

Apoptosis Regulators Bcl-2 and Caspase-3


Department of Anatomy, Institute of Biomedicine and Translational Medicine, University of Tartu, 50411 Tartu, Estonia
Encyclopedia 2022, 2(4), 1624-1636; https://doi.org/10.3390/encyclopedia2040111
Submission received: 30 May 2022
/
Revised: 12 September 2022
/
Accepted: 15 September 2022
/
Published: 21 September 2022
(This article belongs to the Section Medicine & Pharmacology)
Definition
:Apoptosis, programmed cell death, has a central role in developmental biology and in maintaining the equilibrium of renewing tissues. A founding member of the Bcl-2 family of regulatory proteins for apoptosis is Bcl-2, which is encoded by the BCL2 gene. Caspase-3 shares typical features with all caspases, including the role of acting as a crucial mediator of apoptosis.
1. Introduction
1.1. Apoptosis
A multicellular organism consists of highly organized cells. The number of cells in this community is tightly regulated, by controlling not only the rate of cell division, but also the rate of cell death. If cells are no longer needed, they activate the intracellular biochemical events which lead to characteristic cell changes and the death program. The cell changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and mRNA decay, and the process is called programmed cell death (PCD); although, it is more commonly referred to as apoptosis (from Ancient Greek, ἀπόπτωσις, apoptosis: “falling off”, as leaves from a tree) [1,2]. Unlike necrosis, a form of traumatic cell death resulting from acute cell damage, the controlled and highly regulated process of apoptosis provides benefits over the life cycle of the body. During apoptosis, cell fragments, also known as apoptotic bodies, are produced. The apoptotic bodies can be engulfed and removed by phagocytes before the contents of the cell can flow into and damage the surrounding cells [1].
In developmental biology, apoptosis plays a key role in the physiological process of selective cell deletion that is required to maintain a steady state of the cell in a constantly renewing tissue [3,4]. In various processes, such as embryonic development, the proper development and functioning of the immune system, normal cell metabolism, hormone-dependent atrophy, and chemically induced cell death, apoptosis is considered an essential component. In addition to its importance as a biological phenomenon, inappropriate apoptosis is regarded to be a factor in dysfunctional health conditions (neurodegenerative diseases, autoimmune disorders, ischemic damage, etc.) [5,6]. Defects in apoptotic processes have been associated with a number of diseases. For example, in cancer, uncontrolled cell proliferation is associated with insufficient apoptosis, and atrophy has been observed to be caused by excessive apoptosis. Because of the great therapeutic potential seen in the ability of apoptosis to modulate cell life or death, modern research continually focuses on the elucidation and analysis of signaling pathways that control cell cycle arrest and apoptosis. There are two well-known groups of features which characterize apoptosis. The first group of features are morphological: condensation of chromatin, reduction of cell volume, and nuclear fragmentation, which results in the formation of apoptotic bodies, also known as “little sealed sacs”. Another feature is the cleavage of DNA by Ca2+/Mg2+-dependent endonuclease into oligonucleosome-long fragments that can be identified by gel electrophoresis as a ladder pattern [7].
Phenomena considered to be endogenous activators of apoptosis include the absence of growth factors, the deficiency of the trophic hormone, glucocorticoid therapy, and the ablation of matrix attachment [8]. Exogenous activators are traumatic physical stimulators, such as radiation, toxins, chemotherapeutics, and infective pathogens such as bacterial toxins and viruses. Programmed cell death is controlled by apoptotic regulators, which either block the protective effect of inhibitors having a pro-apoptotic effect, or have an inhibitory, anti-apoptotic influence [9,10]. The genes that control apoptosis were firstly identified by studies in the nematode, C. elegans, the homologues of which function in humans to regulate apoptosis [11]. By encoding their own anti-apoptotic genes which prevent the target cells from passing away earlier, some of the viruses have found a protective method to combat apoptosis.
Apoptosis is a highly regulated process that cannot be stopped once it has started [12]. The initiation of apoptosis goes through one of two pathways: intrinsic or extrinsic pathways [1,13]. In the inner pathway, or as it is more commonly known, the intrinsic or mitochondrial pathway, the cell sensing the cellular stress kills itself. In the outer or so-called extrinsic pathway, the tumor necrosis factor (TNF) pathway, and the first apoptosis signal (Fas) pathway caused by the signals from other cells, the cell kills itself.
1.2. Intrinsic Pathway
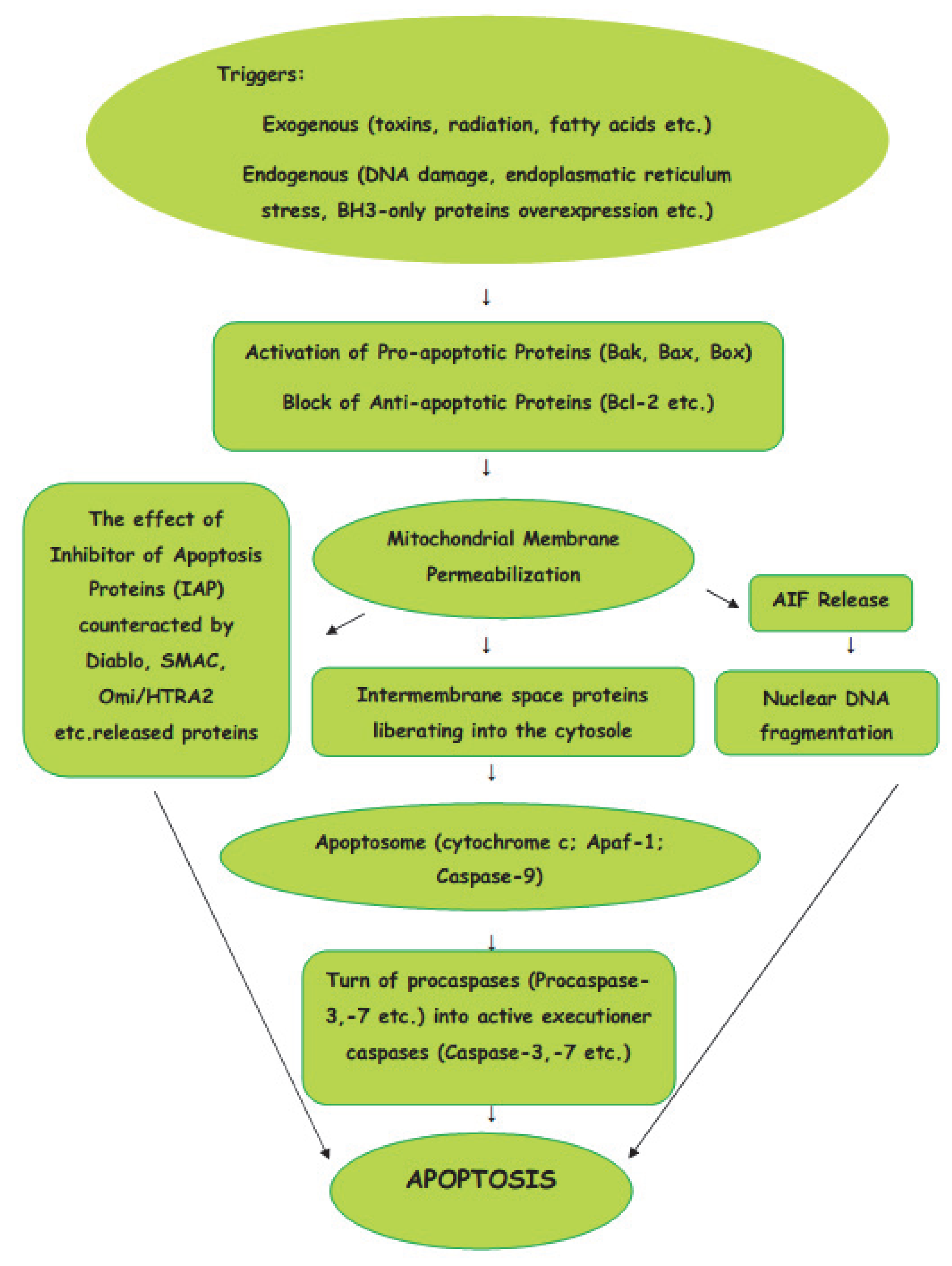
The intrinsic pathway of apoptosis is a cellular response to a change in the intracellular environment caused by stress. The sources of stress that cause cellular changes can be exogenous or endogenous. The exogenous triggers are toxins, radiation, fatty acids, etc. The endogenous stressors include DNA damage, endoplasmic reticulum stress, mitochondrial translocation, p53 activation, overexpression of BH3-only proteins, Ca2+ homeostasis, and/or imbalance in the redox status [14]. As the signals caused by both types of triggers influence mitochondria, the intrinsic pathway of apoptosis is known as the mitochondrial pathway (Figure 1).
Mitochondria are the basic “energy stations” of the cell, where, through oxidative phosphorylation, the energy is converted to ATP molecules.Mitochondria can be affected by signals in a variety of ways, such as by translocation to the outer mitochondrial matrix, or by post-transduction modification of mitochondrial proteins and channel formation. The effects of the signals causes the selective permeability of the mitochondrial inner or outer membrane, which causes the proteins in the intermembrane space to be liberated into the cytosole. The Bcl-2 protein family is involved in the process of the formation of macropores in the outer mitochondrial membrane. The active pro-apoptotic proteins, Bak, Bax, and Bok, undergo hetero–homo oligomerization, leading to the formation of macropores which enable proteins from the mitochondrial intermembrane space to enter the cytosole [15]. From mitochondrial intermembrane space proteins, cytochrome c, after cytosolic release, participates in the formation of a protein complex known as apoptosome, which cleaves Pro-caspase-9 to active Caspase-9. In turn, the activation of Caspase-9 activates the Pro-caspase-3 into effector Caspase-3. Thus, the cascade of caspases is initiated. The effect of IAP (inhibitor of apoptosis proteins), which ordinarily prevents the activation of Caspase-3, may be counteracted by some proteins, such as Arts, Diablo, Second Mitochondria-Derived Activator of Caspase (SMAC), and High-Temperature Requirement Protein A2 (Omi/HTRA2), released from damaged mitochondria. The interaction of IAPs, SMACs, and Omi/HTRA2 of members of the Bcl family is central to the pathway of intrinsic apoptosis. It has been demonstrated recently that Endonuclease-G (EndoG), the nuclease which is specifically activated by apoptotic stimuli, is also capable of inducing nucleosomal fragmentation of DNA independently of DNA Fragmentation Factor (DFF)/Caspase-Activated DNAse (CAD) and Caspase [16].
1.3. Extrinsic Pathway
The signals from the extracellular environment which designate the death of the cell cause the extrinsic pathway of apoptosis (Figure 2).
There are two main theories of the extrinsic pathways of apoptosis: the TNF (tumor necrosis factor-induced) pathway and the Fas (Fas ligand-mediated model) pathway [17,18]. These pathways involve receptors of the TNF receptor family that are associated with external signals, and both pathways induce cell death by activating caspases, which then activate executioner caspases; after which, the cells are killed by degrading proteins indiscriminately.
Death receptors (DRs), the cell surface receptors, transmit apoptotic signals initiated by specific ligands, and have a major role in instructive apoptosis.
1.3.1. TNF Pathway
The TNF pathway involves receptor systems. The Tumor Necrosis Factor Receptor family includes cell surface receptors, death receptors (DRs), which convey apoptotic signals initiated by particular ligands with a major role in instructive apoptosis [19]. Apoptosis of the cell in the TNF pathway is caused by the activation of death caspases activated by the DRs. The apoptotic signal is transmitted from DRs to the death machinery by adapter molecules, such as Daxx containing Death Domains, Fas-Associated via Death Domain (FADD), and Tumor Necrosis Factor Receptor-1-Associated Death Domain (TRADD).
1.3.2. Fas Pathway
The Fas pathway is triggered by cytotoxic stress. The Fas receptor is a transmembrane protein belonging to the TNF family which binds the FasL (Fas ligand) [17]. When the interaction occurs between FasL and Fas, the death-inducing signaling complex (DISC), consisting of Fas, FADD, and Procaspase-8, is formed, and by interaction with the regulator FLICE Inhibitory Protein (FLIP), the co-factor function of FADD is blocked. Following recruitment by FADD, the oligomerization of Procaspase-8 activates it by self-cleavage; after which, Caspase-3 and Caspase-7 are activated by Caspase-8, causing cell apoptosis.
1.4. Other Pathways of Apoptosis
In addition to the caspase-dependent intrinsic and extrinsic apoptotic pathways, the caspase-independent pathway, mediated by apoptosis-inducing factor (AIF), has been described [20,21]. Apart from apoptotic activation by caspase or by AIF-mediated pathways, studies have demonstrated that apoptosis can also be induced by dual pathways: via the activation of AIF and caspase cascade simultaneously [22]. It has been shown that the modulation of mitochondrial activity may lead to the activation of cell death signals, which results in the release of cytochrome c and AIF in the cytoplasm [23,24]. Cytochrome c binds to the apoptotic protease-activating factor and to deoxy-ATP, activating Caspase-9 and causing the activation of the caspase cascade. This kind of signaling pathway is responsible for the hydrolysis of several key cytoplasmic proteins required for cell survival.
Recently, a nuclear pathway associated with apoptosis has been proposed [25]. Precisely, ZIP Kinase, after initiating apoptosis from nuclear Promyelocytic Leukemia Oncogenic Domains, combines with Daxx and Prostate Apoptosis Response Protein-4, a nuclear, caspase-independent apoptosis pathway.
Additionally, as the Jun N-terminal kinases (JNK) can also promote apoptosis by increasing the expression of pro-apoptotic genes through the transactivation of c-Jun/AP1-dependent or p53/73 protein-dependent mechanisms, the pathway is known as the JNK-mediated apoptosis pathway [26].
Some members of the Bcl-2 family of proteins inhibit apoptosis, whereas other factors, such as Fas receptors and caspases, promote apoptosis. The Bcl-2 family proteins have been found to include members that either promote or inhibit apoptosis [27]. In apoptosis, pro- and anti-apoptotic signaling proteins both have essential roles [28].
2. Apoptosis Regulators
2.1. Bcl-2
The Bcl-2 family comprises many proteins that share domains of Bcl-2 homology, and the proteins in this family have a general structure consisting of a hydrophobic α-helix surrounded by amphipathic α-helixes [29]. The Bcl-2 family consists of pro- and anti-apoptotic members (Table 1), and controls apoptosis through its members that either inhibit or promote apoptosis [30].
The Bcl-2 family of proteins have a similar general structure, consisting of a hydrophobic α-helix encircled by amphipathic α-helixes. Proteins of the Bcl-2 family, including pro- and anti-apoptotic members of the family, regulate mitochondrial outer membrane permeability, which plays an important role in the control of apoptosis.
In the Bcl-2 family, all proteins contain either the domains, BH1, BH2, and BH3, which are Bcl-2 homology (BH) domains, or the BH4 domain [31,32]. These BH domains are known to be vital for the function of the proteins, dividing pro-apoptotic Bcl-2 proteins into those with only the BH3 domain, so-called pro-apoptotic BH3-only proteins (Bim, Bid, etc.), and into proteins with multiple BH domains—the pro-apoptotic pore-formers (Bax, Bak, Bok) [33]. All Bcl-2 family proteins contain the BH3 domain, which is among the four BH domains associated with the interactions between these proteins, and is required for dimeration with other Bcl-2 family proteins, which is essential for their killing activity. The multi-Bcl-2 homology domain proteins that contain all four BH domains are all anti-apoptotic proteins and pore-forming pro-apoptotic proteins. These proteins take on a very conserved tertiary structure, forming a hydrophobic BH3 domain binding region which acts for other family members’ BH3 domains as a receptor. BH1, BH2, and BH3 are three functionally important regions of the Bcl-2 homology which are in close spatial vicinity, thus forming a gap that can be a binding point for the other Bcl-2 family members. Among the anti-apoptotic proteins, the dominant regulator of apoptosis in mammalian cells is Bcl-x [34,35]. Though the long form of Bcl-x, i.e., Bcl-xL, shows cellular repressor activity, its short isoforms, i.e., Bcl-xβ and Bcl-xS, promote cell death.
There are many theories about how pro- or anti-apoptotic activity is exhibited by the Bcl-2 family [36]. It has been shown that the Bcl-2 family members’ primary task is to directly control the permeability of mitochondrial membranes by regulating the release of apoptogenic factors from the intermembranous space into the cytosole [37,38,39]. It is also believed that pro-apoptotic proteins of the Bcl-2 family may induce, and anti-apoptotic members may inhibit, cytochrome c liberation into the cytosol; after which, the activation of Caspase-3 and Caspase-9 leads to apoptosis. Apoptogenic factors known to be released include endonuclease G and AIF [20], cytochrome c, Smac Diablo [40], and heat shock protein 60 [41]. Cytochrome c and Smac Diablo are involved in the activation of caspases. In inducing caspase-independent apoptotic changes, the caspase-independent apoptotic changes in nuclear endonuclease and AIF are assumed to be important. Anti-apoptotic members of the Bcl-2 family inhibit the release of the apoptogenic factors, whereas pro-apoptotic members promote it [42]. The activation of Bcl-2, Mcl-1, and Bid proteins has also suggested a possible role of Rho proteins, small (2121 kDa) signaling proteins belonging to the Ras superfamily [43]. It has been noted that the inhibition of Rho increases protein levels of the pro-apoptotic Bid, and decreases the expression of anti-apoptotic Mcl-1 and Bcl-2 proteins. However, the inhibition of Rho activates Caspase-3- and Caspase-9-dependent apoptosis in cultured human endothelial cells, but does not affect Baxi or FLIP levels.
The “return point” of the apoptotic pathway is determined by mitochondrial outer membrane permeabilization (MOMP), which leads to the release of cytochrome c [44]. As the MOMP is regulated by the Bcl-2 family proteins, the cellular commitment to apoptosis is thereby determined. Primarily directed between pro- and anti-apoptotic members of the B-cell lymphoma 2 (Bcl-2) family, MOMP is a largely organized process [45]. The intrinsic apoptotic pathway is controlled by a balance of pro-apoptotic proteins which carry a single BH3 domain and apoptotic proteins belonging to the Bcl-2 family. Bad, Bid, and Bax, the pro-apoptotic proteins which reside in healthy cells’ cytosol, relocate to the mitochondrial outer membrane, which causes the mitochondria to lose membrane potential and initiate apoptosis.
It has been shown that to prevent mitochondrial pore formation, cytochrome c release, and the initiation of apoptosis, Bcl-2 acts by heterodimerization with proapoptotic members of the Bcl-2 family [46]. The intrinsic (internal) pathway of apoptosis is regulated by the Bcl-2 family, being activated by the mitochondrial disorder, and followed by the release of cytochrome c. The initiators of the internal pathway include cytotoxic drugs and UV irradiation. The “apoptosome” (a large quaternary protein which is formed during apoptosis) is formed by the interaction of the cytosolic protein, Apaf-1, cytochrome c, d-ATP/ATP, and Pro-caspase-9, followed by the initiation of the caspase cascade.
Studies suggesting mechanisms by which Bcl-2 may prevent cell death include the local inhibition of free radical production and ion conduction channel formation in membranes [47]. Extensive oxidative damage in ischemic neuronal death suggests that the Bcl-2’s antioxidant mechanism would be beneficial in cases where oxidative damage is a significant component as a trigger of apoptosis.
The anti-apoptotic protein, Bcl-xL, has two central hydrophobic helices surrounded by five amphipathic helices [48], having a similar structure to certain bacterial toxins. Minn et al. [49] showed that Bcl-xL can be incorporated into membranes to form an ionic conduction channel which is cation-selective at physiological pH. The consequences of ischemia include the possibility that Bcl-xL may regulate intracellular calcium sequestration, which is an essential component in the excitotoxic apoptosis mechanism.
Bcl-2-associated proteins belong to the apoptotic nuclear machinery, which has been conserved in species as largely diverse from each another as Caenorhabditis elegans and mammals. Proteins associated with Bcl-2 can either promote or inhibit apoptosis, and whether the cell is alive or dying is determined by the interaction between proteins belonging to opposing factions. From this perspective, the best-understood pathway is the example of the C. elegans worm, where genetic studies have shown in detail that two of the Bcl-2-associated proteins—pro-apoptotic Egl-1 and survival-promoting Ced-9—are indispensable to controlling the evolution of the programmed cell deaths of the somatic cells [50]. The expression of a lethal trigger, Egl-1, is caused by injury signals. When Egl-1 binds to Ced-9, an ortholog of the C. elegans Bcl-2, Ced-4 adapter protein from Ced-9 is released and, upon release, the protein, Ced-4, binds to and activates Ced-3 caspase, causing cell death.
Apoptosis Regulator Bcl-2
Encoded by the BCL2 gene, Bcl-2 (B-cell CLL/lymphoma 2) is the founding member of the Bcl-2 family of regulator proteins which inhibit or induce apoptosis [51,52,53]. Damage to the BCL2 gene has been detected as a reason for chronic lymphocytic leukemia, as well as many other different types of cancers [54,55], autoimmunity, and neurodegenerative diseases [56]. It also causes resistance to the treatment of cancers [57]. As knowledge of the mechanisms of Bcl-2 proteins that affect programmed cell death may contribute to the development of new cancer, autoimmune, and neurological disease therapies, continuous research in this field is essential. Bcl-2 is particularly important as an apoptotic inhibitor in the development of liver tissue and liver pathology in acute and chronic hepatitis [58,59,60]. In patients with cirrhosis caused by chronic viral hepatitis C, Bcl-2 has been found to be localized in hepatocytes, and the localization of Bcl-2 in hepatic cholesterol proliferation areas has been reported in patients with chronic hepatitis B and C [61,62]. Studies have shown Bcl-2 activity in hepatocytes and Kupffer cells, as well as sinusoidal endotheliocytes in central, middle, and portal zones of liver lobules with positively-stained lymphocytes and cholangiocytes in portal zones during chronic hepatitis B, indicating the significance of the anti-apoptotic protein, Bcl-2, in regulating apoptosis not only in hepatocytes, but also in other cellular components of the liver in patients with chronic viral hepatitis B [63]. The expression of Bcl-2 has been detected in different types of hepatocytes in mono- and binucleated hepatocytes during chronic hepatitis B.
In animal cells, Bcl-2 proteins have been found on the outer mitochondrial membrane, which is supposed to form a complex with the voltage-gated anion channel porin (VDAC) [64]. Interaction between Bcl-2 with VDAC3- or VDAC1-derived peptides has been shown to protect against cell death by inhibiting cytochrome c release. Bcl-2 family proteins have also been detected in the perinuclear envelope and found to be broadly located in many tissues. The ability of the Bcl-2 family proteins to form oligomeric pores in artificial lipid bilayers has been documented.
When liberated from mitochondria, apoptogenic factors (Smac/Diablo homologue, cytochrome c homologue, etc.) activate caspases, which, in turn, induce programmed cell death [65].
2.2. Caspases
Caspases (cysteine aspartases, or cysteine-dependent aspartate-directed proteases, or cysteine-aspartic proteases), belonging to the family of proteases, are significant in the regulation of apoptosis and inflammatory processes and in development [66,67], as the intrinsic or extrinsic pathways of apoptosis both induce cell death by activating caspases [11]. In total, 14 types of caspases have been found, divided into three main subtypes: initiator, executioner, and inflammatory caspases, with initiator and executioner caspases participating in apoptosis, and inflammatory caspases participating in pyroptosis [68]. Besides the three subtypes of caspases, Caspase-14 has been detected as having a role in death-receptor- and granzyme-B-induced apoptosis [69,70] (Table 2).
Initially, caspases are produced in an inactive form, and are thereafter activated by various apoptotic signals of pro-caspases [59,71]. It is supposed that caspases regulate the last steps of apoptosis [72]. The other important roles of caspases are necroptosis and pyroptosis (inflammatory caspases) [73]. Caspases also participate in cell proliferation and differentiation, tumor suppression, neural development, etc. The development of tumors is caused by mutations in caspases which respond to their changes by inducing cell death in abnormally growing cells [74]. The insufficient activation of caspases involved with processing inflammatory signals increases an organism’s susceptibility to infection. During liver diseases, an increased activation of caspases has been noted [75,76,77]. For instance, the activation of caspases has been detected in the serum of patients with chronic hepatitis C virus (HCV), as well as in patients with non-alcoholic fatty liver disease [78,79].
As caspases influence cell death and disease, they have been used as a target in the research of drug development; for example, caspases have been used in cancer therapy to destroy unwanted cells [80]. Caspases are synthesized as zymogens, which are inactive pro-caspases that are activated only after an appropriate trigger. In the biological environment, pro-caspase dimers are combined to form a heterotetrametric enzyme [81]. Often, during the activation of caspases, the multiprotein complexes form apoptosomes during intrinsic apoptosis; death-inducing signaling complexes (DISC) during extrinsic apoptosis; and inflammasomes, as a result of pyroptosis.
Caspase-3
Caspase-3 is a member of the family of cysteine-aspartic acid proteases, and is encoded by the CASP3 gene. Apart from being found in various mammals, the orthologs of CASP3 [82,83,84] have also been detected in teleosts, lissamphibians, lizards, and birds. A number of typical features shared by all currently known caspases are also characteristic of Caspase-3.
Following the occurrence of apoptotic signaling, Pro-caspase-3, which has been previously found in an inactive form, is cleaved by an initiator caspase [85]. With Caspase-3 playing a central role, the extrinsic activation initiates the caspase cascade specific to the apoptotic pathway [86,87].
During the intrinsic activation, to process the Caspase-3 zymogen, the mitochondrial cytochrome c collaborates with apoptosis-activating factor 1 (Apaf-1), ATP, and Caspase-9 [88,89]. These molecules, adequate for the in vitro activation of Caspase-3, need additional regulatory proteins for the in vivo activation of Caspase-3 [90]. The inhibition of caspase has been described as occurring through another protein family, precisely through the inhibitor of apoptosis proteins (IAP), including XIAP, ML-IAP, c-IAP1, and c-IAP2 [91]. The initiator, Caspase-9, which is directly involved in the activation of the executioner, Caspase-3, is tied and inhibited by XIAP [90]. In this cascade, Caspase-3 inhibits the activity of XIAP, cleaving Caspase-9 at a certain site, thus making it impossible for XIAP to bind and inhibit Caspase-9 activities [92].
The over-activation of Caspase-3 may result in excessive apoptosis; for example, it has been noted in various neurodegenerative diseases, such as Alzheimer’s disease, where the loss of neural cells occurs [74]. As a sign of a recent myocardial infarction, there are also signs of high levels of a fragment of Caspase-3, p17, found in the bloodstream [93]. Recently, it has been discovered that Caspase-3 could have a role in embryonic and hematopoietic stem cell differentiation [94,95]. According to studies including DNA fragmentation and chromatin condensation, a cascade of enzymatic changes leading to morphological manifestations of programmed cell death is caused by the activation of Caspase-3 [96,97]. In mono (chronic hepatitis B (HBV)) and mixed infections (tuberculosis (TB), chronic hepatitis C (HCV), human immunodeficiency virus (HIV)), the expression of Caspase-3 in liver biopsies has been detected, in addition to hepatocytes, in Kupffer cells and endothelial cells, in the bile ducts’ epithelial cells, as well as in intralobular focal necrosis and in lymphocytic infiltration in the portal areas [98,99]. It has been noted that during these infections, the expression of Caspase-3 depends on the amount of inflammation, fibrosis, and necrosis, indicating the correlation between the degree of histological activity and Caspase-3 activity. It has been shown that the expression of Caspase-3 differs in mono- and binucleated hepatocytes. Furthermore, in biopsies of patients with mono (HBV) and mixed infections (TB, HCV, HIV), considerable differences have been proven by quantitative analysis to occur in the expression of Caspase-3, indicating a high level of apoptosis during infections. It has been noted that the level of Caspase-3 is remarkably elevated during chronic hepatitis in the case of strong histological necroinflammatory activity, and is particularly severe in patients with co-infections.
3. Conclusions
Apoptosis, programmed cell death, is a conserved pathway that, according to its basic principles, appears to be functional in all metazoans [17]. Insufficient apoptosis can manifest as cancer or autoimmunity, whereas accelerated cell death is evident in acute and chronic degenerative diseases, immunodeficiency, and infertility [100]. In order to maintain tissue homeostasis, apoptosis, a form of cell death, allows the removal of damaged cells. Regulated apoptosis is critical to development and tissue homeostasis. Apoptosis is mediated by two pathways, extrinsic and intrinsic, regulated up-stream of the mitochondria by the coordinated action of various molecules, including proteins of the Bcl-2 family [13,101]. Although the Bcl-2 family of proteins is involved in various intracellular pathways associated with cell survival, the most prominent function of the proteins is the regulation of the apoptosis’ initiation of internal, intrinsic (mitochondrial) pathways [102,103]. The Bcl-2 family is composed of anti-apoptotic and pro-apoptotic (BH3-only and multidomain) proteins [104]; interactions between these proteins control the process of the release of cytochrome c from mitochondria by modulating sensitivity to cell death signals [105]. Caspases are crucial mediators of apoptosis [88]. Among them, Caspase-3 is a frequently activated protease, which is activated in the cell both by the intrinsic and extrinsic pathways of apoptosis [83]. Known also as an executioner caspase, Caspase-3 carries out proteolysis, which leads to apoptosis. The dysregulation of Caspase-3-mediated apoptosis may lead to serious diseases in organisms (neurodegenerative diseases, myocardial infarction, etc.) [106,107]. Considerable differences in the expression of Caspase-3 in liver biopsies of the patients with mono (HBV) and mixed infections (tuberculosis, HCV, HIV) have been found by quantitative analysis, proving that the expression of Caspase-3 depends on the degree of inflammation, fibrosis, and necrosis [98]. Caspase-3 is also thought to be a marker of early liver disease [75]. The identification of new regulators of apoptosis and the monitoring of their effects may contribute to the diagnosis of diseases, as well as to the selection of an effective therapeutic strategy in the future [108,109].
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Apoptosis: Programmed cell death eliminates unwanted cells. In Molecular Biology of the Cell, 5th ed.; Alberts, B., Ed.; Garland Science: New York, NY, USA, 2008; p. 1115. [Google Scholar]
- Green, D. Means to an End: Apoptosis and Other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2011. [Google Scholar]
- Ucker, P.S. Death by suicide: One way to go in mammalian cellular development. New Biol. 1991, 3, 103–109. [Google Scholar] [PubMed]
- Wyllie, A.H. Cell death: A new classification separating apoptosis from necrosis. In Cell Death in Biology and Pathology; Bowen, I.D., Lockshin, R.A., Eds.; Chapman and Hall: London, UK, 1981; pp. 9–34. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H. Apoptosis (the 1992 Frank Rose Memorial Lecture). Br. J. Cancer 1993, 67, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Chaudhry, G.E. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L. A boom time for necrobiology. Curr. Biol. 1993, 3, 877–878. [Google Scholar] [CrossRef]
- Milliman, C.L.; Korsmeyer, S.J.; Wang, K.; Yin, X.M.; Chao, D.T. BID: A Novel BH3 Domain-only Death Agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar] [CrossRef]
- Arvanitis, M.; Li, D.D.; Lee, K.; Mylonakis, E. Apoptosis in C. elegans: Lessons for cancer and immunity. Front. Cell. Infect. Microbiol. 2013, 3, 67. [Google Scholar] [CrossRef]
- Raychaudhuri, S. A minimal model of signalling network elucidates cell-to cell stochastic variability in apoptosis. PLoS ONE 2010, 5, e11930. [Google Scholar] [CrossRef] [Green Version]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef]
- Wang, Z.; Figueiredo-Pereira, C.; Oudot, C.; Vieira, H.L.A.; Brenner, C. Mitochondrion: A common organelle for distinct cell deaths? Int. Rev. Cell Mol. Biol. 2017, 331, 245–287. [Google Scholar] [PubMed]
- Urbani, A.; Prosdocimi, E.; Carrer, A.; Checchetto, V.; Szabò, I. Mitochondrial ion channels of the inner membrane and their regulation in cell death signaling. Front. Cell Dev. Biol. 2021, 8, 620081. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Mansour, A.; Van Obberghen-Schilling, E.; Chambard, J.C. Raf-1 activation prevents caspase 9 processing downstream of apoptosome formation. J. Signal Transduct. 2011, 2011, 834948. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Azzwali, A.A.A.; Azab, A.E. Apoptosis: Insight into Stages, Extrinsic and Intrinsic Pathways. Clin. Med. 2019, 7, 80–82. [Google Scholar]
- Muntané, J. Harnessing tumor necrosis factor receptors to enhance antitumor activities of drugs. Chem. Res. Toxicol. 2011, 24, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Aral, K.; Aral, C.A.; Kapila, Y. The role of caspase-8, caspase-9, and apoptosis inducing factor in periodontal disease. J. Periodontol. 2019, 90, 288–294. [Google Scholar] [CrossRef]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and-7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, J.; Guo, Z.; Shi, X.; Zhang, Y.; Zhang, L.; Yu, Q.; Han, L. Effect of Oxidative Stress on AIF-mediated Apoptosis and Bovine Muscle Tenderness during Postmortem Aging. J. Food Sci. 2020, 85, 77–85. [Google Scholar] [CrossRef]
- Talanian, R.V.; Allen, H. Roles of caspases in inflammation and apoptosis: Prospects as drug discovery targets. Annu. Rep. Med. Chem. 1998, 33, 273–282. [Google Scholar]
- Bojarska-Junak, A.; Sieklucka, M.; Hus, I.; Wąsik-Szczepanek, E.; Kusz, M.L.; Surdacka, A.; Chocholska, S.; Dmoszyńska, A.; Roliński, J. Assessment of the pathway of apoptosis involving PAR-4, DAXX and ZIPK proteins in CLL patients and its relationship with the principal prognostic factors. Folia Histochem. Cytobiol. 2011, 49, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.K.; Chang, S.N.; Vadlamudi, Y.; Park, J.G.; Kang, S.C. Synergistic therapy with tangeretin and 5-fluorouracil accelerates the ROS/JNK mediated apoptotic pathway in human colorectal cancer cell. Food Chem. Toxicol. 2020, 143, 111529. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Brenner, C.; Marzo, I.; Susin, S.A.; Kroemer, G. Subcellular and submitochondrial mode of action of Bcl-2 like oncoproteins. Oncogene 1998, 16, 2265–2282. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Zukowska-Szczechowska, E. Caspases and apoptosis: Die and let live. Wiad. Lec. 2002, 55, 100–106. [Google Scholar]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Hussar, P.; Žuravskaja, M.; Kärner, M. Apoptosis regulator BCL-2. Pap. Anthropol. 2013, XXII, 63–67. [Google Scholar] [CrossRef]
- Reed, J.C.; Zha, H.; Aime-Sempe, C.; Takayama, S.; Wang, H.G. Structure-function analysis of Bcl-2 family proteins. Regulators of programmed cell death. Adv. Exp. Med. Biol. 1996, 406, 99–112. [Google Scholar] [CrossRef]
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.; Andrews, D. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. Bcl-x, a bcl-2 related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Bcl-2 family: Life-or-death switch. FEBS Lett. 2000, 466, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Green, D. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef]
- Martinou, J.-C.; Green, R.D. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibtion. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro- caspase -3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999, 18, 2040–2048. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Bcl-2 Family of Proteins: Life-or-Death Switch in Mitochondria. Biosci. Rep. 2002, 22, 47–58. [Google Scholar] [CrossRef]
- Hippenstiel, S.; Schmeck, B.; N’Guessan, P.D.; Seybold, J.; Krüll, M.; Preissner, K.; Eichel-Streiber, C.V.; Suttorp, N. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L830–L838. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.T.; Letai, A. BH3-only proteins and their effects on cancer. Adv. Exp. Med. Biol 2010, 687, 49–63. [Google Scholar] [CrossRef]
- MacManus, J.P.; Matthew, D. Gene expression induced by cerebral ischemia: An apoptotic perspective. J. Cereb. Blood Flow Metab. 1997, 17, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.-L.; et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef]
- Minn, A.J.; Velez, P.; Schendel, S.L.; Liang, H.; Muchmore, S.W.; Fesik, S.W.; Fill, M.; Thompson, C.B. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature 1997, 385, 353–357. [Google Scholar] [CrossRef]
- Horvitz, H.R. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999, 59, 1701s–1706s. [Google Scholar]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t (14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Cleary, M.L.; Smith, S.D.; Sklar, J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the (14;18) translocation. Cell 1986, 47, 19–28. [Google Scholar] [CrossRef]
- Willis, S.; Day, C.L.; Hinds, M.G.; Huang, D.C.S. The Bcl-2-regulated apoptotic pathway. J Cell Sci. 2003, 116, 4053–4056. [Google Scholar] [CrossRef]
- Otake, Y.; Soundararajan, S.; Sengupta, T.K.; Kio, E.A.; Smith, J.C.; Pineda-Roman, M.; Stuart, R.K.; Spicer, E.K.; Fernandes, D.J. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood 2007, 109, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, L.; Wojciechowski, P.; Kazmierczak, M.; Lewandowski, K. Transcript-Level Dysregulation of BCL2 Family Genes in Acute Myeloblastic Leukemia. Cancers 2021, 13, 3175. [Google Scholar] [CrossRef] [PubMed]
- Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A.; Jarskog, L.F. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr. Res. 2006, 81, 47–63. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Richardson, A.; Kaye, S.B. Pharmacological inhibition of the Bcl-2 family of apoptosis regulators as cancer therapy. Curr. Mol. Pharmacol. 2008, 1, 244–254. [Google Scholar] [CrossRef]
- Charlotte, F.; L’Herminé, A.; Martin, N.; Geleyn, Y.; Nollet, M.; Gaulard, P.; Zafrani, E.S. Immunohistochemical detection of bcl-2 protein in normal and pathological human liver. Am. J. Pathol. 1994, 144, 460–465. [Google Scholar]
- Patel, T.; Gores, G.J. Apoptosis and hepatobiliary disease. Hepatology 1995, 21, 1725–1741. [Google Scholar] [CrossRef]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef]
- Frommel, T.O.; Yong, S.; Zarling, E.J. Immunohistochemical evaluation of Bcl-2 gene family expression in liver of hepatitis C and cirrhotic patients: A novel mechanism to explain the high incidence of hepatocarcinoma in cirrhotics. Am. J. Gastroenterol. 1999, 94, 178–182. [Google Scholar] [CrossRef]
- Tsamandas, A.C.; Thomopoulos, K.; Zolota, V.; Kourelis, T.; Karatzas, T.; Ravazoula, P.; Tepetes, K.; Petsas, T.; Karavias, D.; Karatza, C.; et al. Potential Role of Bcl-2 and Bax mRNA and Protein Expression in Chronic Hepatitis Type B and C: A Clinicopathologic Study. Mod. Pathol. 2003, 16, 1273–1288. [Google Scholar] [CrossRef]
- Tokin, I.I.; Hussar, P.; Tokin, I.B.; Filimonova, G.; Zuravaskaja, M.; Tisler, A.; Järveots, T. Immunohiostochemical evaluation of expression of Bcl-2 in chronic viral hepatitis B. BJVM 2012, 15, 66. [Google Scholar]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent Anion Channel 1-based Peptides Interact with Bcl-2 to Prevent Antiapoptotic Activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B.; Montessuit, S.; Lauper, S.; Eskes, R.; Martinou, J.C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000, 345 Pt 2, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W.; Shi, Y. Structural biology. Controlling the caspases. Science 2001, 294, 1477–1478. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies Within. Science 1998, 281, 1312–1326. [Google Scholar] [CrossRef]
- Galluzzi, L.; López-Soto, A.; Kumar, S.; Kroemer, G. Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 2016, 44, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.; Srinivasula, S.M.; Hegde, R.; Mukattash, R.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification and characterization of murine caspase-14, a new member of the caspase family. Cancer Res. 1998, 58, 5201–5205. [Google Scholar]
- Markiewicz, A.; Sigorski, D.; Markiewicz, M.; Owczarczyk-Saczonek, A.; Placek, W. Caspase-14—From Biomolecular Basics to Clinical Approach. A Review of Available Data. Int. J. Mol. Sci. 2021, 22, 5575. [Google Scholar] [CrossRef]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384 Pt 2, 201–232. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Goodsell, D.S. The molecular perspective: Caspases. Oncologist 2000, 5, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Ruck, P.; Schulze-Osthoff, K. In situ monitoring of caspase activation in hepatobiliary diseases. Cell Death Differ. 2000, 7, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Lügering, A.; Poremba, C.; Lügering, N.; Held, J.; Domschke, W.; Schulze-Osthoff, K. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology 2001, 34 Pt 1, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Ruck, P.; Gregor, M.; Schulze-Osthoff, K. Detection of elevated caspase activation and early apoptosis in liver diseases. Eur. J. Cell Biol. 2001, 80, 230–239. [Google Scholar] [CrossRef]
- Bantel, H.; Lügering, A.; Heidemann, J.; Volkmann, X.; Poremba, C.; Strassburg, C.P.; Manns, M.P.; Schulze-Osthoff, K. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology 2004, 40, 1078–1087. [Google Scholar] [CrossRef]
- Wieckowska, A.; Zein, N.N.; Yerian, L.M.; Lopez, A.R.; McCullough, A.J.; Feldstein, A.E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006, 44, 27–33. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Lahm, A.; Paradisi, A.; Green, D.R.; Melino, G. Death fold domain interaction in apoptosis. Cell Death Differ. 2003, 10, 10–12. [Google Scholar] [CrossRef]
- Gu, J.; Zhan, A.J.; Jiang, J.L.; Chen, Y.; Xu, J.; Ye, L.; Mao, M.G. Conserved function of Pacific cod Caspase-3 in apoptosis. Gene 2020, 732, 144370. [Google Scholar] [CrossRef]
- Salvesen, G.S. Caspases: Opening the boxes and interpreting the arrows. Cell Death Differ. 2002, 9, 3–5. [Google Scholar] [CrossRef]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.; Pop, C.; Scott, F.L.; Drag, M.; Swartz, P.; Mattos, C.; Salvesen, G.S.; Clark, A.C. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem. J. 2009, 424, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.K.; Smyth, M.J.; Stennicke, H.R.; Salvesen, G.S.; Duriez, P.; Poirier, G.G.; Hannun, Y.A. Zinc is a potent inhibitor of the apoptotic protease, caspase -3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 1997, 272, 18530–18533. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Katunuma, N.; Matsui, A.; Le, Q.; Utsumi, K.; Salvesen, G.; Ohashi, A. Novel procaspase-3 activating cascade mediated by lysoapoptases and its biological significances in apoptosis. Adv. Enzym. Regul. 2001, 41, 237–250. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Wang, X. Mitochondrial activation of apoptosis. Cell 2004, 116 (Suppl. S2), S57–S59. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2665–2672. [Google Scholar] [CrossRef]
- Denault, J.-B.; Eckelman, B.P.; Shin, H.; Pop, C.; Salvesen, G.S. Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)–mediated inhibition of caspase 9. Biochem. J. 2007, 405, 11–19. [Google Scholar] [CrossRef]
- Agosto, M.; Azrin, M.; Singh, K.; Jaffe, A.S.; Liang, B.T. Serum Caspase-3 p17 Fragment Is Elevated in Patients With ST-Segment Elevation Myocardial Infarction: A Novel Observation. J. Am. Coll. Cardiol. 2011, 57, 220–221. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Megeney, L.A. Rehabilitation of a Contract Killer: Caspase-3 Directs Stem Cell Differentiation. Cell Stem Cell 2008, 2, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol. Appl. Biochem. 2021, 69, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 Is Required for DNA Fragmentation and Morphological Changes Associated with Apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Tokin, I.I.; Tokin, I.B.; Filimonova, G.F.; Hussar, P. Comparative Immunohistochemical Detection of Caspase-3 Activation in Liver Biopsy Specimens of Patients with Mono and Mixed Infection. AJPNM 2013, 1, 92–97. [Google Scholar]
- Rusin, P.; Jabłońska, K. Disturbances in the Mechanism of Apoptosis as One of the Causes of the Development of Cancer Diseases. Studia Ecol. Bioethicae 2020, 18, 63–73. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Skommer, J.; Brittain, T.; Raychaudhuri, S. Bcl-2 inhibits apoptosis by increasing the time-to-death and intrinsic cell-to-cell variations in the mitochondrial pathway of cell death. Apoptosis 2010, 15, 1223–1233. [Google Scholar] [CrossRef]
- Hatok, J.; Racay, P. Bcl-2 family proteins: Master regulators of cell survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef]
- Suraweera, C.D.; Banjara, S.; Hinds, M.G.; Kvansakul, M. Metazoans and Intrinsic Apoptosis: An Evolutionary Analysis of the Bcl-2 Family. Int. J. Mol. Sci. 2022, 23, 3691. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domaineither sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Guedes, J.P.; Baptista, V.; Santos-Pereira, C.; Sousa, M.J.; Manon, S.; Chaves, S.R.; Côrte-Real, M. Acetic acid triggers cytochrome c release in yeast heterologously expressing human Bax. Apoptosis 2022, 27, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Mason, B.L.; Diaz, A.P.; Jha, M.K.; Soares, J.C.; Trivedi, M.H.; Quevedo, J. Dysregulation of mitochondrial dynamics, mitophagy and apoptosis in major depressive disorder: Does inflammation play a role? Mol. Psychiatry 2021, 27, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Sarić, N.; Hashimoto-Torii, K.; Jevtović-Todorović, V.; Ishibashi, N. Nonapoptotic caspases in neural development and in anesthesia-induced neurotoxicity. Trends Neurosci. 2022, 45, 446–458. [Google Scholar] [CrossRef]
- Christgen, S.; Tweedell, R.E.; Kanneganti, T.-D. Programming inflammatory cell death for therapy. Pharmacol. Ther. 2021, 232, 108010. [Google Scholar] [CrossRef]
- Ersoy, T.; Ozmen, O. Immunohistochemical detection of caspase 3 and proliferating cell nuclear antigen in the intestines of dogs naturally infected with parvovirus. Vet. Res. Forum 2022, 1, 127–131. [Google Scholar] [CrossRef]
Figure 1.
Intrinsic (mitochondrial) pathway of apoptosis.

Figure 2.
Extrinsic pathway of apoptosis.


{kind=link}
{kind=link}
Table 1.
Bcl-2 family pro-and anti-apoptotic members.
| Pro-apoptotic members | Pro-apoptotic proteins (pore-formers): Bax Bcl-2-like protein 4 Bak Bcl-2 homologous antagonist killer Bok Bcl-2-related ovarian killer Mtd Matador |
| Pro-apoptotic (BH3-only) proteins: Bad Bcl-2-associated death promoter Bid BH3 interacting domain death agonist Bik Bcl-2-interacting killer Bim Bcl-2-like protein 11 Bmf Bcl-2-modifying factor Egl-1 Caenorhabditis elegans gene egl-1 Hrk Activator of apoptosis harakiri Puma p53 upregulated modulator of apoptosis etc. | |
| Anti-apoptotic members | Bcl-2 B-cell CLL/lymphoma 2 Bcl-xL B-cell lymphoma-extra large Mcl-1 MCL1, myeloid cell leukemia 1 Bcl-w Bcl-2-like protein 2, B-cell lymphoma-w CED-9 C. elegans gene ced-9 Bfl-1/A1 Burkholderia lethal factor 1 etc. |
Table 2.
Division of caspases.
| Inflammatory type of caspases | Caspase-1, -4, -5, -11, -12, -13 |
| Initiator type of caspases | Caspase-2, -8, -9, -10 |
| Executioner type of caspases | Caspase-3, -6, -7 |
| Other type of caspases | Caspase-14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624-1636. https://doi.org/10.3390/encyclopedia2040111
AMA Style
Hussar P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia. 2022; 2(4):1624-1636. https://doi.org/10.3390/encyclopedia2040111
Chicago/Turabian StyleHussar, Piret. 2022. "Apoptosis Regulators Bcl-2 and Caspase-3" Encyclopedia 2, no. 4: 1624-1636. https://doi.org/10.3390/encyclopedia2040111