
Immunoinformatic Approaches to Identify Immune Epitopes and Design an Epitope-Based Subunit Vaccine against Emerging Tilapia Lake Virus (TiLV)

 ,
,  ,
,
Abstract
:1. Introduction
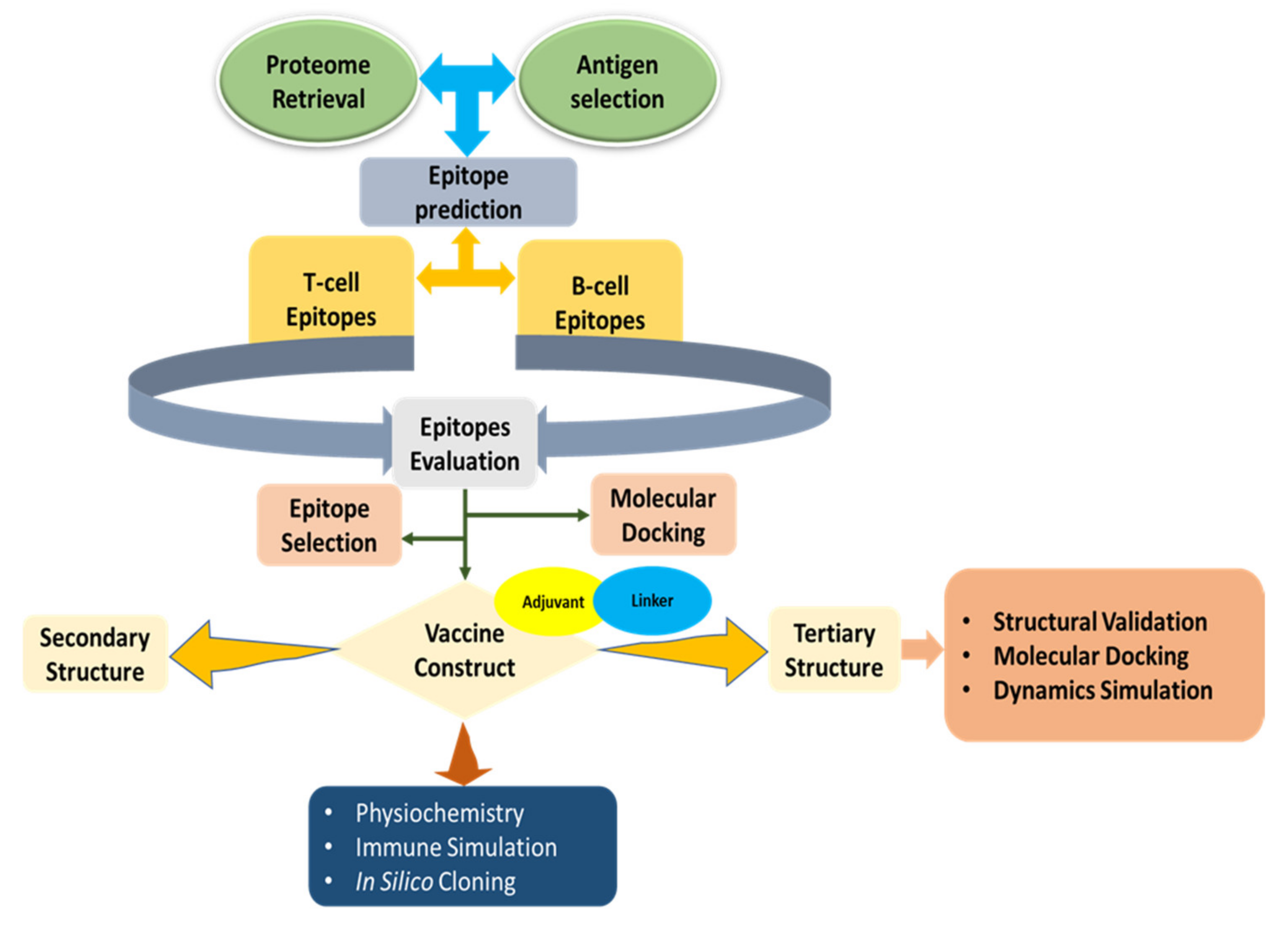
2. Methods
2.1. Antigen Selection and Retrieval of the Proteome
2.2. CTLs Epitope Prediction and Evaluation
2.3. HTLs Epitope Prediction and Assessment
2.4. Epitopes of Linear B-Lymphocytes: Prediction and Evaluation
2.5. Molecular Docking and Peptide Modeling
2.6. Development of an Epitope-Based Vaccination
2.7. Evaluation of Physicochemical and Immunological Factors
2.8. 3D Structure Prediction
2.9. Validation, 3D Structure Refinement, and Homology Modeling
2.10. Disulfide Engineering of the Toccine
2.11. Studies on Molecular Docking
2.12. Molecular Dynamics Simulation
2.13. Simulation of Immune Response
3. Results
3.1. Antigenicity Prediction
3.2. Prediction of Potential CTL Epitopes
3.3. Prediction of Best HTL Epitopes
3.4. Potential LBL Epitopes
3.5. Epitopes with Neighboring Alleles Molecular Docking Study
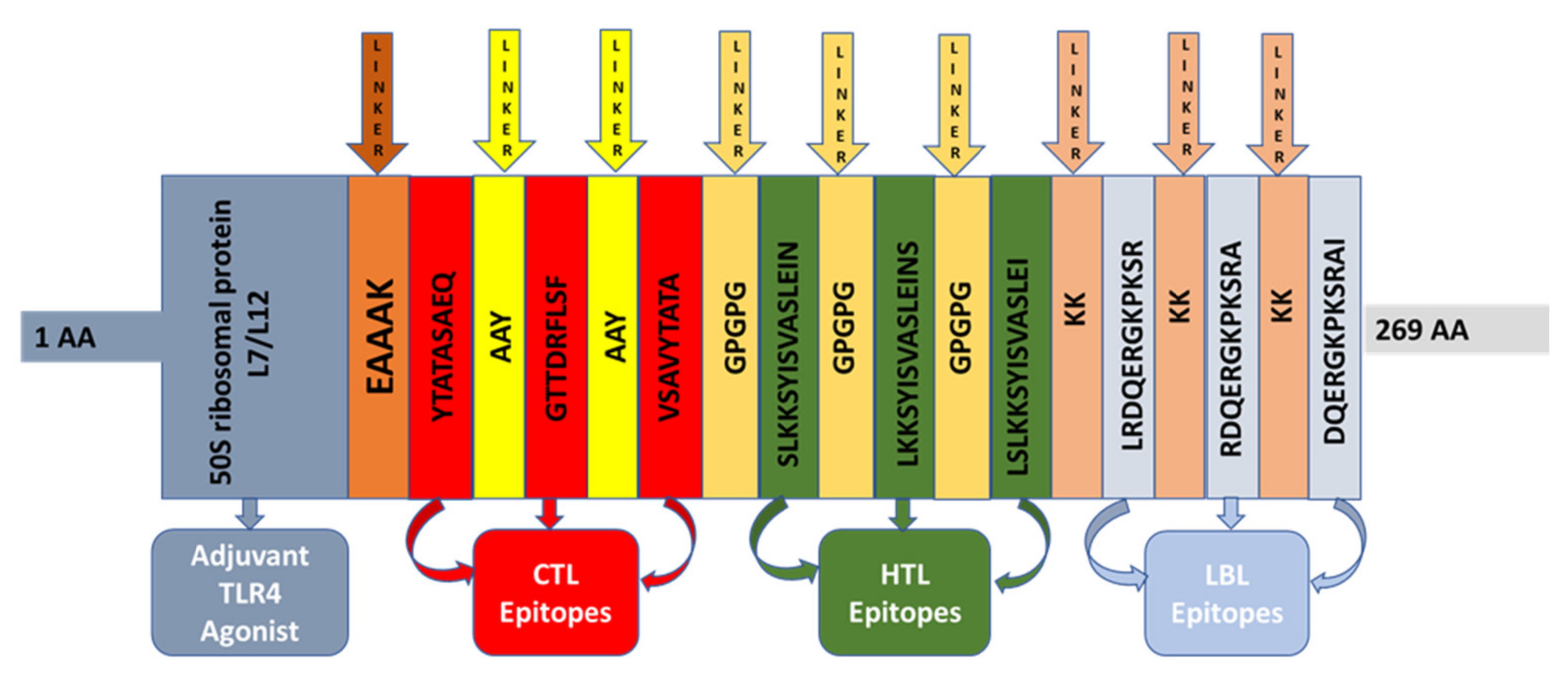
3.6. Core Properties and Structure of a Vaccine
3.7. Prediction of the Physicochemical and Immunological Features of the Vaccine
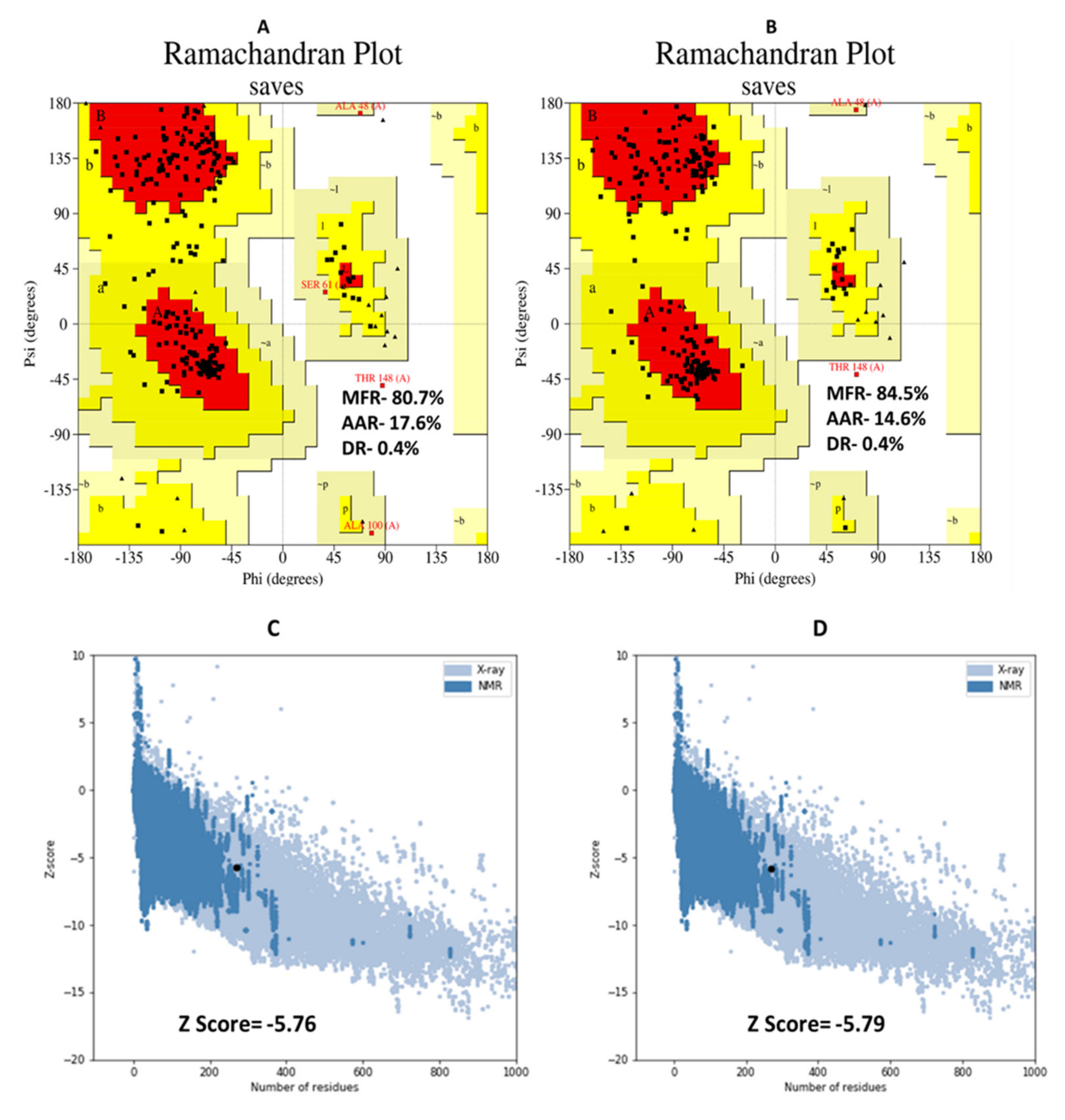
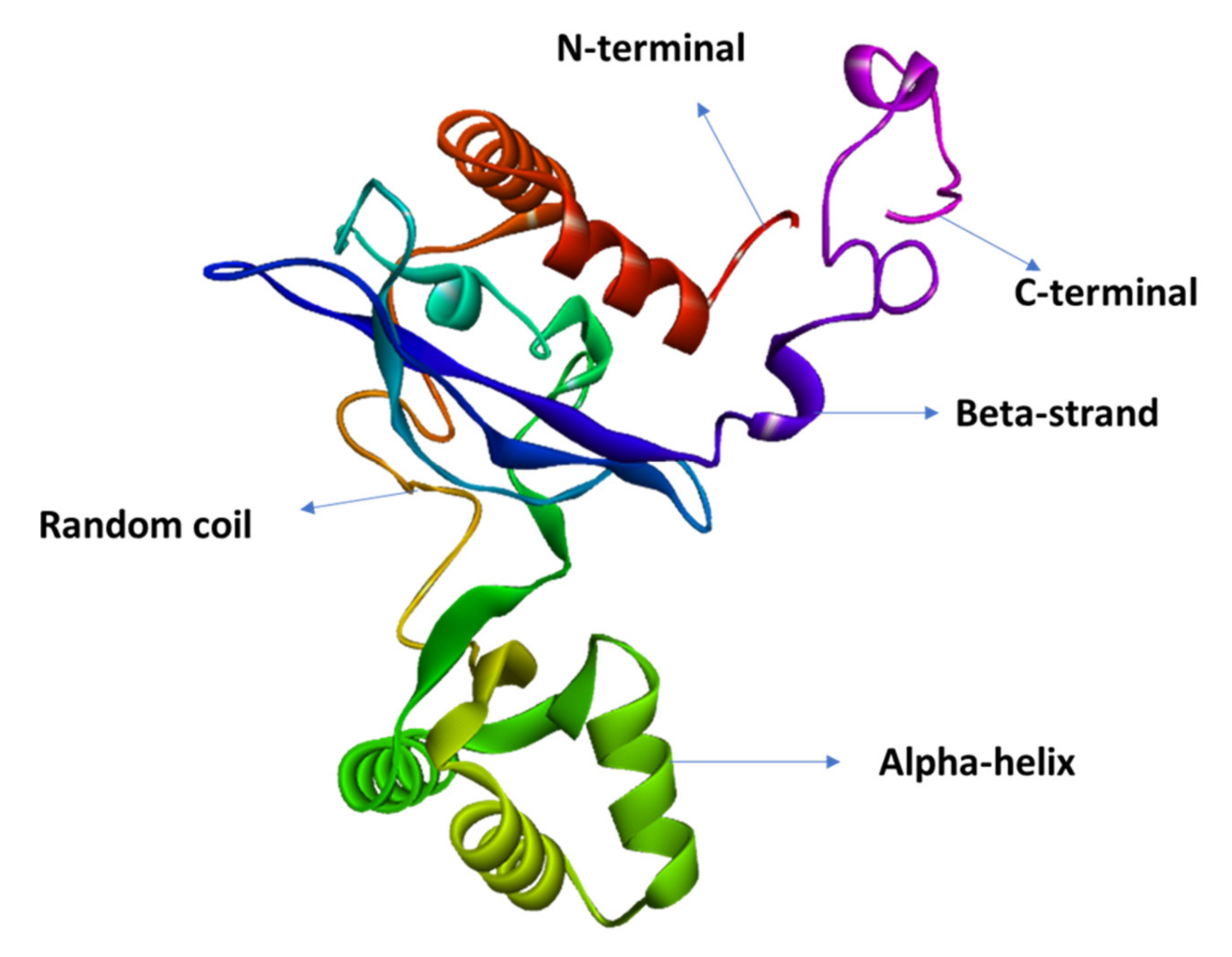
3.8. D Structure, Refinement, and Validation
3.9. Vaccine Disulfide Engineering
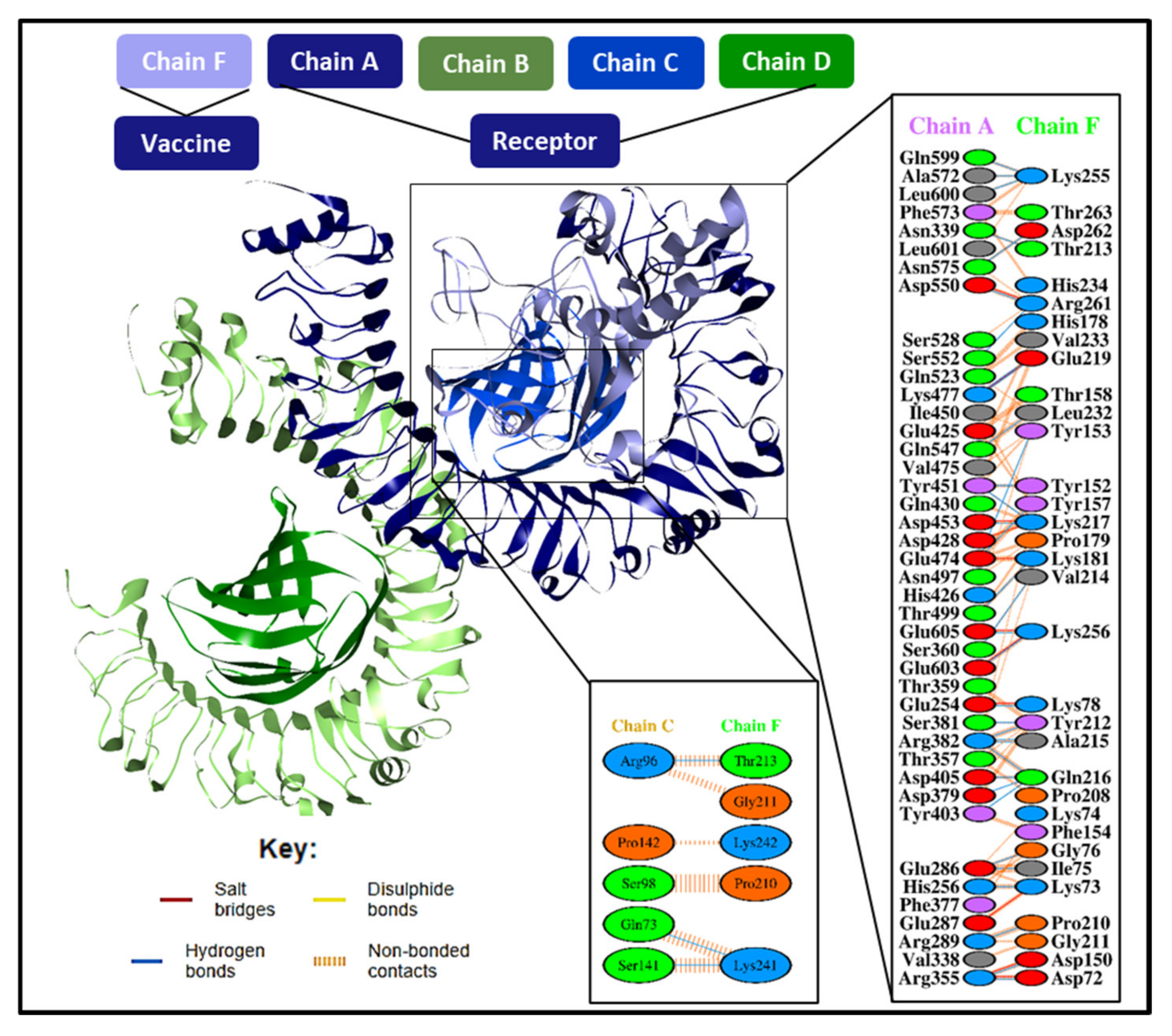
3.10. Molecular Docking Studies
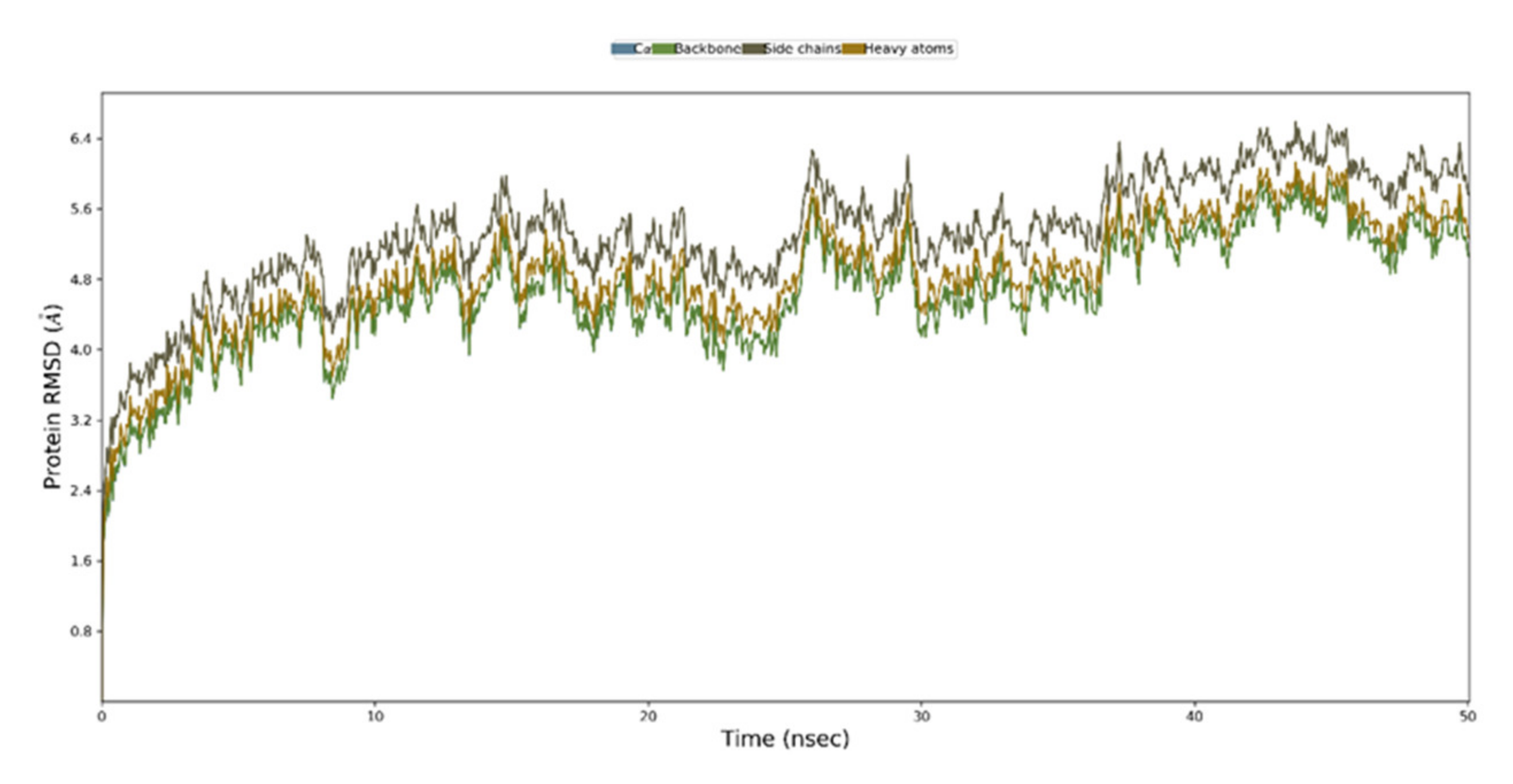
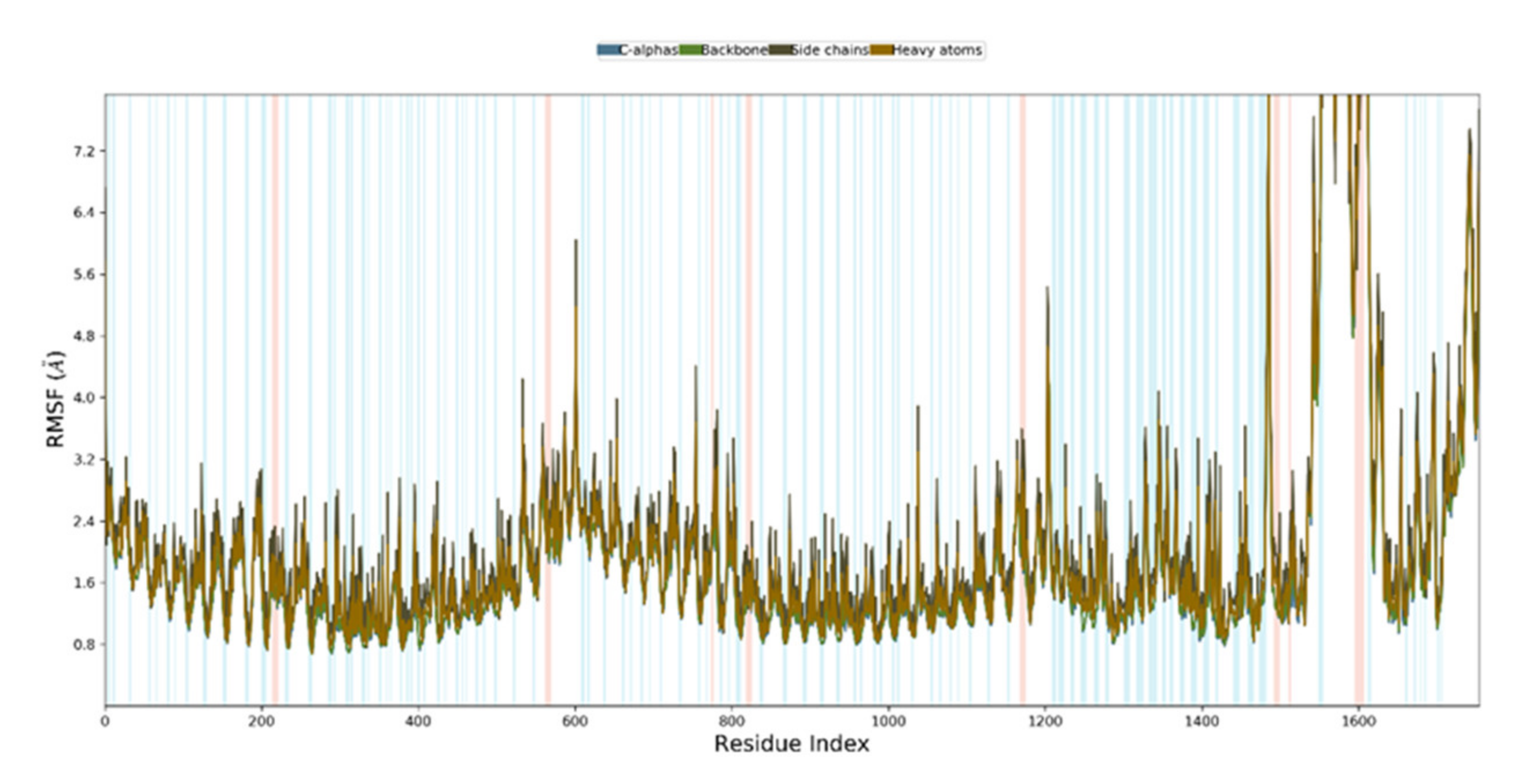
3.11. Molecular Dynamics Simulation
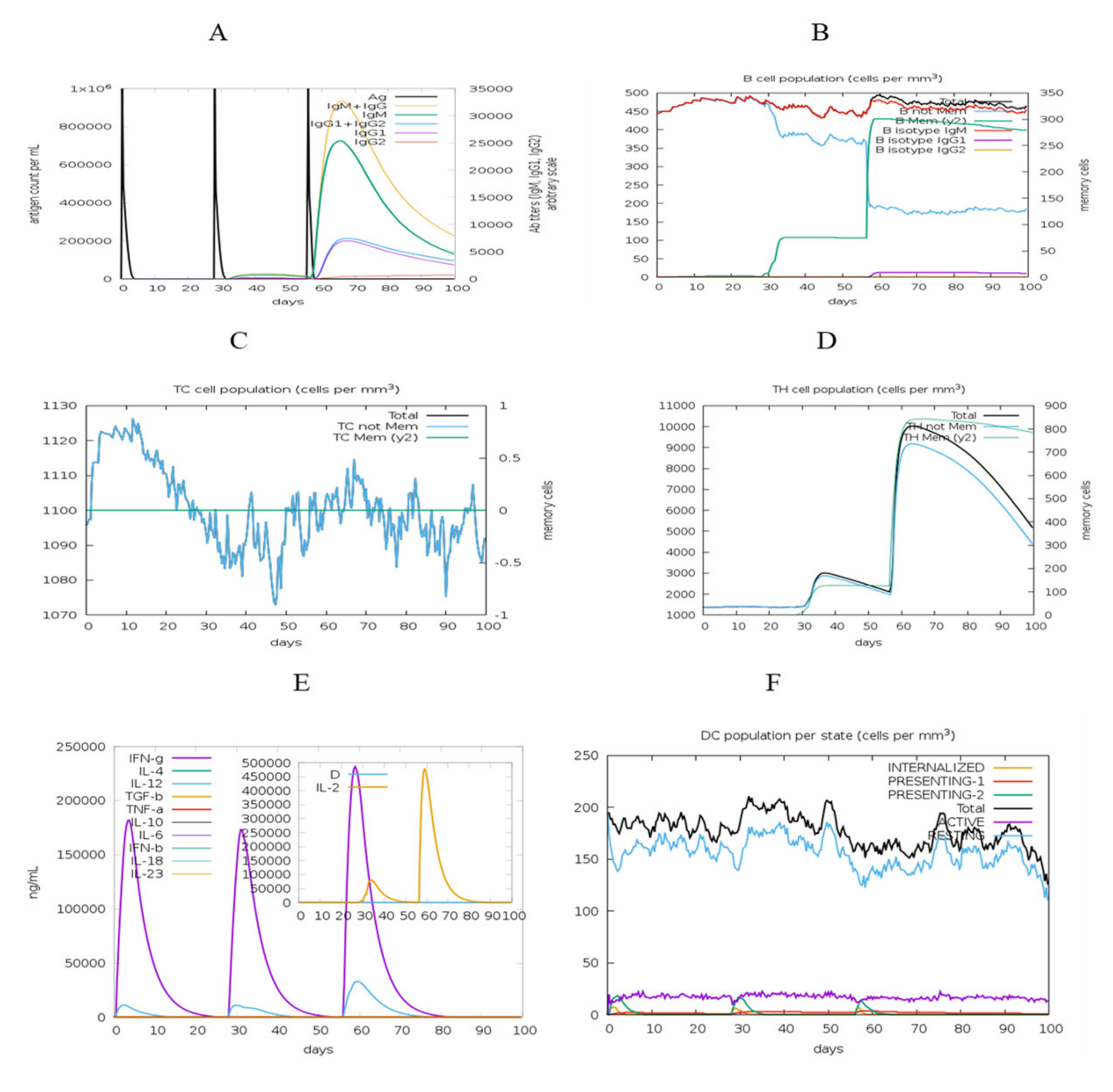
3.12. Immune Response Simulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Surachetpong, W.; Janetanakit, T.; Nonthabenjawan, N.; Tattiyapong, P.; Sirikanchana, K.; Amonsin, A. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015–2016. Emerg. Infect. Dis. 2017, 23, 1031–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, Y.; Velazquez, J.; Hernandez, L.; Swain, J.K.; Rodríguez, A.R.; Martínez, R.; García, C.; Ramos, Y.; Estrada, M.P.; Carpio, Y. Promiscuous T cell epitopes boosts specific IgM immune response against a P0 peptide antigen from sea lice in different teleost species. Fish Shellfish Immunol. 2019, 92, 322–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashfaq, H.; Soliman, H.; Fajmann, S.; Sexl, V.; El-Matbouli, M.; Saleh, M. Kinetics of CD4-1+ lymphocytes in brown trout after exposure to viral haemorrhagic septicaemia virus. J. Fish Dis. 2021, 44, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Fischer, U.; Dijkstra, J.M.; Hasegawa, S.; Somamoto, T.; Okamoto, N.; Ototake, M. Cytotoxic T cell function in fish. Dev. Comp. Immunol. 2002, 26, 131–139. [Google Scholar] [CrossRef]
- Adams, A. Progress, challenges and opportunities in fish vaccine development. Fish Shellfish Immunol. 2019, 90, 210–214. [Google Scholar] [CrossRef]
- Muñoz-Medina, J.E.; Sánchez-Vallejo, C.J.; Méndez-Tenorio, A.; Monroy-Muñoz, I.E.; Angeles-Martínez, J.; Coy-Arechavaleta, A.S.; Santacruz-Tinoco, C.E.; Gonzalez-Ibarra, J.; Aguiano-Hernandez, Y.-M.; Gonzalez-Bonilla, C.R.; et al. In Silico Identification of Highly Conserved Epitopes of Influenza A H1N1, H2N2, H3N2, and H5N1 with Diagnostic and Vaccination Potential. Biomed Res. Int. 2015, 2015, 813047. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.T.; Morshed, M.M.; Hassan, F. A Computational Approach for Designing a Universal Epitope-Based Peptide Vaccine Against Nipah Virus. Interdiscip. Sci. Comput. Life Sci. 2015, 7, 177–185. [Google Scholar] [CrossRef]
- Anwar, S.; Mourosi, J.T.; Khan, M.F.; Hosen, M.J. Prediction of Epitope-Based Peptide Vaccine Against the Chikungunya Virus by Immuno-informatics Approach. Curr. Pharm. Biotechnol. 2020, 21, 325–340. [Google Scholar] [CrossRef]
- Dash, R.; Das, R.; Junaid, M.; Akash, F.C.; Islam, A.; Hosen, S.Z. In silico-based vaccine design against Ebola virus glycoprotein. Adv. Appl. Bioinform. Chem. 2017, 10, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhang, J.; Li, S.; Sun, J.; Teng, Y.; Wu, M.; Li, J.; Hu, N.; Wang, H.; Hu, Y. Epitope-Based Vaccine Target Screening against Highly Pathogenic MERS-CoV: An In Silico Approach Applied to Emerging Infectious Diseases. PLoS ONE 2015, 10, e0144475. [Google Scholar] [CrossRef] [Green Version]
- Grimholt, U. MHC and Evolution in Teleosts. Biology 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkstra, J.M.; Grimholt, U.; Leong, J.; Koop, B.F.; Hashimoto, K. Comprehensive analysis of MHC class II genes in teleost fish genomes reveals dispensability of the peptide-loading DM system in a large part of vertebrates. BMC Evol. Biol. 2013, 13, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Dijkstra, J.M. Major Histocompatibility Complex (MHC) Genes and Disease Resistance in Fish. Cells 2019, 8, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stosik, M.; Tokarz-Deptuła, B.; Deptuła, W. Major histocompatibility complex in Osteichthyes. J. Vet.-Res. 2020, 64, 127–136. [Google Scholar] [CrossRef]
- Bolnick, D.; Snowberg, L.; Stutz, W.; Caporaso, G.; Lauber, C.; Knight, R.; Lauber, C.; Caporaso, J.G. Major Histocompatibility Complex class IIb polymorphism influences gut microbiota composition and diversity. Mol. Ecol. 2014, 23, 4831–4845. [Google Scholar] [CrossRef]
- Marana, M.H.; Jørgensen, L.V.G.; Skov, J.; Chettri, J.K.; Mattsson, A.H.; Dalsgaard, I.; Kania, P.W.; Buchmann, K. Subunit vaccine candidates against Aeromonas salmonicida in rainbow trout Oncorhynchus mykiss. PLoS ONE 2017, 12, e0171944. [Google Scholar] [CrossRef] [Green Version]
- Mahendran, R.; Jeyabaskar, S.; Sitharaman, G.; Michael, R.D.; Paul, A.V. Computer-aided vaccine designing approach against fish pathogens Edwardsiella tarda and Flavobacterium columnare using bioinformatics software. Drug Des. Devel. Ther. 2016, 10, 1703–1714. [Google Scholar] [CrossRef] [Green Version]
- Pereira, U.; Soares, S.; Blom, J.; Leal, C.; Ramos, R.; Guimarães, L.C.; Oliveira, L.; Almeida, S.; Hassan, S.; Santos, A.; et al. In silico prediction of conserved vaccine targets in Streptococcus agalactiae strains isolated from fish, cattle, and human samples. Genet. Mol. Res. 2013, 12, 2902–2912. [Google Scholar] [CrossRef]
- Pumchan, A.; Krobthong, S.; Roytrakul, S.; Sawatdichaikul, O.; Kondo, H.; Hirono, I.; Areechon, N.; Unajak, S. Novel Chimeric Multiepitope Vaccine for Streptococcosis Disease in Nile Tilapia (Oreochromis niloticus Linn.). Sci. Rep. 2020, 10, 603. [Google Scholar] [CrossRef]
- Madonia, A.; Melchiorri, C.; Bonamano, S.; Marcelli, M.; Bulfon, C.; Castiglione, F.; Galeotti, M.; Volpatti, D.; Mosca, F.; Tiscar, P.-G.; et al. Computational modeling of immune system of the fish for a more effective vaccination in aquaculture. Bioinformatics 2017, 33, 3065–3071. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Pathak, D.C.; Mannan, M.U.; Kaushik, V. In-silico designing of an epitope-based vaccine against the seven banded grouper nervous necrosis virus affecting fish species. Netw. Modeling Anal. Health Inform. Bioinform. 2021, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2018, 234, 8509–8521. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14 (Suppl. 6), S4. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, G.; Usmani, S.S.; Dhanda, S.; Kaur, H.; Singh, S.; Sharma, M.; Raghava, G.P.S. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, srep42851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nain, Z.; Abdullah, F.; Rahman, M.M.; Karim, M.M.; Khan, S.A.; Bin Sayed, S.; Mahmud, S.; Rahman, S.M.R.; Sheam, M.; Haque, Z.; et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [Green Version]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef]
- Nain, Z.; Karim, M.M.; Sen, M.K.; Adhikari, U.K. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans—An integrated vaccinomics approach. Mol. Immunol. 2020, 120, 146–163. [Google Scholar] [CrossRef]
- Olejnik, J.; Hume, A.; Mühlberger, E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog. 2018, 14, e1007390. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdellrazeq, G.S.; Fry, L.M.; Elnaggar, M.M.; Bannantine, J.P.; Schneider, D.A.; Chamberlin, W.M.; Mahmoud, A.H.; Park, K.-T.; Hulubei, V.; Davis, W.C. Simultaneous cognate epitope recognition by bovine CD4 and CD8 T cells is essential for primary expansion of antigen-specific cytotoxic T-cells following ex vivo stimulation with a candidate Mycobacterium avium subsp. paratuberculosis peptide vaccine. Vaccine 2020, 38, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2008; pp. 531–552. [Google Scholar]
- Gordon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus pre-diction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- Xu, J.; McPartlon, M.; Li, J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 2021, 3, 601–609. [Google Scholar] [CrossRef]
- Nugent, T.; Cozzetto, D.; Jones, D.T. Evaluation of predictions in the CASP10 model refinement category. Proteins: Struct. Funct. Bioinform. 2014, 82 (Suppl. 2), 98–111. [Google Scholar] [CrossRef] [Green Version]
- Delano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Pokhrel, S.; Bouback, T.A.; Samad, A.; Nur, S.M.; Alam, R.; Abdullah-Al-Mamun, M.; Nain, Z.; Imon, R.R.; Talukder, K.; Tarwq, I.M.; et al. Spike protein recognizer receptor ACE2 targeted identification of potential natural antiviral drug candidates against SARS-CoV. Int. J. Biol. Macromol. 2021, 191, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bouback, T.A.; Pokhrel, S.; Albeshri, A.; Aljohani, A.M.; Samad, A.; Alam, R.; Hossen, M.S.; Al-Ghamdi, K.; Talukder, M.; Kabir, E.; et al. Pharmacophore-based virtual screening, quantum mechanics calculations, and molecular dynamics simulation approaches identified potential natural antiviral drug candidates against MERS-CoV S1-NTD. Molecules 2021, 26, 4961. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the Interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [Green Version]
- Thawornwattana, Y.; Dong, H.T.; Phiwsaiya, K.; Sangsuriya, P.; Senapin, S.; Aiewsakun, P. Tilapia lake virus (TiLV): Genomic epidemiology and its early origin. Transbound. Emerg. Dis. 2021, 68, 435–444. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [Green Version]
- Bol, K.F.; Aarntzen, E.H.J.G.; Pots, J.M.; Nordkamp, M.A.M.O.; Van De Rakt, M.W.M.M.; Scharenborg, N.M.; De Boer, A.J.; Van Oorschot, T.G.M.; Croockewit, S.A.J.; Blokx, W.A.M.; et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol. Immunother. 2016, 65, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An Overview of Novel Adjuvants Designed for Improving Vaccine Efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epitope | C-Score | Antigenicity | Immunogenicity | Toxicity | Allergenicity |
|---|---|---|---|---|---|
| YTATASAEQ | 0.8770 | 0.5345 | Positive | Negative | Negative |
| GTTDRFLSF | 0.5838 | 0.7173 | Positive | Negative | Negative |
| VSAVYTATA | 0.5343 | 0.6773 | Positive | Negative | Negative |
| Epitope | Antigenicity | IFNᵧ | IL4 | IL10 | Toxicity | Allergenicity |
|---|---|---|---|---|---|---|
| SLKKSYISVASLEIN | 1.0072 | Positive | Inducer | Inducer | Negative | Negative |
| LKKSYISVASLEINS | 0.8496 | Positive | Inducer | Inducer | Negative | Negative |
| LSLKKSYISVASLEI | 1.2590 | Positive | Inducer | Inducer | Negative | Negative |
| Epitope | Probability | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|
| LRDQERGKPKSR | 0.8157 | 1.9529 | Negative | Negative |
| RDQERGKPKSRA | 0.7908 | 1.7915 | Negative | Negative |
| DQERGKPKSRAI | 0.7332 | 1.3894 | Negative | Negative |
| T-Cell Epitope | HLA Allele | Epitope Affinity (kcal/mol) | Control Affinity (kcal/mol) | Number of Hydrogen Bonds (CHB) | Residues Involved in CHB Networks (n) |
|---|---|---|---|---|---|
| YTATASAEQ | HLA-A*0201 | −7.2 | −9.2 | 9(7) | Gln69, Trp149, Thr7, Ile8, Met19, Ile1, Ala2, Ile7, Tyr74 (9) |
| GTTDRFLSF | HLA-A*0201 | −7.1 | −8.2 | 9(8) | Lys80, Tyr84, Lys146, Val2, Thr7, Val9, Lys66, Asn77, Thr143 (9) |
| VSAVYTATA | HLA-B*3501 | −9.0 | −8.2 | 18(14) | Tyr7, Arg2, Asp9, Glu63, Lys66, Arg69, Asn77, Asn77, Lys80, Tyr84, Tyr99, Thr143, Lys146, Trp147, Glu15, Glu152, Tyr159, Tyr171 (18) |
| SLKKSYISVASLEIN | DRB1*03:01 | −6.9 | −7.5 | 9(7) | Arg71, Thr77, Asn82, Ala12, Thr13, Val14, Val1, Glu6, Ser4 (9) |
| LKKSYISVASLEINS | DRB5*01:01 | −7.0 | −7.7 | 12(10) | Tyr7, Asp9, Asp9, Ser24, Glu63, Lys66, Arg69, Arg69, Tyr99, Glu152, Glu152, Gln155 (12) |
| LSLKKSYISVASLEI | DRB4*01:01 | −6.9 | −7.3 | 10(8) | Ser63, Glu85, Asn72, His328, Trp7, Ala15, Phe17, Tyr8, Ile17, Ile3 (10) |
| Characteristics | Finding | Remark |
|---|---|---|
| Number of amino acids | 269 | Suitable |
| Aliphatic index of vaccine | 85.06 | Thermostable |
| Grand average of hydropathicity (GRAVY) | −0.292 | Hydrophilic |
| Antigenicity | 0.7053 | Antigenic |
| Immunogenicity | Positive | Immunogenic |
| Allergenicity | No | Non-allergen |
| Solubility | 0.901123 | Soluble |
| Characteristics | SOMPA Server | PSIPRED Server | ||
|---|---|---|---|---|
| Amino Acid | % | AA | % | |
| α-helix | 131 | 48.70% | 105 | 39.033% |
| β-strand | 38 | 14.13% | 47 | 17.47% |
| Random coil | 81 | 30.11% | 117 | 43.494% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, S.I.; Mahfuj, S.; Alam, M.A.; Ara, Y.; Sanjida, S.; Mou, M.J. Immunoinformatic Approaches to Identify Immune Epitopes and Design an Epitope-Based Subunit Vaccine against Emerging Tilapia Lake Virus (TiLV). Aquac. J. 2022, 2, 186-202. https://doi.org/10.3390/aquacj2020010
Islam SI, Mahfuj S, Alam MA, Ara Y, Sanjida S, Mou MJ. Immunoinformatic Approaches to Identify Immune Epitopes and Design an Epitope-Based Subunit Vaccine against Emerging Tilapia Lake Virus (TiLV). Aquaculture Journal. 2022; 2(2):186-202. https://doi.org/10.3390/aquacj2020010
Chicago/Turabian StyleIslam, Sk Injamamul, Sarower Mahfuj, Md. Ashraful Alam, Yeasmin Ara, Saloa Sanjida, and Moslema Jahan Mou. 2022. "Immunoinformatic Approaches to Identify Immune Epitopes and Design an Epitope-Based Subunit Vaccine against Emerging Tilapia Lake Virus (TiLV)" Aquaculture Journal 2, no. 2: 186-202. https://doi.org/10.3390/aquacj2020010