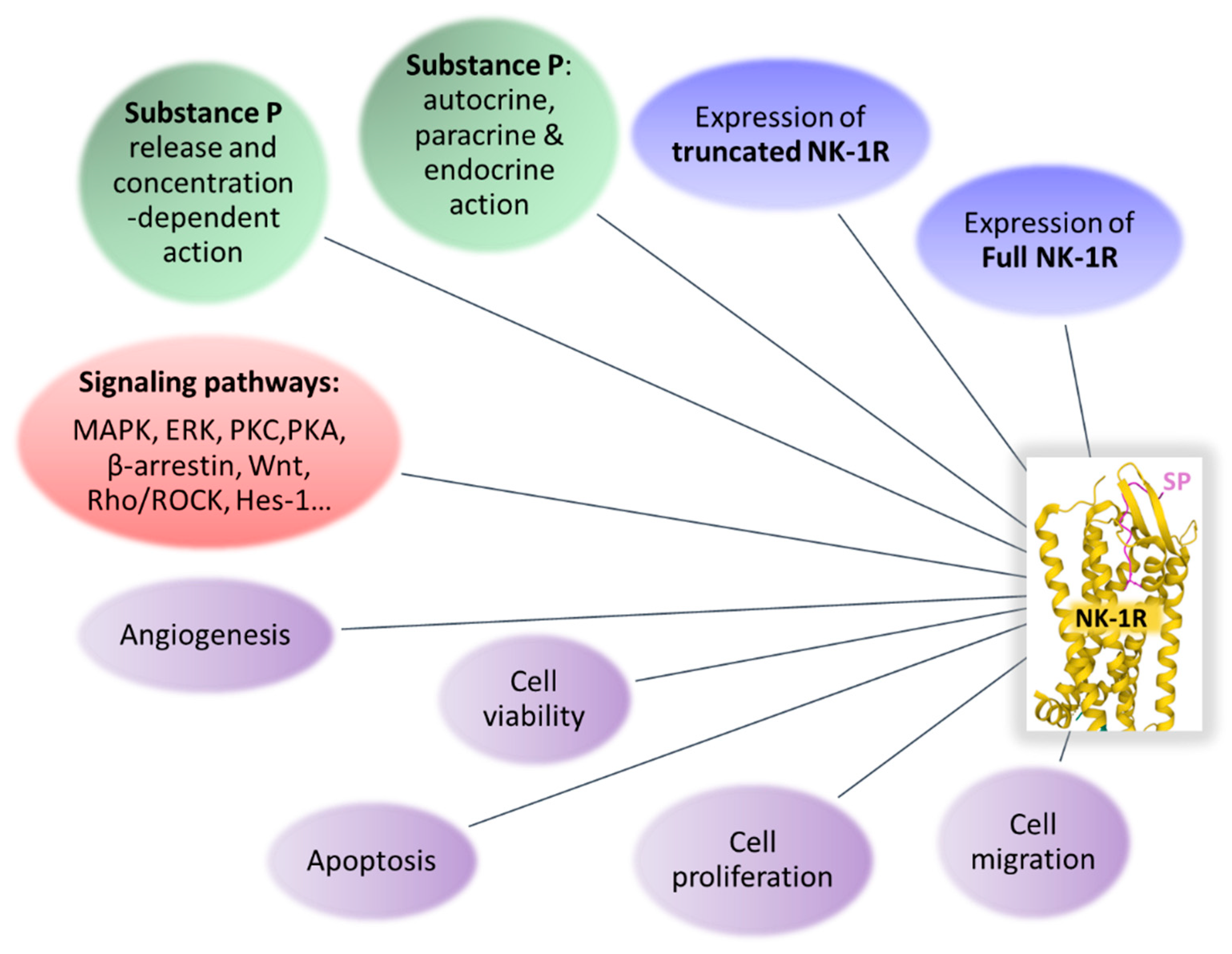
| NK-1R: regulates anti-apoptotic and tumor cell proliferation/migration signaling pathways and angiogenesis [34] |
| NK-1R: overexpressed in tumor cells [25] |
| Higher NK-1R level in tumor cells: related to cancer stage, tumor-node metastasis, poor prognosis, larger tumor size, and higher invasion/metastatic potential [1,72,73,75,92,93] |
| SP: increases NK-1R expression but not NK-2R/NK-3R expression [77,133] |
| NK-1R: involved in the viability of tumor cells [15] |
| Tumor cells: higher truncated NK-1R level and lower full-length form level than normal cells [61] |
| Full-length NK-1R: involved in NK-1R desensitization, internalization, and endocytosis [18] |
| Truncated NK-1R: oncogenic isoform mediating tumor growth and malignancy [18] |
| Activation of truncated NK-1R: increases metastasis and tumor cell proliferation; full-length activation decreases both mechanisms [55] |
| Patients with cancer: higher serum SP level and number of NK-1Rs [73,75] |
| NK-1R mRNA expression: lower in benign tissues than in malignant ones [48,61,62] |
| SP exerts a proliferative action on cancer cells expressing NK-1R. Many human cancer cell lines express NK-1R; a common antitumor strategy, irrespective of the tumor type, can be applied using NK-1R antagonists (aprepitant) [17,77,78] |
| Aprepitant: broad-spectrum antitumor agent [1,11,13,67,70,77,82,91,144,165,166,168,169] |
| Aprepitant: blocks proliferation/migration of cancer cells, promotes apoptosis, and exerts anti-Warburg/anti-angiogenic effects [77] |
| Aprepitant promotes apoptotic mechanisms by increasing mitochondrial reactive oxygen species [1,25,171]. |
| Aprepitant inhibits the Wnt canonical pathway, increases membrane stabilization of β-catenin, blocks the G2/M-phase cell cycle, increases the sensitization of cancer cells to cytotoxic action, and activates caspase-3-dependent apoptotic cascade [25,90,137,172,173,174] |
| NF-κB pathway overactivation: decreases the antitumor effect of aprepitant, and NF-κB activation by SP increases NK-1R expression; this activation is suppressed when NK-1R is blocked [123,133]. |
| Aprepitant exerted the highest antitumor action when tumor cells expressed higher truncated NK-1R levels [70] |
| NK-1R: involved in chemotherapy-induced side effects (hepatotoxicity, neurotoxicity, nephrotoxicity, and cardiotoxicity) [1,9] |
| Combination therapy of chemotherapy/radiotherapy with aprepitant: tumor radio and chemo sensitization counteract the side effects exerted by chemotherapy/radiotherapy and promotes a synergic antitumor action [1,9,183,184,185,186,187] |
| Single nucleotide variants (SNPs): their pathological significance has not been analyzed |
| To know the molecular mechanisms involved in NK-1R overexpression |
| To know how cancer cells express more truncated than full-length isoforms |
| No information on the formation of NK-1R dimers/oligomers has been provided: its capacity to heterodimerize is possible |
| To confirm whether the truncated form prolongs SP response |
| To know how the total number of full-length/truncated NK-1R isoforms is involved in cancer progression and the antitumor efficacy of NK-1R antagonists |
| To know the physiological significance of the presence of SP/full-length NK-1R form in the nucleus and the truncated NK-1R form in the cytoplasm |
| To confirm whether NK-1R regulates the balance between oxidant/antioxidant components of the redox system |
| To know why SP did not exert a proliferative action in specific tumor cells and how SP exerted an antimetastatic effect in some cases |
| The roles played by SP in the tumor microenvironment must be elucidated |
| To confirm whether the overexpression of NK-1R by cancer cells is a prognostic biomarker and whether an increased serum SP level is a predictive factor indicating a high risk of developing cancer or tumor development |
| Aprepitant administration before/after surgical procedures has been suggested to prevent recurrence and metastasis: this must be confirmed |
| To confirm whether patients with pancreatic ductal adenocarcinoma and high levels of NK-1R show a better overall survival |
| To know the function–structure relationships between NK-1R and SP for designing new antitumor drugs |
| Crystal structures of NK-1R bound to antagonists are crucial for designing compounds with specific characteristics related to their binding and specificity properties; these studies must be developed |
| Strategies to increase the bioavailability of aprepitant must be performed |
| To know how NK-1R antagonists decrease the number of PD-1-positive cells |
| Aprepitant is a promising antitumor agent against rhabdoid tumors, either alone or in combination therapy (chemotherapy): this must be confirmed |
| Combination strategy (chemotherapy or radiotherapy + aprepitant): a promising approach for fewer sequelae and higher cure rates in patients with cancer: this must be confirmed |
| Phase I and II dose escalation clinical trials with high doses of aprepitant are needed to learn about drug-drug interactions, tolerability, safety, and efficacy: aprepitant can be administered alone (20–40 mg/kg/day for a long time) or in combination therapy with chemotherapy or radiotherapy |
| Antiemetic aprepitant drug: a crucial antitumor agent candidate targeting the NK-1R. Its repurposing as a new therapeutic strategy to overcome cancer is urgently needed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coveñas, R.; Rodríguez, F.D.; Muñoz, M. The Neurokinin-1 Receptor: A Promising Antitumor Target. Receptors 2022, 1, 72-97. https://doi.org/10.3390/receptors1010005
Coveñas R, Rodríguez FD, Muñoz M. The Neurokinin-1 Receptor: A Promising Antitumor Target. Receptors. 2022; 1(1):72-97. https://doi.org/10.3390/receptors1010005
Chicago/Turabian StyleCoveñas, Rafael, Francisco D. Rodríguez, and Miguel Muñoz. 2022. "The Neurokinin-1 Receptor: A Promising Antitumor Target" Receptors 1, no. 1: 72-97. https://doi.org/10.3390/receptors1010005