
The View of Pediatric Nephrotic Syndrome as a Podocytopathy
 ,
,
Abstract
:1. Introduction
2. Pathophysiological Mechanisms of the Podocytopathies
2.1. Pathophysiology of Nephrotic Syndrome and the Glomerular Filtration
2.2. Podocyte Foot Processes: Update on Molecular Anatomy and Effacement
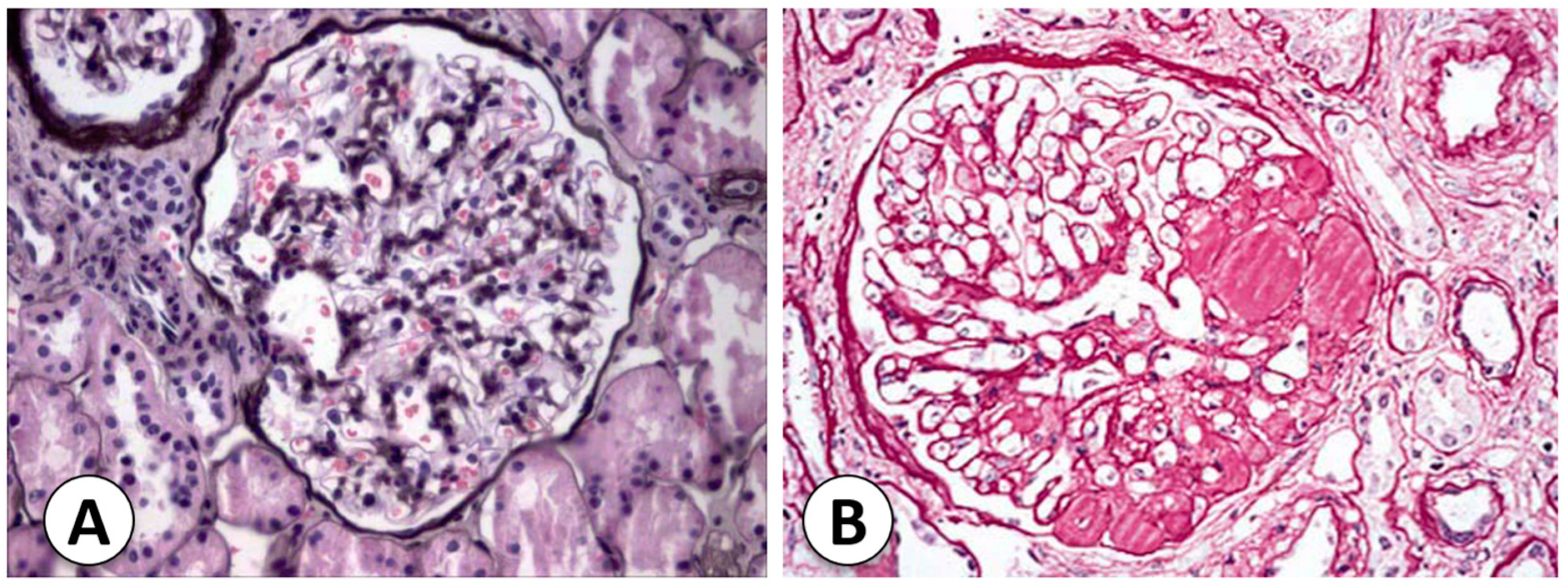
2.3. Histopathology of Common Types of Nephrotic Syndrome in Childhood
2.3.1. Congenital Nephrotic Syndrome (Finnish Type)
2.3.2. Diffuse Mesangial Sclerosis
2.3.3. Focal Segmental Glomerular Sclerosis
2.4. Histological Classifications for Focal Segmental Glomerular Sclerosis
2.4.1. Cellular Focal Segmental Glomerular Sclerosis
2.4.2. Primary Focal Segmental Glomerular Sclerosis with Mesangial Hypercellularity
2.4.3. Familial Focal Segmental Glomerular Sclerosis
2.4.4. Secondary Focal Segmental Glomerular Sclerosis
2.4.5. Collapsing Glomerulopathy
2.4.6. C1q Nephropathy
2.5. Hypotheses Linking Glomerulosclerosis to Podocyte-Derived Alterations
2.5.1. Immune-Mediated
2.5.2. Systemic Circulating Factors

{kind=link}
{kind=link}
{kind=link}
| Nephrotic Syndrome | Characteristics | Causes | References |
|---|---|---|---|
| Congenital nephrotic syndrome (Finnish type) | Congenital NS is an autosomal-recessive disorder with heavy proteinuria in the neonatal period. A renal biopsy shows many glomeruli with mesangial hypercellularity and hyperlobulated capillary tufts. Proximal and distal tubules may present microcystic dilatation, and the podocytes present FP effacement. | The mutated gene related to Finnish nephropathy (FN) is NPHS1, mapped to the long arm of chromosome 19 (19q13.1), which codes for nephrin. | [2] |
| Diffuse mesangial sclerosis | It presents nonspecific mesangial IgM, C3, and C1q deposits, detected via immunofluorescence. Electron microscopy shows podocyte hypertrophy and irregular FP effacement. Late stages of DMS exhibit thickened capillary loops, a decrease in capillary lumens, and the formation of a sclerotic mass in the mesangium. | WT1 gene mutations in exons 8 or 9 are possible causes. | [2,18] |
| Focal segmental glomerular sclerosis | FSGS is related to the presence of segmental sclerotic lesions within the glomeruli. | Some molecules: cardiotrophin-like cytokine factor 1, apoA1b (an isoform of ApoA1), anti-CD40 antibody, and serum urine-type plasminogen activator receptor (suPAR). More than 50 genes are potential factors for monogenic forms of FSGS. Drugs and anabolic steroid abuse are other factors. | [2,6,24,25,26,27,29,30,31,32] |
| Cellular FSGS | It presents hypercellularity and endocapillary proliferation with luminal obliteration of capillaries. | ||
| Primary FSGS with mesangial hypercellularity | This type is associated with mesangial hypercellularity in the non-sclerotic glomeruli. | ||
| Familial FSGS | It exhibits loss of podocin staining, detected using immunohistochemistry methods. | Mutations in the NPHS2 gene, which codes for podocin. | [2,18] |
| Secondary FSGS | This type presents mesangial hypercellularity, mesangial IgA, and basement membrane abnormalities. | IgA nephropathy, hereditary nephritis (Alport’s syndrome), and lupus nephritis. | [2,51,52,53] |
| Collapsing glomerulopathy | It is related to the implosive collapse of the capillary loops with alterations of the basement membrane and hypertrophy and hyperplasia of podocytes. | COQ2 mutations that cause mitochondrial disorders. HIV, malaria, visceral leishmaniasis, cytomegalovirus, parvovirus B19, hepatitis C virus, dengue virus, and Zika virus are possible factors. | [2,55,56,57,58,59] |
2.5.3. Genetic Variants
3. Molecular and Genetic Markers
3.1. Genetic Mutations
3.2. Molecular Markers
3.2.1. MicroRNAs
3.2.2. Proteomics
3.2.3. Inflammatory Markers, Cytokines, and Chemokines
3.2.4. Other Potential Markers
3.3. Metabolomics
3.4. Biomarkers Associated with Histological Features
3.4.1. Transforming Growth Factor Beta (TGF-β)
3.4.2. CD44
3.4.3. CD80
3.4.4. Extracellular Vesicles
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Noone, D.G.; Iijima, K.; Parekh, R. Idiopathic Nephrotic Syndrome in Children. Lancet 2018, 392, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S. Pathology of Podocytopathies Causing Nephrotic Syndrome in Children. Front. Pediatr. 2016, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.S.; Amaker, B.H.; Chan, J.C.M. Nephrotic Syndrome: Pathogenesis and Management. Pediatr. Rev. 2002, 23, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Banh, T.H.M.; Hussain-Shamsy, N.; Patel, V.; Vasilevska-Ristovska, J.; Borges, K.; Sibbald, C.; Lipszyc, D.; Brooke, J.; Geary, D.; Langlois, V.; et al. Ethnic Differences in Incidence and Outcomes of Childhood Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A.; Symons, J.M. Nephrotic Syndrome in Childhood. Lancet 2003, 362, 629–639. [Google Scholar] [CrossRef]
- Mundel, P.; Shankland, S.J. Podocyte Biology and Response to Injury. J. Am. Soc. Nephrol. 2002, 13, 3005–3015. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal Segmental Glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef]
- Downie, M.L.; Gallibois, C.; Parekh, R.S.; Noone, D.G. Nephrotic Syndrome in Infants and Children: Pathophysiology and Management. Paediatr. Int. Child. Health 2017, 37, 248–258. [Google Scholar] [CrossRef]
- Pollak, M.R.; Quaggin, S.E.; Hoenig, M.P.; Dworkin, L.D. The Glomerulus: The Sphere of Influence. Clin. J. Am. Soc. Nephrol. 2014, 9, 1461–1469. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Nishinakamura, R. Podocyte Development, Disease, and Stem Cell Research. Kidney Int. 2019, 96, 1077–1082. [Google Scholar] [CrossRef]
- Donoviel, D.B.; Freed, D.D.; Vogel, H.; Potter, D.G.; Hawkins, E.; Barrish, J.P.; Mathur, B.N.; Turner, C.A.; Geske, R.; Montgomery, C.A.; et al. Proteinuria and Perinatal Lethality in Mice Lacking NEPH1, a Novel Protein with Homology to NEPHRIN. Mol. Cell Biol. 2001, 21, 4829–4836. [Google Scholar] [CrossRef] [PubMed]
- Blaine, J.; Dylewski, J. Regulation of the Actin Cytoskeleton in Podocytes. Cells 2020, 9, 1700. [Google Scholar] [CrossRef]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Young Choi, H.; Hyung Chang, J.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin Protects Against Proteinuria by Disrupting Cdc42:IRSp53:Mena Signaling Complexes in Kidney Podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Garola, R.E.; Whiting, J.M.; Alon, U.S. Synaptopodin Expression in Idiopathic Nephrotic Syndrome of Childhood. Kidney Int. 2001, 59, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.W.; Venkatachalam, M.A.; Cotran, R.S. Glomerular Epithelium: Structural Alterations Induced by Polycations. Science 1975, 189, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Kerjaschki, D. Polycation-Induced Dislocation of Slit Diaphragms and Formation of Cell Junctions in Rat Kidney Glomeruli: The Effects of Low Temperature, Divalent Cations, Colchicine, and Cytochalasin B. Lab. Investig. 1978, 39, 430–440. [Google Scholar] [PubMed]
- da Silva Filha, R.; Burini, K.; Pires, L.G.; Brant Pinheiro, S.V.; Simões E Silva, A.C. Idiopathic Nephrotic Syndrome in Pediatrics: An Up-to-Date. Curr. Pediatr. Rev. 2022, 18, 251–264. [Google Scholar] [CrossRef]
- Liapis, H. Molecular Pathology of Nephrotic Syndrome in Childhood: A Contemporary Approach to Diagnosis. Pediatr. Dev. Pathol. 2008, 11, 154–163. [Google Scholar] [CrossRef]
- Kriz, W.; Elger, M.; Nagata, M.; Kretzler, M.; Uiker, S.; Koeppen-Hageman, I.; Tenschert, S.; Lemley, K.V. The Role of Podocytes in the Development of Glomerular Sclerosis. Kidney Int. Suppl. 1994, 45, S64–S72. [Google Scholar]
- Kriz, W.; Endlich, K. Hypertrophy of Podocytes: A Mechanism to Cope with Increased Glomerular Capillary Pressures? Kidney Int. 2005, 67, 373–374. [Google Scholar] [CrossRef]
- Königshausen, E.; Sellin, L. Circulating Permeability Factors in Primary Focal Segmental Glomerulosclerosis: A Review of Proposed Candidates. Biomed. Res. Int. 2016, 2016, 3765608. [Google Scholar] [CrossRef] [PubMed]
- Gallon, L.; Leventhal, J.; Skaro, A.; Kanwar, Y.; Alvarado, A. Resolution of Recurrent Focal Segmental Glomerulosclerosis after Retransplantation. N. Engl. J. Med. 2012, 366, 1648–1649. [Google Scholar] [CrossRef] [PubMed]
- Uffing, A.; Pérez-Sáez, M.J.; Mazzali, M.; Manfro, R.C.; Bauer, A.C.; de Drumond, F.S.; O’Shaughnessy, M.M.; Cheng, X.S.; Chin, K.-K.; Ventura, C.G.; et al. Recurrence of FSGS after Kidney Transplantation in Adults. Clin. J. Am. Soc. Nephrol. CJASN 2020, 15, 247. [Google Scholar] [CrossRef] [PubMed]
- Savin, V.J.; Sharma, M.; Zhou, J.; Gennochi, D.; Fields, T.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Domen, J.; Tormo, A.; et al. Renal and Hematological Effects of CLCF-1, a B-Cell-Stimulating Cytokine of the IL-6 Family. J. Immunol. Res. 2015, 2015, 714964. [Google Scholar] [CrossRef]
- Lopez-Hellin, J.; Cantarell, C.; Jimeno, L.; Sanchez-Fructuoso, A.; Puig-Gay, N.; Guirado, L.; Vilariño, N.; Gonzalez-Roncero, F.M.; Mazuecos, A.; Lauzurica, R.; et al. A Form of Apolipoprotein A-I Is Found Specifically in Relapses of Focal Segmental Glomerulosclerosis Following Transplantation. Am. J. Transplant. 2013, 13, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Delville, M.; Sigdel, T.K.; Wei, C.; Li, J.; Hsieh, S.-C.; Fornoni, A.; Burke, G.W.; Bruneval, P.; Naesens, M.; Jackson, A.; et al. A Circulating Antibody Panel for Pretransplant Prediction of FSGS Recurrence after Kidney Transplantation. Sci. Transl. Med. 2014, 6, 256ra136. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.-K.; Saleem, M.; et al. Circulating Urokinase Receptor as a Cause of Focal Segmental Glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.J.H.; Deegens, J.K.J.; Wetzels, J.F.M. Serum suPAR in Patients with FSGS: Trash or Treasure? Pediatr. Nephrol. 2013, 28, 1041–1048. [Google Scholar] [CrossRef]
- Rood, I.M.; Deegens, J.K.J.; Wetzels, J.F.M. Genetic Causes of Focal Segmental Glomerulosclerosis: Implications for Clinical Practice. Nephrol. Dial. Transplant. 2012, 27, 882–890. [Google Scholar] [CrossRef]
- Santín, S.; Bullich, G.; Tazón-Vega, B.; García-Maset, R.; Giménez, I.; Silva, I.; Ruíz, P.; Ballarín, J.; Torra, R.; Ars, E. Clinical Utility of Genetic Testing in Children and Adults with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2011, 6, 1139–1148. [Google Scholar] [CrossRef]
- Markowitz, G.S.; Appel, G.B.; Fine, P.L.; Fenves, A.Z.; Loon, N.R.; Jagannath, S.; Kuhn, J.A.; Dratch, A.D.; D’Agati, V.D. Collapsing Focal Segmental Glomerulosclerosis Following Treatment with High-Dose Pamidronate. J. Am. Soc. Nephrol. 2001, 12, 1164–1172. [Google Scholar] [CrossRef]
- Herlitz, L.C.; Markowitz, G.S.; Farris, A.B.; Schwimmer, J.A.; Stokes, M.B.; Kunis, C.; Colvin, R.B.; D’Agati, V.D. Development of Focal Segmental Glomerulosclerosis after Anabolic Steroid Abuse. J. Am. Soc. Nephrol. 2010, 21, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Rheault, M.N.; Gbadegesin, R.A. The Genetics of Nephrotic Syndrome. J. Pediatr. Genet. 2016, 5, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hinkes, B.; Wiggins, R.C.; Gbadegesin, R.; Vlangos, C.N.; Seelow, D.; Nürnberg, G.; Garg, P.; Verma, R.; Chaib, H.; Hoskins, B.E.; et al. Positional Cloning Uncovers Mutations in PLCE1 Responsible for a Nephrotic Syndrome Variant That May Be Reversible. Nat. Genet. 2006, 38, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D. Pathobiology of Focal Segmental Glomerulosclerosis: New Developments. Curr. Opin. Nephrol. Hypertens. 2012, 21, 243–250. [Google Scholar] [CrossRef]
- Paranhos, R.M.; De Souza Figueiredo, G.A.; De Abreu, G.R.; Ferreira, G.C.; Fonseca, G.G.; Simões E Silva, A.C. Immunoglobulin A Nephropathy in Paediatrics: An up-to-Date. Nephrology 2022, 27, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, A.L.; Bitencourt, L.; Paranhos, R.M.; Leitáo, C.A.; Ferreira, G.C.; Simões E Silva, A.C. Alport Syndrome: A Comprehensive Review on Genetics, Pathophysiology, Histology, Clinical and Therapeutic Perspectives. Curr. Med. Chem. 2021, 28, 5602–5624. [Google Scholar] [CrossRef]
- Pinheiro, S.V.B.; Dias, R.F.; Fabiano, R.C.G.; de Araujo, S.A.; Silva, A.C.S.E. Pediatric Lupus Nephritis. J. Bras. Nefrol. 2019, 41, 252–265. [Google Scholar] [CrossRef]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 Nephropathy: A Newly Described Inherited Mitochondriopathy with Primary Renal Involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef]
- Albaqumi, M.; Barisoni, L. Current Views on Collapsing Glomerulopathy. J. Am. Soc. Nephrol. 2008, 19, 1276–1281. [Google Scholar] [CrossRef]
- Tanji, N.; Ross, M.D.; Tanji, K.; Bruggeman, L.A.; Markowitz, G.S.; Klotman, P.E.; D’Agati, V.D. Detection and Localization of HIV-1 DNA in Renal Tissues by in Situ Polymerase Chain Reaction. Histol. Histopathol. 2006, 21, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Albaqumi, M.; Soos, T.J.; Barisoni, L.; Nelson, P.J. Collapsing Glomerulopathy. J. Am. Soc. Nephrol. 2006, 17, 2854–2863. [Google Scholar] [CrossRef] [PubMed]
- Coventry, S.; Shoemaker, L.R. Collapsing Glomerulopathy in a 16-Year-Old Girl with Pulmonary Tuberculosis: The Role of Systemic Inflammatory Mediators. Pediatr. Dev. Pathol. 2004, 7, 166–170. [Google Scholar] [CrossRef]
- de Araújo, S.A.; Cordeiro, T.M.E.; Belisário, A.R.; Araújo, R.F.; Marinho, P.E.S.; Kroon, E.G.; de Oliveira, D.B.; Teixeira, M.M.; Simões E Silva, A.C. First Report of Collapsing Variant of Focal Segmental Glomerulosclerosis Triggered by Arbovirus: Dengue and Zika Virus Infection. Clin. Kidney J. 2019, 12, 355–361. [Google Scholar] [CrossRef]
- Jennette, J.C.; Hipp, C.G. C1q Nephropathy: A Distinct Pathologic Entity Usually Causing Nephrotic Syndrome. Am. J. Kidney Dis. 1985, 6, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.S.; Schwimmer, J.A.; Stokes, M.B.; Nasr, S.; Seigle, R.L.; Valeri, A.M.; D’Agati, V.D. C1q Nephropathy: A Variant of Focal Segmental Glomerulosclerosis. Kidney Int. 2003, 64, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, R.C. The Spectrum of Podocytopathies: A Unifying View of Glomerular Diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef]
- de Pereira, W.F.; Brito-Melo, G.E.A.; Guimarães, F.T.L.; Carvalho, T.G.R.; Mateo, E.C.; Simões e Silva, A.C. The Role of the Immune System in Idiopathic Nephrotic Syndrome: A Review of Clinical and Experimental Studies. Inflamm. Res. 2014, 63, 1–12. [Google Scholar] [CrossRef]
- Lin, C.Y.; Hsu, H.C. Histopathological and Immunological Studies in Spontaneous Remission of Nephrotic Syndrome after Intercurrent Measles Infection. Nephron 1986, 42, 110–115. [Google Scholar] [CrossRef]
- Audard, V.; Larousserie, F.; Grimbert, P.; Abtahi, M.; Sotto, J.-J.; Delmer, A.; Boue, F.; Nochy, D.; Brousse, N.; Delarue, R.; et al. Minimal Change Nephrotic Syndrome and Classical Hodgkin’s Lymphoma: Report of 21 Cases and Review of the Literature. Kidney Int. 2006, 69, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Kofman, T.; Zhang, S.-Y.; Copie-Bergman, C.; Moktefi, A.; Raimbourg, Q.; Francois, H.; Karras, A.; Plaisier, E.; Painchart, B.; Favre, G.; et al. Minimal Change Nephrotic Syndrome Associated with Non-Hodgkin Lymphoid Disorders: A Retrospective Study of 18 Cases. Medicine 2014, 93, 350–358. [Google Scholar] [CrossRef]
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in Podocytes Is Associated with Nephrotic Syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.W.; Chandar, J.; Sageshima, J.; Ortigosa-Goggins, M.; Amarapurkar, P.; Mitrofanova, A.; Defreitas, M.J.; Katsoufis, C.P.; Seeherunvong, W.; Centeno, A.; et al. Benefit of B7-1 Staining and Abatacept for Treatment-Resistant Post-Transplant Focal Segmental Glomerulosclerosis in a Predominantly Pediatric Cohort: Time for a Reappraisal. Pediatr. Nephrol. 2023, 38, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Delville, M.; Baye, E.; Durrbach, A.; Audard, V.; Kofman, T.; Braun, L.; Olagne, J.; Nguyen, C.; Deschênes, G.; Moulin, B.; et al. B7-1 Blockade Does Not Improve Post-Transplant Nephrotic Syndrome Caused by Recurrent FSGS. J. Am. Soc. Nephrol. 2016, 27, 2520–2527. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, R.; Sharma, M.; McCarthy, E.T.; Swan, S.K.; Ellis, E.; Lovell, H.; Warady, B.; Gunwar, S.; Chonko, A.M.; et al. Circulating Factor Associated with Increased Glomerular Permeability to Albumin in Recurrent Focal Segmental Glomerulosclerosis. N. Engl. J. Med. 1996, 334, 878–883. [Google Scholar] [CrossRef]
- Kemper, M.J.; Wolf, G.; Müller-Wiefel, D.E. Transmission of Glomerular Permeability Factor from a Mother to Her Child. N. Engl. J. Med. 2001, 344, 386–387. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, P.E.C. Vascular Permeability Factors in Steroid-Sensitive Nephrotic Syndrome and Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2003, 18 (Suppl. S6), vi21–vi25. [Google Scholar] [CrossRef] [PubMed]
- Clement, L.C.; Macé, C.; Avila-Casado, C.; Joles, J.A.; Kersten, S.; Chugh, S.S. Circulating Angiopoietin-like 4 Links Proteinuria with Hypertriglyceridemia in Nephrotic Syndrome. Nat. Med. 2014, 20, 37–46. [Google Scholar] [CrossRef]
- McCarthy, E.T.; Sharma, M.; Savin, V.J. Circulating Permeability Factors in Idiopathic Nephrotic Syndrome and Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2115–2121. [Google Scholar] [CrossRef]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef]
- Trautmann, A.; Bodria, M.; Ozaltin, F.; Gheisari, A.; Melk, A.; Azocar, M.; Anarat, A.; Caliskan, S.; Emma, F.; Gellermann, J.; et al. Spectrum of Steroid-Resistant and Congenital Nephrotic Syndrome in Children: The PodoNet Registry Cohort. Clin. J. Am. Soc. Nephrol. 2015, 10, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Gbadegesin, R.A.; Adeyemo, A.; Webb, N.J.A.; Greenbaum, L.A.; Abeyagunawardena, A.; Thalgahagoda, S.; Kale, A.; Gipson, D.; Srivastava, T.; Lin, J.-J.; et al. HLA-DQA1 and PLCG2 Are Candidate Risk Loci for Childhood-Onset Steroid-Sensitive Nephrotic Syndrome. J. Am. Soc. Nephrol. 2015, 26, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Lagueruela, C.C.; Buettner, T.L.; Cole, B.R.; Kissane, J.M.; Robson, A.M. HLA Extended Haplotypes in Steroid-Sensitive Nephrotic Syndrome of Childhood. Kidney Int. 1990, 38, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Shih, N.Y.; Li, J.; Karpitskii, V.; Nguyen, A.; Dustin, M.L.; Kanagawa, O.; Miner, J.H.; Shaw, A.S. Congenital Nephrotic Syndrome in Mice Lacking CD2-Associated Protein. Science 1999, 286, 312–315. [Google Scholar] [CrossRef]
- Gigante, M.; Pontrelli, P.; Montemurno, E.; Roca, L.; Aucella, F.; Penza, R.; Caridi, G.; Ranieri, E.; Ghiggeri, G.M.; Gesualdo, L. CD2AP Mutations Are Associated with Sporadic Nephrotic Syndrome and Focal Segmental Glomerulosclerosis (FSGS). Nephrol. Dial. Transplant. 2009, 24, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Löwik, M.M.; Groenen, P.J.T.A.; Pronk, I.; Lilien, M.R.; Goldschmeding, R.; Dijkman, H.B.; Levtchenko, E.N.; Monnens, L.A.; van den Heuvel, L.P. Focal Segmental Glomerulosclerosis in a Patient Homozygous for a CD2AP Mutation. Kidney Int. 2007, 72, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Philippe, A.; Nevo, F.; Esquivel, E.L.; Reklaityte, D.; Gribouval, O.; Tête, M.-J.; Loirat, C.; Dantal, J.; Fischbach, M.; Pouteil-Noble, C.; et al. Nephrin Mutations Can Cause Childhood-Onset Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2008, 19, 1871–1878. [Google Scholar] [CrossRef]
- Li, X.; Chuang, P.Y.; D’Agati, V.D.; Dai, Y.; Yacoub, R.; Fu, J.; Xu, J.; Taku, O.; Premsrirut, P.K.; Holzman, L.B.; et al. Nephrin Preserves Podocyte Viability and Glomerular Structure and Function in Adult Kidneys. J. Am. Soc. Nephrol. 2015, 26, 2361–2377. [Google Scholar] [CrossRef]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally Cloned Gene for a Novel Glomerular Protein—Nephrin—Is Mutated in Congenital Nephrotic Syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Bouchireb, K.; Boyer, O.; Gribouval, O.; Nevo, F.; Huynh-Cong, E.; Morinière, V.; Campait, R.; Ars, E.; Brackman, D.; Dantal, J.; et al. NPHS2 Mutations in Steroid-Resistant Nephrotic Syndrome: A Mutation Update and the Associated Phenotypic Spectrum. Hum. Mutat. 2014, 35, 178–186. [Google Scholar] [CrossRef]
- Boute, N.; Gribouval, O.; Roselli, S.; Benessy, F.; Lee, H.; Fuchshuber, A.; Dahan, K.; Gubler, M.C.; Niaudet, P.; Antignac, C. NPHS2, Encoding the Glomerular Protein Podocin, Is Mutated in Autosomal Recessive Steroid-Resistant Nephrotic Syndrome. Nat. Genet. 2000, 24, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Ebarasi, L.; Ashraf, S.; Bierzynska, A.; Gee, H.Y.; McCarthy, H.J.; Lovric, S.; Sadowski, C.E.; Pabst, W.; Vega-Warner, V.; Fang, H.; et al. Defects of CRB2 Cause Steroid-Resistant Nephrotic Syndrome. Am. J. Hum. Genet. 2015, 96, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Identification and Characterization of Crumbs Homolog 2 Gene at Human Chromosome 9q33.3. Int. J. Oncol. 2004, 24, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Cil, O.; Perwad, F. Monogenic Causes of Proteinuria in Children. Front. Med. 2018, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Liang, X.; Chen, J.; Hong, J.; Li, L.; He, Q.; Cai, X. History and Progression of Fat Cadherins in Health and Disease. Onco. Targets Ther. 2016, 9, 7337–7343. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 Mutations Cause a Glomerulotubular Nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.; Hanby, A.M.; Poulsom, R.; Jones, T.A.; Sheer, D.; Chin, W.G.; Da, S.M.; Zhao, Q.; Beverley, P.C.; Owen, M.J. Molecular Cloning and Tissue Expression of FAT, the Human Homologue of the Drosophila Fat Gene That Is Located on Chromosome 4q34-Q35 and Encodes a Putative Adhesion Molecule. Genomics 1995, 30, 207–223. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Comparative Integromics on FAT1, FAT2, FAT3 and FAT4. Int. J. Mol. Med. 2006, 18, 523–528. [Google Scholar] [CrossRef]
- Yun, S.; Hong, W.-P.; Choi, J.H.; Yi, K.S.; Chae, S.-K.; Ryu, S.H.; Suh, P.-G. Phospholipase C-Epsilon Augments Epidermal Growth Factor-Dependent Cell Growth by Inhibiting Epidermal Growth Factor Receptor down-Regulation. J. Biol. Chem. 2008, 283, 341–349. [Google Scholar] [CrossRef]
- Gbadegesin, R.; Hinkes, B.G.; Hoskins, B.E.; Vlangos, C.N.; Heeringa, S.F.; Liu, J.; Loirat, C.; Ozaltin, F.; Hashmi, S.; Ulmer, F.; et al. Mutations in PLCE1 Are a Major Cause of Isolated Diffuse Mesangial Sclerosis (IDMS). Nephrol. Dial. Transplant. 2008, 23, 1291–1297. [Google Scholar] [CrossRef]
- Kanda, S.; Harita, Y.; Shibagaki, Y.; Sekine, T.; Igarashi, T.; Inoue, T.; Hattori, S. Tyrosine Phosphorylation–Dependent Activation of TRPC6 Regulated by PLC-Γ1 and Nephrin: Effect of Mutations Associated with Focal Segmental Glomerulosclerosis. Mol. Biol. Cell 2011, 22, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Kim, E.Y.; Hagmann, H.; Benzing, T.; Dryer, S.E. Opposing Effects of Podocin on the Gating of Podocyte TRPC6 Channels Evoked by Membrane Stretch or Diacylglycerol. Am. J. Physiol. Cell Physiol. 2013, 305, C276–C289. [Google Scholar] [CrossRef] [PubMed]
- Gigante, M.; Caridi, G.; Montemurno, E.; Soccio, M.; d’Apolito, M.; Cerullo, G.; Aucella, F.; Schirinzi, A.; Emma, F.; Massella, L.; et al. TRPC6 Mutations in Children with Steroid-Resistant Nephrotic Syndrome and Atypical Phenotype. Clin. J. Am. Soc. Nephrol. 2011, 6, 1626–1634. [Google Scholar] [CrossRef]
- D’Esposito, M.; Strazzullo, M.; Cuccurese, M.; Spalluto, C.; Rocchi, M.; D’Urso, M.; Ciccodicola, A. Identification and Assignment of the Human Transient Receptor Potential Channel 6 Gene TRPC6 to Chromosome 11q21-->q22. Cytogenet. Cell Genet. 1998, 83, 46–47. [Google Scholar] [CrossRef]
- Feng, D.; Kumar, M.; Muntel, J.; Gurley, S.B.; Birrane, G.; Stillman, I.E.; Ding, L.; Wang, M.; Ahmed, S.; Schlondorff, J.; et al. Phosphorylation of ACTN4 Leads to Podocyte Vulnerability and Proteinuric Glomerulosclerosis. J. Am. Soc. Nephrol. 2020, 31, 1479–1495. [Google Scholar] [CrossRef]
- Dai, S.; Wang, Z.; Pan, X.; Chen, X.; Wang, W.; Ren, H.; Feng, Q.; He, J.C.; Han, B.; Chen, N. ACTN4 Gene Mutations and Single Nucleotide Polymorphisms in Idiopathic Focal Segmental Glomerulosclerosis. Nephron Clin. Pract. 2009, 111, c87–c94. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; Correia, L.A.; Tong, H.Q.; Mathis, B.J.; Rodríguez-Pérez, J.C.; Allen, P.G.; Beggs, A.H.; et al. Mutations in ACTN4, Encoding Alpha-Actinin-4, Cause Familial Focal Segmental Glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef]
- Gbadegesin, R.A.; Hall, G.; Adeyemo, A.; Hanke, N.; Tossidou, I.; Burchette, J.; Wu, G.; Homstad, A.; Sparks, M.A.; Gomez, J.; et al. Mutations in the Gene That Encodes the F-Actin Binding Protein Anillin Cause FSGS. J. Am. Soc. Nephrol. 2014, 25, 1991–2002. [Google Scholar] [CrossRef]
- Hall, G.; Lane, B.M.; Khan, K.; Pediaditakis, I.; Xiao, J.; Wu, G.; Wang, L.; Kovalik, M.E.; Chryst-Stangl, M.; Davis, E.E.; et al. The Human FSGS-Causing ANLN R431C Mutation Induces Dysregulated PI3K/AKT/mTOR/Rac1 Signaling in Podocytes. J. Am. Soc. Nephrol. 2018, 29, 2110–2122. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Identification and Characterization of ARHGAP24 and ARHGAP25 Genes in Silico. Int. J. Mol. Med. 2004, 14, 333–338. [Google Scholar] [CrossRef]
- Akilesh, S.; Suleiman, H.; Yu, H.; Stander, M.C.; Lavin, P.; Gbadegesin, R.; Antignac, C.; Pollak, M.; Kopp, J.B.; Winn, M.P.; et al. Arhgap24 Inactivates Rac1 in Mouse Podocytes, and a Mutant Form Is Associated with Familial Focal Segmental Glomerulosclerosis. J. Clin. Investig. 2011, 121, 4127–4137. [Google Scholar] [CrossRef]
- Gupta, I.R.; Baldwin, C.; Auguste, D.; Ha, K.C.H.; El Andalousi, J.; Fahiminiya, S.; Bitzan, M.; Bernard, C.; Akbari, M.R.; Narod, S.A.; et al. ARHGDIA: A Novel Gene Implicated in Nephrotic Syndrome. J. Med. Genet. 2013, 50, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Tommerup, N.; Wirth, J.; Leffers, H.; Zimmer, J.; Back, E.; Weissenbach, J.; Scherer, G. A Somatic Cell Hybrid Panel for Distal 17q: GDIA1 Maps to 17q25.3. Cytogenet. Cell Genet. 1997, 76, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, E.S.; Higgs, H.N. INF2 Is a WASP Homology 2 Motif-Containing Formin That Severs Actin Filaments and Accelerates Both Polymerization and Depolymerization. J. Biol. Chem. 2006, 281, 26754–26767. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Huynh Cong, E.; Arrondel, C.; Tête, M.-J.; Montjean, R.; et al. INF2 Mutations in Charcot-Marie-Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Iatropoulos, P.; Donadelli, R.; Calabria, A.; Maranta, R.; Cassis, P.; Buelli, S.; Tomasoni, S.; Piras, R.; Krendel, M.; et al. MYO1E Mutations and Childhood Familial Focal Segmental Glomerulosclerosis. N. Engl. J. Med. 2011, 365, 295–306. [Google Scholar] [CrossRef]
- Krendel, M.; Kim, S.V.; Willinger, T.; Wang, T.; Kashgarian, M.; Flavell, R.A.; Mooseker, M.S. Disruption of Myosin 1e Promotes Podocyte Injury. J. Am. Soc. Nephrol. 2009, 20, 86–94. [Google Scholar] [CrossRef]
- Hasson, T.; Skowron, J.F.; Gilbert, D.J.; Avraham, K.B.; Perry, W.L.; Bement, W.M.; Anderson, B.L.; Sherr, E.H.; Chen, Z.Y.; Greene, L.A.; et al. Mapping of Unconventional Myosins in Mouse and Human. Genomics 1996, 36, 431–439. [Google Scholar] [CrossRef]
- Gee, H.Y.; Zhang, F.; Ashraf, S.; Kohl, S.; Sadowski, C.E.; Vega-Warner, V.; Zhou, W.; Lovric, S.; Fang, H.; Nettleton, M.; et al. KANK Deficiency Leads to Podocyte Dysfunction and Nephrotic Syndrome. J. Clin. Investig. 2015, 125, 2375–2384. [Google Scholar] [CrossRef]
- Sarkar, S.; Roy, B.C.; Hatano, N.; Aoyagi, T.; Gohji, K.; Kiyama, R. A Novel Ankyrin Repeat-Containing Gene (Kank) Located at 9p24 Is a Growth Suppressor of Renal Cell Carcinoma. J. Biol. Chem. 2002, 277, 36585–36591. [Google Scholar] [CrossRef]
- Pei, Q.; Ni, W.; Yuan, Y.; Yuan, J.; Zhang, X.; Yao, M. HSP70 Ameliorates Septic Lung Injury via Inhibition of Apoptosis by Interacting with KANK2. Biomolecules 2022, 12, 410. [Google Scholar] [CrossRef]
- Anjanappa, R.M.; Nayak, S.; Moily, N.S.; Manduva, V.; Nadella, R.K.; Viswanath, B.; Reddy, Y.C.J.; Jain, S.; Anand, A. A Linkage and Exome Study Implicates Rare Variants of KANK4 and CAP2 in Bipolar Disorder in a Multiplex Family. Bipolar Disord. 2020, 22, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q Biosynthesis in Health and Disease. Biochim. Biophys. Acta 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 Mutations Promote Steroid-Resistant Nephrotic Syndrome through CoQ10 Biosynthesis Disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef]
- Forsgren, M.; Attersand, A.; Lake, S.; Grünler, J.; Swiezewska, E.; Dallner, G.; Climent, I. Isolation and Functional Expression of Human COQ2, a Gene Encoding a Polyprenyl Transferase Involved in the Synthesis of CoQ. Biochem. J. 2004, 382, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of Coenzyme Q10 Deficiency. Mol. Syndromol. 2014, 5, 156–162. [Google Scholar] [CrossRef]
- Gigante, M.; Diella, S.; Santangelo, L.; Trevisson, E.; Acosta, M.J.; Amatruda, M.; Finzi, G.; Caridi, G.; Murer, L.; Accetturo, M.; et al. Further Phenotypic Heterogeneity of CoQ10 Deficiency Associated with Steroid Resistant Nephrotic Syndrome and Novel COQ2 and COQ6 Variants. Clin. Genet. 2017, 92, 224–226. [Google Scholar] [CrossRef]
- Park, E.; Ahn, Y.H.; Kang, H.G.; Yoo, K.H.; Won, N.H.; Lee, K.B.; Moon, K.C.; Seong, M.-W.; Gwon, T.R.; Park, S.S.; et al. COQ6 Mutations in Children With Steroid-Resistant Focal Segmental Glomerulosclerosis and Sensorineural Hearing Loss. Am. J. Kidney Dis. 2017, 70, 139–144. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 Mutations in Human Patients Produce Nephrotic Syndrome with Sensorineural Deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef]
- Li, Y.; Lin, S.; Li, L.; Tang, Z.; Hu, Y.; Ban, X.; Zeng, T.; Zhou, Y.; Zhu, Y.; Gao, S.; et al. PDSS2 Deficiency Induces Hepatocarcinogenesis by Decreasing Mitochondrial Respiration and Reprogramming Glucose Metabolism. Cancer Res. 2018, 78, 4471–4481. [Google Scholar] [CrossRef]
- Gasser, D.L.; Winkler, C.A.; Peng, M.; An, P.; McKenzie, L.M.; Kirk, G.D.; Shi, Y.; Xie, L.X.; Marbois, B.N.; Clarke, C.F.; et al. Focal Segmental Glomerulosclerosis Is Associated with a PDSS2 Haplotype and, Independently, with a Decreased Content of Coenzyme Q10. Am. J. Physiol. Renal Physiol. 2013, 305, F1228–F1238. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic Bases and Clinical Manifestations of Coenzyme Q10 (CoQ 10) Deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Mariyama, M.; Leinonen, A.; Mochizuki, T.; Tryggvason, K.; Reeders, S.T. Complete Primary Structure of the Human Alpha 3(IV) Collagen Chain. Coexpression of the Alpha 3(IV) and Alpha 4(IV) Collagen Chains in Human Tissues. J. Biol. Chem. 1994, 269, 23013–23017. [Google Scholar] [CrossRef] [PubMed]
- Hostikka, S.L.; Eddy, R.L.; Byers, M.G.; Höyhtyä, M.; Shows, T.B.; Tryggvason, K. Identification of a Distinct Type IV Collagen Alpha Chain with Restricted Kidney Distribution and Assignment of Its Gene to the Locus of X Chromosome-Linked Alport Syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1606–1610. [Google Scholar] [CrossRef] [PubMed]
- Lemmink, H.H.; Schröder, C.H.; Monnens, L.A.; Smeets, H.J. The Clinical Spectrum of Type IV Collagen Mutations. Hum. Mutat. 1997, 9, 477–499. [Google Scholar] [CrossRef]
- Mochizuki, T.; Lemmink, H.H.; Mariyama, M.; Antignac, C.; Gubler, M.C.; Pirson, Y.; Verellen-Dumoulin, C.; Chan, B.; Schröder, C.H.; Smeets, H.J. Identification of Mutations in the Alpha 3(IV) and Alpha 4(IV) Collagen Genes in Autosomal Recessive Alport Syndrome. Nat. Genet. 1994, 8, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.E.; Mariyama, M.; Yang-Feng, T.L.; Reeders, S.T. Sequence and Localization of a Partial cDNA Encoding the Human Alpha 3 Chain of Type IV Collagen. Am. J. Hum. Genet. 1991, 49, 545–554. [Google Scholar]
- Wang, D.; Shan, C.; Jing, X.; Zhang, Q.; Chang, H.; Lin, Y. Clinical Features and Familial Mutations in an Autosomal-Inherited Alport Syndrome Patient With the Presentation of Nephrotic Syndrome. Front. Pediatr. 2021, 9, 678633. [Google Scholar] [CrossRef]
- Takada, Y.; Murphy, E.; Pil, P.; Chen, C.; Ginsberg, M.H.; Hemler, M.E. Molecular Cloning and Expression of the cDNA for Alpha 3 Subunit of Human Alpha 3 Beta 1 (VLA-3), an Integrin Receptor for Fibronectin, Laminin, and Collagen. J. Cell Biol. 1991, 115, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin A3 Mutations with Kidney, Lung, and Skin Disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef]
- Jones, S.D.; van der Flier, A.; Sonnenberg, A. Genomic Organization of the Human Alpha 3 Integrin Subunit Gene. Biochem. Biophys. Res. Commun. 1998, 248, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Pulkkinen, L.; Murrell, D.; Rico, J.; Lucky, A.W.; Garzon, M.; Stevens, C.A.; Robertson, S.; Pfendner, E.; Uitto, J. Epidermolysis Bullosa with Congenital Pyloric Atresia: Novel Mutations in the Beta 4 Integrin Gene (ITGB4) and Genotype/Phenotype Correlations. Pediatr. Res. 2001, 49, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Kambham, N.; Tanji, N.; Seigle, R.L.; Markowitz, G.S.; Pulkkinen, L.; Uitto, J.; D’Agati, V.D. Congenital Focal Segmental Glomerulosclerosis Associated with Beta4 Integrin Mutation and Epidermolysis Bullosa. Am. J. Kidney Dis. 2000, 36, 190–196. [Google Scholar] [CrossRef]
- Ashton, G.H.; Sorelli, P.; Mellerio, J.E.; Keane, F.M.; Eady, R.A.; McGrath, J.A. Alpha 6 Beta 4 Integrin Abnormalities in Junctional Epidermolysis Bullosa with Pyloric Atresia. Br. J. Dermatol. 2001, 144, 408–414. [Google Scholar] [CrossRef]
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Müntefering, H.; Fenski, R.; Pitz, S.; Schumacher, V.; Royer-Pokora, B.; Wühl, E.; et al. Human Laminin Beta2 Deficiency Causes Congenital Nephrosis with Mesangial Sclerosis and Distinct Eye Abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Wewer, U.M.; Gerecke, D.R.; Durkin, M.E.; Kurtz, K.S.; Mattei, M.G.; Champliaud, M.F.; Burgeson, R.E.; Albrechtsen, R. Human Beta 2 Chain of Laminin (Formerly S Chain): cDNA Cloning, Chromosomal Localization, and Expression in Carcinomas. Genomics 1994, 24, 243–252. [Google Scholar] [CrossRef]
- Isojima, T.; Harita, Y.; Furuyama, M.; Sugawara, N.; Ishizuka, K.; Horita, S.; Kajiho, Y.; Miura, K.; Igarashi, T.; Hattori, M.; et al. LMX1B Mutation with Residual Transcriptional Activity as a Cause of Isolated Glomerulopathy. Nephrol. Dial. Transplant. 2014, 29, 81–88. [Google Scholar] [CrossRef]
- Dreyer, S.D.; Zhou, G.; Baldini, A.; Winterpacht, A.; Zabel, B.; Cole, W.; Johnson, R.L.; Lee, B. Mutations in LMX1B Cause Abnormal Skeletal Patterning and Renal Dysplasia in Nail Patella Syndrome. Nat. Genet. 1998, 19, 47–50. [Google Scholar] [CrossRef]
- Iannotti, C.A.; Inoue, H.; Bernal, E.; Aoki, M.; Liu, L.; Donis-Keller, H.; German, M.S.; Permutt, M.A. Identification of a Human LMX1 (LMX1.1)-Related Gene, LMX1.2: Tissue-Specific Expression and Linkage Mapping on Chromosome 9. Genomics 1997, 46, 520–524. [Google Scholar] [CrossRef]
- Herold, A.; Suyama, M.; Rodrigues, J.P.; Braun, I.C.; Kutay, U.; Carmo-Fonseca, M.; Bork, P.; Izaurralde, E. TAP (NXF1) Belongs to a Multigene Family of Putative RNA Export Factors with a Conserved Modular Architecture. Mol. Cell Biol. 2000, 20, 8996–9008. [Google Scholar] [CrossRef]
- Esposito, T.; Lea, R.A.; Maher, B.H.; Moses, D.; Cox, H.C.; Magliocca, S.; Angius, A.; Nyholt, D.R.; Titus, T.; Kay, T.; et al. Unique X-Linked Familial FSGS with Co-Segregating Heart Block Disorder Is Associated with a Mutation in the NXF5 Gene. Hum. Mol. Genet. 2013, 22, 3654–3666. [Google Scholar] [CrossRef] [PubMed]
- Bétous, R.; Glick, G.G.; Zhao, R.; Cortez, D. Identification and Characterization of SMARCAL1 Protein Complexes. PLoS ONE 2013, 8, e63149. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, L.I.; Huang, C.; Northrop, J.L.; Deguchi, K.; Clewing, J.M.; Armstrong, D.L.; Boerkoel, C.F. Schimke Immuno-Osseous Dysplasia: A Cell Autonomous Disorder? Am. J. Med. Genet. A 2006, 140, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.A.; Eisen, J.A.; Mohrenweiser, H.W. Cloning and Characterization of HARP/SMARCAL1: A Prokaryotic HepA-Related SNF2 Helicase Protein from Human and Mouse. Genomics 2000, 65, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-D.; Wagner, N.; Schley, G.; Theres, H.; Scholz, H. The Wilms’ Tumor Suppressor Wt1 Encodes a Transcriptional Activator of the Class IV POU-Domain Factor Pou4f2 (Brn-3b). Gene 2003, 305, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Bruening, W.; Li, F.P.; Haber, D.A.; Glaser, T.; Housman, D.E. WT1 Mutations Contribute to Abnormal Genital System Development and Hereditary Wilms’ Tumour. Nature 1991, 353, 431–434. [Google Scholar] [CrossRef]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L. Germline Mutations in the Wilms’ Tumor Suppressor Gene Are Associated with Abnormal Urogenital Development in Denys-Drash Syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef]
- Little, M.H.; Williamson, K.A.; Mannens, M.; Kelsey, A.; Gosden, C.; Hastie, N.D.; van Heyningen, V. Evidence That WT1 Mutations in Denys-Drash Syndrome Patients May Act in a Dominant-Negative Fashion. Hum. Mol. Genet. 1993, 2, 259–264. [Google Scholar] [CrossRef]
- Salazar, H.; Kanbour, A.; Burgess, F. Ultrastructure and Observations on the Histogenesis of Mesotheliomas, “Adenomatoid Tumors”, of the Female Genital Tract. Cancer 1972, 29, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Rose, E.A.; Glaser, T.; Jones, C.; Smith, C.L.; Lewis, W.H.; Call, K.M.; Minden, M.; Champagne, E.; Bonetta, L.; Yeger, H. Complete Physical Map of the WAGR Region of 11p13 Localizes a Candidate Wilms’ Tumor Gene. Cell 1990, 60, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Prunetti, L.; Arraf, A.; Haas, D.; Bacusmo, J.M.; Hu, J.F.; Ta-Shma, A.; Dedon, P.C.; de Crécy-Lagard, V.; Elpeleg, O. tRNA N6-Adenosine Threonylcarbamoyltransferase Defect Due to KAE1/TCS3 (OSGEP) Mutation Manifest by Neurodegeneration and Renal Tubulopathy. Eur. J. Hum. Genet. 2017, 25, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Tsai, J.D.; Lin, S.P.; Tzen, C.Y.; Shen, E.Y.; Shih, C.S. Galloway-Mowat Syndrome: A Glomerular Basement Membrane Disorder? Pediatr. Nephrol. 2001, 16, 653–657. [Google Scholar] [CrossRef]
- Seki, Y.; Ikeda, S.; Kiyohara, H.; Ayabe, H.; Seki, T.; Matsui, H. Sequencing Analysis of a Putative Human O-Sialoglycoprotein Endopeptidase Gene (OSGEP) and Analysis of a Bidirectional Promoter between the OSGEP and APEX Genes. Gene 2002, 285, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Rao, J.; Mollet, G.; Schapiro, D.; Daugeron, M.-C.; Tan, W.; Gribouval, O.; Boyer, O.; Revy, P.; Jobst-Schwan, T.; et al. Mutations in KEOPS-Complex Genes Cause Nephrotic Syndrome with Primary Microcephaly. Nat. Genet. 2017, 49, 1529–1538. [Google Scholar] [CrossRef]
- Liu, T.-L.; Lin, S.-P.; Zenker, M.; Chen, T.-Y.; Chang, J.-H.; Lin, C.-C.; Tsai, J.-D. X-Linked Recessive Galloway-Mowat Syndrome 2 Caused by a Specific LAGE3 Variant. Pediatr. Neonatol. 2023, 64, 208–209. [Google Scholar] [CrossRef]
- Jinks, R.N.; Puffenberger, E.G.; Baple, E.; Harding, B.; Crino, P.; Fogo, A.B.; Wenger, O.; Xin, B.; Koehler, A.E.; McGlincy, M.H.; et al. Recessive Nephrocerebellar Syndrome on the Galloway-Mowat Syndrome Spectrum Is Caused by Homozygous Protein-Truncating Mutations of WDR73. Brain 2015, 138, 2173–2190. [Google Scholar] [CrossRef]
- Colin, E.; Huynh Cong, E.; Mollet, G.; Guichet, A.; Gribouval, O.; Arrondel, C.; Boyer, O.; Daniel, L.; Gubler, M.-C.; Ekinci, Z.; et al. Loss-of-Function Mutations in WDR73 Are Responsible for Microcephaly and Steroid-Resistant Nephrotic Syndrome: Galloway-Mowat Syndrome. Am. J. Hum. Genet. 2014, 95, 637–648. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Fahiminiya, S.; Sorfazlian, N.; Almuriekhi, M.; Nawaz, Z.; Nadaf, J.; Khadija, K.A.; Zaineddin, S.; Kamel, H.; Majewski, J.; et al. Nonsense Mutation in the WDR73 Gene Is Associated with Galloway-Mowat Syndrome. J. Med. Genet. 2015, 52, 381–390. [Google Scholar] [CrossRef]
- Abe, Y.; Matsumoto, S.; Wei, S.; Nezu, K.; Miyoshi, A.; Kito, K.; Ueda, N.; Shigemoto, K.; Hitsumoto, Y.; Nikawa, J.; et al. Cloning and Characterization of a P53-Related Protein Kinase Expressed in Interleukin-2-Activated Cytotoxic T-Cells, Epithelial Tumor Cell Lines, and the Testes. J. Biol. Chem. 2001, 276, 44003–44011. [Google Scholar] [CrossRef]
- Grandi, P.; Dang, T.; Pané, N.; Shevchenko, A.; Mann, M.; Forbes, D.; Hurt, E. Nup93, a Vertebrate Homologue of Yeast Nic96p, Forms a Complex with a Novel 205-kDa Protein and Is Required for Correct Nuclear Pore Assembly. Mol. Biol. Cell 1997, 8, 2017–2038. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Sadowski, C.E.; Kohl, S.; Lovric, S.; Astrinidis, S.A.; Pabst, W.L.; Gee, H.Y.; Ashraf, S.; Lawson, J.A.; Shril, S.; et al. Mutations in Nuclear Pore Genes NUP93, NUP205 and XPO5 Cause Steroid-Resistant Nephrotic Syndrome. Nat. Genet. 2016, 48, 457–465. [Google Scholar] [CrossRef]
- Boehmer, T.; Enninga, J.; Dales, S.; Blobel, G.; Zhong, H. Depletion of a Single Nucleoporin, Nup107, Prevents the Assembly of a Subset of Nucleoporins into the Nuclear Pore Complex. Proc. Natl. Acad. Sci. USA 2003, 100, 981–985. [Google Scholar] [CrossRef]
- Miyake, N.; Tsukaguchi, H.; Koshimizu, E.; Shono, A.; Matsunaga, S.; Shiina, M.; Mimura, Y.; Imamura, S.; Hirose, T.; Okudela, K.; et al. Biallelic Mutations in Nuclear Pore Complex Subunit NUP107 Cause Early-Childhood-Onset Steroid-Resistant Nephrotic Syndrome. Am. J. Hum. Genet. 2015, 97, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Weinberg-Shukron, A.; Renbaum, P.; Kalifa, R.; Zeligson, S.; Ben-Neriah, Z.; Dreifuss, A.; Abu-Rayyan, A.; Maatuk, N.; Fardian, N.; Rekler, D.; et al. A Mutation in the Nucleoporin-107 Gene Causes XX Gonadal Dysgenesis. J. Clin. Investig. 2015, 125, 4295–4304. [Google Scholar] [CrossRef] [PubMed]
- Marquez, J.; Bhattacharya, D.; Lusk, C.P.; Khokha, M.K. Nucleoporin NUP205 Plays a Critical Role in Cilia and Congenital Disease. Dev. Biol. 2021, 469, 46–53. [Google Scholar] [CrossRef]
- Brownawell, A.M.; Macara, I.G. Exportin-5, a Novel Karyopherin, Mediates Nuclear Export of Double-Stranded RNA Binding Proteins. J. Cell Biol. 2002, 156, 53–64. [Google Scholar] [CrossRef]
- Kozyraki, R.; Kristiansen, M.; Silahtaroglu, A.; Hansen, C.; Jacobsen, C.; Tommerup, N.; Verroust, P.J.; Moestrup, S.K. The Human Intrinsic Factor-Vitamin B12 Receptor, Cubilin: Molecular Characterization and Chromosomal Mapping of the Gene to 10p within the Autosomal Recessive Megaloblastic Anemia (MGA1) Region. Blood 1998, 91, 3593–3600. [Google Scholar] [CrossRef]
- Nykjaer, A.; Fyfe, J.C.; Kozyraki, R.; Leheste, J.R.; Jacobsen, C.; Nielsen, M.S.; Verroust, P.J.; Aminoff, M.; de la Chapelle, A.; Moestrup, S.K.; et al. Cubilin Dysfunction Causes Abnormal Metabolism of the Steroid Hormone 25(OH) Vitamin D(3). Proc. Natl. Acad. Sci. USA 2001, 98, 13895–13900. [Google Scholar] [CrossRef]
- Kalantry, S.; Manning, S.; Haub, O.; Tomihara-Newberger, C.; Lee, H.G.; Fangman, J.; Disteche, C.M.; Manova, K.; Lacy, E. The Amnionless Gene, Essential for Mouse Gastrulation, Encodes a Visceral-Endoderm-Specific Protein with an Extracellular Cysteine-Rich Domain. Nat. Genet. 2001, 27, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Atienza-Manuel, A.; Castillo-Mancho, V.; De Renzis, S.; Culi, J.; Ruiz-Gómez, M. Endocytosis Mediated by an Atypical CUBAM Complex Modulates Slit Diaphragm Dynamics in Nephrocytes. Development 2021, 148, dev199894. [Google Scholar] [CrossRef] [PubMed]
- Gräsbeck, R. Imerslund-Gräsbeck Syndrome (Selective Vitamin B(12) Malabsorption with Proteinuria). Orphanet J. Rare Dis. 2006, 1, 17. [Google Scholar] [CrossRef]
- Christ, A.; Herzog, K.; Willnow, T.E. LRP2, an Auxiliary Receptor That Controls Sonic Hedgehog Signaling in Development and Disease. Dev. Dyn. 2016, 245, 569–579. [Google Scholar] [CrossRef]
- Kantarci, S.; Al-Gazali, L.; Hill, R.S.; Donnai, D.; Black, G.C.M.; Bieth, E.; Chassaing, N.; Lacombe, D.; Devriendt, K.; Teebi, A.; et al. Mutations in LRP2, Which Encodes the Multiligand Receptor Megalin, Cause Donnai-Barrow and Facio-Oculo-Acoustico-Renal Syndromes. Nat. Genet. 2007, 39, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, B.P.; Lundgren, S.; Johansson, M.; Hjälm, G.; Akerström, G.; Gustavsson, I.; Rask, L. In Situ Hybridization Mapping of a 500-kDa Calcium-Sensing Protein Gene (LRP2) to Human Chromosome Region 2q31-->q32.1 and Porcine Chromosome Region 15q22-->q24. Cytogenet. Cell Genet. 1995, 71, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Ault, B.H. Factor H and the Pathogenesis of Renal Diseases. Pediatr. Nephrol. 2000, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Katz, Y.; Schlesinger, M.; Carmi, R.; Shalev, H.; Haider, N.; Beck, G.; Sheffield, V.C.; Landau, D. Complement Factor H Gene Mutation Associated with Autosomal Recessive Atypical Hemolytic Uremic Syndrome. Am. J. Hum. Genet. 1999, 65, 1538–1546. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Wiech, T.; Stea, E.D.; Skerka, C. CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J. Am. Soc. Nephrol. 2020, 31, 241–256. [Google Scholar] [CrossRef]
- Kömpf, J.; Luckenbach, C.; Kloor, D.; Schunter, F.; Wernet, P.; Ritter, H. Human Factor H (Beta 1H-Globulin): Linkage Analysis. Hum. Genet. 1988, 79, 181–182. [Google Scholar] [CrossRef]
- Ozaltin, F.; Li, B.; Rauhauser, A.; An, S.-W.; Soylemezoglu, O.; Gonul, I.I.; Taskiran, E.Z.; Ibsirlioglu, T.; Korkmaz, E.; Bilginer, Y.; et al. DGKE Variants Cause a Glomerular Microangiopathy That Mimics Membranoproliferative GN. J. Am. Soc. Nephrol. 2013, 24, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.C.; Price, J.A.; Bobby, P.L.; Pettenati, M.J.; Shashi, V.; Von Kap Herr, C.; Van Dyke, T.E. Cytogenetic Assignment and Physical Mapping of the Human DGKE Gene to Chromosome 17q22. Genomics 1999, 56, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Schollen, E.; Pardon, E.; Veiga-Da-Cunha, M.; Jaeken, J.; Cassiman, J.J.; Van Schaftingen, E. Mutations in PMM2, a Phosphomannomutase Gene on Chromosome 16p13, in Carbohydrate-Deficient Glycoprotein Type I Syndrome (Jaeken Syndrome). Nat. Genet. 1997, 16, 88–92. [Google Scholar] [CrossRef]
- Thakor, J.M.; Parmar, G.; Mistry, K.N.; Gang, S.; Rank, D.N.; Joshi, C.G. Mutational Landscape of TRPC6, WT1, LMX1B, APOL1, PTPRO, PMM2, LAMB2 and WT1 Genes Associated with Steroid Resistant Nephrotic Syndrome. Mol. Biol. Rep. 2021, 48, 7193–7201. [Google Scholar] [CrossRef] [PubMed]
- Banderali, G.; Salvatici, E.; Rovelli, V.; Jaeken, J. PMM2-CDG and Nephrotic Syndrome: A Case Report. Clin. Case Rep. 2022, 10, e05347. [Google Scholar] [CrossRef]
- Ozaltin, F.; Ibsirlioglu, T.; Taskiran, E.Z.; Baydar, D.E.; Kaymaz, F.; Buyukcelik, M.; Kilic, B.D.; Balat, A.; Iatropoulos, P.; Asan, E.; et al. Disruption of PTPRO Causes Childhood-Onset Nephrotic Syndrome. Am. J. Hum. Genet. 2011, 89, 139–147. [Google Scholar] [CrossRef]
- Dai, W.; Xiang, W.; Han, L.; Yuan, Z.; Wang, R.; Ma, Y.; Yang, Y.; Cai, S.; Xu, Y.; Mo, S.; et al. PTPRO Represses Colorectal Cancer Tumorigenesis and Progression by Reprogramming Fatty Acid Metabolism. Cancer Commun. 2022, 42, 848–867. [Google Scholar] [CrossRef]
- Heybrock, S.; Kanerva, K.; Meng, Y.; Ing, C.; Liang, A.; Xiong, Z.-J.; Weng, X.; Ah Kim, Y.; Collins, R.; Trimble, W.; et al. Lysosomal Integral Membrane Protein-2 (LIMP-2/SCARB2) Is Involved in Lysosomal Cholesterol Export. Nat. Commun. 2019, 10, 3521. [Google Scholar] [CrossRef]
- Berkovic, S.F.; Dibbens, L.M.; Oshlack, A.; Silver, J.D.; Katerelos, M.; Vears, D.F.; Lüllmann-Rauch, R.; Blanz, J.; Zhang, K.W.; Stankovich, J.; et al. Array-Based Gene Discovery with Three Unrelated Subjects Shows SCARB2/LIMP-2 Deficiency Causes Myoclonus Epilepsy and Glomerulosclerosis. Am. J. Hum. Genet. 2008, 82, 673–684. [Google Scholar] [CrossRef]
- Badhwar, A.; Berkovic, S.F.; Dowling, J.P.; Gonzales, M.; Narayanan, S.; Brodtmann, A.; Berzen, L.; Caviness, J.; Trenkwalder, C.; Winkelmann, J.; et al. Action Myoclonus-Renal Failure Syndrome: Characterization of a Unique Cerebro-Renal Disorder. Brain 2004, 127, 2173–2182. [Google Scholar] [CrossRef]
- Xiao, C.; Ahn, H.; Kibrom, S.; Toro, C. SCARB2-Related Action Myoclonus—Renal Failure Syndrome. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Freije, J.M.; Blay, P.; Pendás, A.M.; Cadiñanos, J.; Crespo, P.; López-Otín, C. Identification and Chromosomal Location of Two Human Genes Encoding Enzymes Potentially Involved in Proteolytic Maturation of Farnesylated Proteins. Genomics 1999, 58, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Fryns, J.-P.; Auchus, R.J.; Garg, A. Zinc Metalloproteinase, ZMPSTE24, Is Mutated in Mandibuloacral Dysplasia. Hum. Mol. Genet. 2003, 12, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Thiel, C.; Lübbehusen, J.; Dorland, B.; de Koning, T.; von Figura, K.; Lehle, L.; Körner, C. Deficiency of GDP-Man:GlcNAc2-PP-Dolichol Mannosyltransferase Causes Congenital Disorder of Glycosylation Type Ik. Am. J. Hum. Genet. 2004, 74, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Harshman, L.A.; Ng, B.G.; Freeze, H.H.; Trapane, P.; Dolezal, A.; Brophy, P.D.; Brumbaugh, J.E. Congenital Nephrotic Syndrome in an Infant with ALG1-Congenital Disorder of Glycosylation. Pediatr. Int. 2016, 58, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhao, Y.; Wu, B.; Shu, J.; Yan, D.; Li, D.; Yu, X.; Cai, C. A Novel Variant in ALG1 Gene Associated with Congenital Disorder of Glycosylation: A Case Report and Short Literature Review. Mol. Genet. Genomic Med. 2023, 11, e2197. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Chen, Z.; Choi, W.-I.; Gee, H.Y.; Hildebrandt, F.; Zhou, W. Loss of Epithelial Membrane Protein 2 Aggravates Podocyte Injury via Upregulation of Caveolin-1. J. Am. Soc. Nephrol. 2016, 27, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Ashraf, S.; Wan, X.; Vega-Warner, V.; Esteve-Rudd, J.; Lovric, S.; Fang, H.; Hurd, T.W.; Sadowski, C.E.; Allen, S.J.; et al. Mutations in EMP2 Cause Childhood-Onset Nephrotic Syndrome. Am. J. Hum. Genet. 2014, 94, 884–890. [Google Scholar] [CrossRef]
- Liehr, T.; Kuhlenbäumer, G.; Wulf, P.; Taylor, V.; Suter, U.; Van Broeckhoven, C.; Lupski, J.R.; Claussen, U.; Rautenstrauss, B. Regional Localization of the Human Epithelial Membrane Protein Genes 1, 2, and 3 (EMP1, EMP2, EMP3) to 12p12.3, 16p13.2, and 19q13.3. Genomics 1999, 58, 106–108. [Google Scholar] [CrossRef]
- Tran, P.V.; Haycraft, C.J.; Besschetnova, T.Y.; Turbe-Doan, A.; Stottmann, R.W.; Herron, B.J.; Chesebro, A.L.; Qiu, H.; Scherz, P.J.; Shah, J.V.; et al. THM1 Negatively Modulates Mouse Sonic Hedgehog Signal Transduction and Affects Retrograde Intraflagellar Transport in Cilia. Nat. Genet. 2008, 40, 403–410. [Google Scholar] [CrossRef]
- Davis, E.E.; Zhang, Q.; Liu, Q.; Diplas, B.H.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V.; et al. TTC21B Contributes Both Causal and Modifying Alleles across the Ciliopathy Spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef]
- Awazu, M.; Yamada, M.; Asada, N.; Hashiguchi, A.; Kosaki, K.; Matsumura, K. A Girl with a Mutation of the Ciliary Gene CC2D2A Presenting with FSGS and Nephronophthisis. CEN Case Rep. 2022, 11, 116–119. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Harris, J.E.; Leo, P.J.; Marshall, M.S.; Gardiner, B.; Kinning, E.; Leong, H.Y.; McKenzie, F.; Ong, W.P.; Vodopiutz, J.; et al. Whole Exome Sequencing Is an Efficient, Sensitive and Specific Method for Determining the Genetic Cause of Short-Rib Thoracic Dystrophies. Clin. Genet. 2015, 88, 550–557. [Google Scholar] [CrossRef]
- Prasad, R.; Hadjidemetriou, I.; Maharaj, A.; Meimaridou, E.; Buonocore, F.; Saleem, M.; Hurcombe, J.; Bierzynska, A.; Barbagelata, E.; Bergadá, I.; et al. Sphingosine-1-Phosphate Lyase Mutations Cause Primary Adrenal Insufficiency and Steroid-Resistant Nephrotic Syndrome. J. Clin. Investig. 2017, 127, 942–953. [Google Scholar] [CrossRef]
- Zhou, J.; Saba, J.D. Identification of the First Mammalian Sphingosine Phosphate Lyase Gene and Its Functional Expression in Yeast. Biochem. Biophys. Res. Commun. 1998, 242, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Tarshish, P.; Tobin, J.N.; Bernstein, J.; Edelmann, C.M. Prognostic Significance of the Early Course of Minimal Change Nephrotic Syndrome: Report of the International Study of Kidney Disease in Children. J. Am. Soc. Nephrol. 1997, 8, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, A.C.Q.; Oliveira, E.A.; Fróes, B.P.; Faria, L.D.C.; Pinto, J.S.; Nogueira, M.M.I.; Lima, G.O.; Resende, P.I.; Assis, N.S.; Simões E Silva, A.C.; et al. A Predictive Model of Progressive Chronic Kidney Disease in Idiopathic Nephrotic Syndrome. Pediatr. Nephrol. 2015, 30, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Poggio, E.D.; McClelland, R.L.; Blank, K.N.; Hansen, S.; Bansal, S.; Bomback, A.S.; Canetta, P.A.; Khairallah, P.; Kiryluk, K.; Lecker, S.H.; et al. Systematic Review and Meta-Analysis of Native Kidney Biopsy Complications. Clin. J. Am. Soc. Nephrol. 2020, 15, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Tøndel, C.; Vikse, B.E.; Bostad, L.; Svarstad, E. Safety and Complications of Percutaneous Kidney Biopsies in 715 Children and 8573 Adults in Norway 1988–2010. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 1591. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, C.; Yu, H.; Ding, M.; Zhang, C.; Lu, X.; Zhang, C.-Y.; Zhang, C. Increased Urinary Exosomal microRNAs in Children with Idiopathic Nephrotic Syndrome. EBioMedicine 2019, 39, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Sun, F.; Yu, P.-F.; Li, J.-X.; Yuan, D.; Chang, J.; Lin, S.-H. Differential microRNA Expression in the Serum of Patients with Nephrotic Syndrome and Clinical Correlation Analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 7282–7286. [Google Scholar]
- Bayomy, N.R.; Abo Alfottoh, W.M.; Ali Eldeep, S.A.; Ibrahim Mabrouk Mersal, A.M.S.; Abd El-Bary, H.M.A.; Abd El Gayed, E.M. Mir-142-5p as an Indicator of Autoimmune Processes in Childhood Idiopathic Nephrotic Syndrome and as a Part of MicroRNAs Expression Panels for Its Diagnosis and Prediction of Response to Steroid Treatment. Mol. Immunol. 2022, 141, 21–32. [Google Scholar] [CrossRef]
- Zhang, Y.-R.; Wu, Y.-F.; Wang, H.; Lin, X.-M.; Zhang, X.-M. Role of microRNA-17-5p in the pathogenesis of pediatric nephrotic syndrome and related mechanisms. Zhongguo Dang Dai Er Ke Za Zhi 2020, 22, 958–963. [Google Scholar] [CrossRef]
- Traum, A.Z.; Schachter, A.D. Proteomic Analysis in Pediatric Renal Disease. Semin. Nephrol. 2007, 27, 652–657. [Google Scholar] [CrossRef]
- Decramer, S.; de Peredo, A.G.; Breuil, B.; Mischak, H.; Monsarrat, B.; Bascands, J.-L.; Schanstra, J.P. Urine in Clinical Proteomics. Mol. Cell. Proteom. 2008, 7, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Glomerular Disease: Gain-of-Glycosylation Mutation in ITGA3 Causes Nephrotic Syndrome. Nat. Rev. Nephrol. 2013, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Piyaphanee, N.; Ma, Q.; Kremen, O.; Czech, K.; Greis, K.; Mitsnefes, M.; Devarajan, P.; Bennett, M.R. Discovery and Initial Validation of α 1-B Glycoprotein Fragmentation as a Differential Urinary Biomarker in Pediatric Steroid-Resistant Nephrotic Syndrome. Proteomics Clin. Appl. 2011, 5, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Pleasant, L.; Haffner, C.; Ma, Q.; Haffey, W.D.; Ying, J.; Wagner, M.; Greis, K.D.; Devarajan, P. A Novel Biomarker Panel to Identify Steroid Resistance in Childhood Idiopathic Nephrotic Syndrome. Biomark. Insights 2017, 12, 1177271917695832. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.M.; Elbehidy, R.M.; Abdelhalim, H.S.; Amr, G.E. Soluble Interleukine-2 Receptor and MDR1 Gene Expression Levels as Inflammatory Biomarkers for Prediction of Steroid Response in Children with Nephrotic Syndrome. Iran. J. Kidney Dis. 2011, 5, 154–161. [Google Scholar]
- Peng, Z.; Mao, J.; Chen, X.; Cai, F.; Gu, W.; Fu, H.; Shen, H.; Wang, J.; Jin, X.; Zhu, X.; et al. Serum suPAR Levels Help Differentiate Steroid Resistance from Steroid-Sensitive Nephrotic Syndrome in Children. Pediatr. Nephrol. 2015, 30, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Simões e Silva, A.C.; Pereira, A.B.; Teixeira, M.M.; Teixeira, A.L. Chemokines as Potential Markers in Pediatric Renal Diseases. Dis. Markers 2014, 2014, 278715. [Google Scholar] [CrossRef]
- Segerer, S.; Alpers, C.E. Chemokines and Chemokine Receptors in Renal Pathology. Curr. Opin. Nephrol. Hypertens. 2003, 12, 243–249. [Google Scholar] [CrossRef]
- Araya, C.; Diaz, L.; Wasserfall, C.; Atkinson, M.; Mu, W.; Johnson, R.; Garin, E. T Regulatory Cell Function in Idiopathic Minimal Lesion Nephrotic Syndrome. Pediatr. Nephrol. 2009, 24, 1691–1698. [Google Scholar] [CrossRef]
- Woroniecki, R.P.; Shatat, I.F.; Supe, K.; Du, Z.; Kaskel, F.J. Urinary Cytokines and Steroid Responsiveness in Idiopathic Nephrotic Syndrome of Childhood. Am. J. Nephrol. 2008, 28, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Vianna, H.R.; Soares, C.M.B.M.; Silveira, K.D.; Elmiro, G.S.; Mendes, P.M.; de Sousa Tavares, M.; Teixeira, M.M.; Miranda, D.M.; Simões E Silva, A.C. Cytokines in Chronic Kidney Disease: Potential Link of MCP-1 and Dyslipidemia in Glomerular Diseases. Pediatr. Nephrol. 2013, 28, 463–469. [Google Scholar] [CrossRef]
- Aizawa, T.; Imaizumi, T.; Tsuruga, K.; Watanabe, S.; Yoshida, H.; Kumagai, N.; Ito, E.; Tanaka, H. Urinary Fractalkine and Monocyte Chemoattractant Protein-1 as Possible Predictors of Disease Activity of Childhood Glomerulonephritis. Tohoku J. Exp. Med. 2013, 231, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Chebotareva, N.; Vinogradov, A.; Cao, V.; Gindis, A.; Berns, A.; Alentov, I.; Sergeeva, N. Serum Levels of Plasminogen Activator Urokinase Receptor and Cardiotrophin-like Cytokine Factor 1 in Patients with Nephrotic Syndrome. Clin. Nephrol. 2022, 97, 103–110. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, W.; Guo, Q.; Zou, Y. Screening for Urinary Biomarkers of Steroid-Resistant Nephrotic Syndrome in Children. Exp. Ther. Med. 2013, 5, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Suresh, C.P.; Saha, A.; Kaur, M.; Kumar, R.; Dubey, N.K.; Basak, T.; Tanwar, V.S.; Bhardwaj, G.; Sengupta, S.; Batra, V.V.; et al. Differentially Expressed Urinary Biomarkers in Children with Idiopathic Nephrotic Syndrome. Clin. Exp. Nephrol. 2016, 20, 273–283. [Google Scholar] [CrossRef]
- Agrawal, S.; Merchant, M.L.; Kino, J.; Li, M.; Wilkey, D.W.; Gaweda, A.E.; Brier, M.E.; Chanley, M.A.; Gooding, J.R.; Sumner, S.J.; et al. Predicting and Defining Steroid Resistance in Pediatric Nephrotic Syndrome Using Plasma Proteomics. Kidney Int. Rep. 2020, 5, 66–80. [Google Scholar] [CrossRef]
- Barragry, J.M.; France, M.W.; Carter, N.D.; Auton, J.A.; Beer, M.; Boucher, B.J.; Cohen, R.D. Vitamin-D Metabolism in Nephrotic Syndrome. Lancet 1977, 2, 629–632. [Google Scholar] [CrossRef]
- Selewski, D.T.; Chen, A.; Shatat, I.F.; Pais, P.; Greenbaum, L.A.; Geier, P.; Nelson, R.D.; Kiessling, S.G.; Brophy, P.D.; Quiroga, A.; et al. Vitamin D in Incident Nephrotic Syndrome: A Midwest Pediatric Nephrology Consortium Study. Pediatr. Nephrol. 2016, 31, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Pordal, A.; Haffner, C.; Pleasant, L.; Ma, Q.; Devarajan, P. Urinary Vitamin D-Binding Protein as a Biomarker of Steroid-Resistant Nephrotic Syndrome. Biomark. Insights 2016, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Molecular Characterization and Pattern of Tissue Expression of the Gene for Neutrophil Gelatinase-Associated Lipocalin from Humans. Genomics 1997, 45, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of Neutrophil Gelatinase-Associated Lipocalin as a Novel Early Urinary Biomarker for Ischemic Renal Injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Lacquaniti, A.; Coppolino, G.; Donato, V.; Campo, S.; Fazio, M.R.; Nicocia, G.; Buemi, M. Neutrophil Gelatinase-Associated Lipocalin (NGAL) and Progression of Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Nickolas, T.L.; Forster, C.S.; Sise, M.E.; Barasch, N.; Solá-Del Valle, D.; Viltard, M.; Buchen, C.; Kupferman, S.; Carnevali, M.L.; Bennett, M.; et al. NGAL (Lcn2) Monomer Is Associated with Tubulointerstitial Damage in Chronic Kidney Disease. Kidney Int. 2012, 82, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Piyaphanee, N.; Czech, K.; Mitsnefes, M.; Devarajan, P. NGAL Distinguishes Steroid Sensitivity in Idiopathic Nephrotic Syndrome. Pediatr. Nephrol. 2012, 27, 807–812. [Google Scholar] [CrossRef]
- Dubin, R.F.; Rhee, E.P. Proteomics and Metabolomics in Kidney Disease, Including Insights into Etiology, Treatment, and Prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef]
- Pereira, P.R.; Carrageta, D.F.; Oliveira, P.F.; Rodrigues, A.; Alves, M.G.; Monteiro, M.P. Metabolomics as a Tool for the Early Diagnosis and Prognosis of Diabetic Kidney Disease. Med. Res. Rev. 2022, 42, 1518–1544. [Google Scholar] [CrossRef]
- Sedic, M.; Gethings, L.A.; Vissers, J.P.C.; Shockcor, J.P.; McDonald, S.; Vasieva, O.; Lemac, M.; Langridge, J.I.; Batinić, D.; Pavelić, S.K. Label-Free Mass Spectrometric Profiling of Urinary Proteins and Metabolites from Paediatric Idiopathic Nephrotic Syndrome. Biochem. Biophys. Res. Commun. 2014, 452, 21–26. [Google Scholar] [CrossRef]
- Gooding, J.R.; Agrawal, S.; McRitchie, S.; Acuff, Z.; Merchant, M.L.; Klein, J.B.; Smoyer, W.E.; Sumner, S.J. Midwest Pediatric Nephrology Consortium Predicting and Defining Steroid Resistance in Pediatric Nephrotic Syndrome Using Plasma Metabolomics. Kidney Int. Rep. 2020, 5, 81–93. [Google Scholar] [CrossRef]
- Stone, H.; Magella, B.; Bennett, M.R. The Search for Biomarkers to Aid in Diagnosis, Differentiation, and Prognosis of Childhood Idiopathic Nephrotic Syndrome. Front. Pediatr. 2019, 7, 404. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.K.; Arif, M.; Amjad, N. A Histopathological Outlook on Nephrotic Syndrome: A Pediatric Perspective. Indian. J. Nephrol. 2016, 26, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S. Mechanisms and Consequences of TGF-ß Overexpression by Podocytes in Progressive Podocyte Disease. Cell Tissue Res. 2012, 347, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.Z.; Youssef, D.M.; El-Shal, A.S.; Abdelsalam, A.A. Transforming Growth Factor-Beta in Nephrotic Syndrome and Its Correlation with Albuminemia and Hyperlipidemia. Biochem. Lett. 2019, 15, 186–195. [Google Scholar] [CrossRef]
- Froes, B.P.; de Almeida Araújo, S.; Bambirra, E.A.; Oliveira, E.A.; Simões E Silva, A.C.; Pinheiro, S.V.B. Is CD44 in Glomerular Parietal Epithelial Cells a Pathological Marker of Renal Function Deterioration in Primary Focal Segmental Glomerulosclerosis? Pediatr. Nephrol. 2017, 32, 2165–2169. [Google Scholar] [CrossRef]
- Guimarães, F.T.L.; Ferreira, R.N.; Brito-Melo, G.E.A.; Rocha-Vieira, E.; de Pereira, W.F.; Pinheiro, S.V.B.; Miranda, A.S.; Simões E Silva, A.C. Pediatric Patients With Steroid-Sensitive Nephrotic Syndrome Have Higher Expression of T Regulatory Lymphocytes in Comparison to Steroid-Resistant Disease. Front. Pediatr. 2019, 7, 114. [Google Scholar] [CrossRef]
- Gonzalez Guerrico, A.M.; Lieske, J.; Klee, G.; Kumar, S.; Lopez-Baez, V.; Wright, A.M.; Bobart, S.; Shevell, D.; Maldonado, M.; Troost, J.P.; et al. Urinary CD80 Discriminates Among Glomerular Disease Types and Reflects Disease Activity. Kidney Int. Rep. 2020, 5, 2021–2031. [Google Scholar] [CrossRef]
- Sansom, D.M.; Manzotti, C.N.; Zheng, Y. What’s the Difference between CD80 and CD86? Trends Immunol. 2003, 24, 314–319. [Google Scholar] [CrossRef]
- Eroglu, F.K.; Orhan, D.; İnözü, M.; Duzova, A.; Gulhan, B.; Ozaltin, F.; Topaloglu, R. CD80 Expression and Infiltrating Regulatory T Cells in Idiopathic Nephrotic Syndrome of Childhood. Pediatr. Int. 2019, 61, 1250–1256. [Google Scholar] [CrossRef]
- Eroglu, F.K.; Yazar, V.; Guler, U.; Yıldırım, M.; Yildirim, T.; Gungor, T.; Celikkaya, E.; Karakaya, D.; Turay, N.; Ciftci Dede, E.; et al. Circulating extracellular vesicles of patients with steroid-sensitive nephrotic syndrome have higher RAC1 and induce recapitulation of nephrotic syndrome phenotype in podocytes. Am. J. Physiol. Renal Physiol. 2021, 321, F659–F673. [Google Scholar] [CrossRef] [PubMed]
- Pekkucuksen, N.T.; Liu, L.P.; Aly, R.; Shoemaker, L.R.; Alli, A.A. Extracellular vesicles from focal segmental glomerulosclerosis pediatric patients induce STAT3 activation and mesangial cell proliferation. PLoS ONE 2022, 17, e0274598. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Candiano, G.; Angeletti, A.; Lugani, F.; Panfoli, I. Extracellular Vesicles as Source of Biomarkers in Glomerulonephritis. Int. J. Mol. Sci. 2023, 24, 13894. [Google Scholar] [CrossRef]



| Gene | Function/Description | Mutations and Associated Conditions | Chromosome Location | Mode of Inheritance | References |
|---|---|---|---|---|---|
| Slit-diaphragm-associated mutations | |||||
| CD2AP | Acts as a bridge between the SD and the actin cytoskeleton. Lack of expression causes mesangial cell proliferation, glomerulosclerosis, and NS. | Mutations alter the SD structure. CD2AP-/- mice develop severe NS. | 6p12.3 | Autosomal Recessive and Dominant | [64,65,66] |
| NPHS1 | Encodes nephrin. Mutations account for 40–60% of infants with congenital NS. Malfunction leads to massive protein loss. | Mutations associated with Finnish-type nephrotic syndrome. | 19q13.12 | Autosomal Recessive | [67,68,69] |
| NPHS2 | Encodes podocin. Plays a role in recruiting nephrin to the SD. Mutations lead to congenital/infantile NS and SRNS. | Mutations lead to childhood- or adult-onset SRNS. | 1q25.2 | Autosomal Recessive | [70,71] |
| CRB2 | Encodes the Crumbs cell polarity complex protein | Mutations lead to SRNS with FSGS. | 9q33.3 | Autosomal Recessive | [72,73,74] |
| FAT1 | Encodes the tumor suppressor of the cadherin superfamily. | Mutations lead to a combination of SRNS, tubular ectasia, hematuria, and neurological involvement. | 4q35.2 | Autosomal Recessive | [75,76,77,78] |
| PLCE1 | Encodes the Phospholipase C epsilon 1 (PLCε1) protein. | Mutations in PLCE1 are responsible for a major part of cases of Mesangial Sclerosis. | 10q23.33 | Autosomal Recessive | [79,80] |
| TRPC6 | Encodes the Transient Receptor Potential Cation Channel Subfamily C member 6 receptor. | Mutations lead to late-onset FSGS. | 11q22.1 | Autosomal Dominant | [81,82,83,84] |
| Actin-cytoskeleton-associated mutations | |||||
| ACTN4 | Encodes alpha-actin 4, an actin-binding protein in the FPs of podocytes. | Mutations associated with adult-onset SRNS with FSGS. | 19q13.2 | Autosomal Recessive | [85,86,87] |
| ANLN | Encodes anilin, an actin-binding protein. Mutations cause a reduction in CD2AP binding, dysregulating signaling in podocytes. | Mutations lead to disarrangement in the SD. | 7p14.2 | Autosomal Dominant | [88,89] |
| ARHGAP24 | Encodes the Rho GTPase-activating protein 24. | Mutations associated with familial SRNS in the second and third decades of life. | 4q21.3 | Autosomal Dominant | [90,91] |
| ARHGDIA | Encodes the RhoGDP dissociation inhibitor α. | Mutations associated with congenital NS or SRNS within the first 2 years. | 17q25.3 | Autosomal Recessive | [92,93] |
| INF2 | Encodes inverted formin 2 and regulates actin polymerization. Mutations related to Charcot–Marie–Tooth disease and FSGS. | Mutations related to Charcot–Marie–Tooth disease and FSGS. | 14q32.33 | Autosomal Dominant | [94,95] |
| MYO1E | Encodes non-muscular myosin 1E, an actin-binding molecular motor in the FPs of podocytes. | Mutations lead to SRNS with focal thickening, disorganization, and multilamination of GBM. | 15q22.2 | Autosomal Recessive | [96,97,98] |
| KANK1 | Encodes kidney ankyrin repeat-containing protein 1. | Mutations cause congenital and early childhood onset NS. | 9p24.3 | Autosomal Recessive | [99,100] |
| KANK2 | Encodes kidney ankyrin repeat-containing protein 2. | Mutations cause congenital and early childhood onset NS. | 19p13.2 | Autosomal Recessive | [99,101] |
| KANK4 | Encodes kidney ankyrin repeat-containing protein 4. | Mutations cause congenital and early childhood onset NS. | 1p31.3 | Autosomal Recessive | [99,102] |
| Mitochondrial protein mutations | |||||
| ADCK4 | Has a role in the synthesis of Coenzyme Q10 (CoQ10), a lipid-soluble ubiquinone. | Mutations cause SRNS. | 19q13.1 | Autosomal Recessive | [103,104,105] |
| COQ2 | Encodes parahydroxybenzoate-polyprenyltransferase, involved in the biosynthesis of ubiquinone CoQ10. | Mutations lead to CoQ10 deficiency, resulting in SRNS. | 4q21.23 | Autosomal Recessive | [106,107] |
| COQ6 | The product is a mono-oxygenase required for CoQ10 biosynthesis. Mutations lead to primary CoQ10 deficiency. | Mutations result in primary CoQ10 deficiency, leading to SRNS. | 14q24.3 | Autosomal Recessive | [108,109,110,111] |
| PDSS2 | Encodes decaprenyl diphosphate synthase, subunit 2, a component of a heterotetrameric decaprenyl synthase. | Mutations lead to primary CoQ10 deficiency, resulting in SRNS. | 6q21 | Autosomal Recessive | [112,113,114] |
| Glomerular-basement-membrane-related mutations | |||||
| COL4A3 | Encodes Collagen Type IV alpha-3 chain. | Mutations related to Alport syndrome type 2 and autosomal dominant or recessive modes of inheritance for benign familial hematuria. | 2q36.3 | Autosomal Dominant (Alport syndrome type 3), Autosomal Recessive (Alport syndrome type 2) | [115,116,117,118,119] |
| COL4A4 | Encodes Collagen Type IV alpha-4 chain. | Mutations related to Alport syndrome type 2 and autosomal dominant or recessive modes of inheritance for benign familial hematuria. | 2q36.3 | Autosomal Dominant (Alport syndrome type 3), Autosomal Recessive (Alport syndrome type 2) | [115,116,117,118,119] |
| COL4A5 | Encodes Collagen Type IV alpha-5 chain. | Mutations related to Alport syndrome type 1, exhibiting X-linked dominant mode of inheritance. | Xq22 | X-Linked Dominant | [115,116,118,119,120] |
| ITGA3 | Encodes integrin alpha-3 chain and forms an integrin-family molecule, VLA-3. | Mutations associated with interstitial lung disease, NS, and congenital epidermolysis bullosa. | 17q21.33 | Autosomal Recessive | [121,122,123] |
| ITGB4 | Encodes integrin beta-4 chain and forms the alpha-6-beta-4 integrin molecule. | Mutations associated with congenital FSGS and epidermolysis bullosa with pyloric atresia. | 17q25.1 | Autosomal Recessive | [124,125,126] |
| LAMB2 | Encodes laminin beta-2 chain. Mutations associated with Pierson syndrome. | Mutations cause Pierson syndrome, presenting as congenital NS with mesangial sclerosis and eye abnormalities. | 3p21 | Autosomal Recessive | [127,128] |
| Nuclear transcription factors and protein mutations | |||||
| LMX1B | Encodes LIM homeobox transcription factor. | Mutations cause FSGS and nail–patella syndrome. | 9q32-34.1 | Autosomal Dominant | [129,130,131] |
| NXF5 | Belongs to a multigene family of nuclear RNA export factors. Role in FSGS is not well understood. | Mutations might contribute to FSGS, but their significance is unknown. | X chromosome | X-Linked Recessive | [132,133] |
| SMARCAL1 | Encodes SWI/SNF-related matrix-associated, actin-dependent regulator of chromatin. | Mutations result in Schimke immunosseous dysplasia and NS. | 2q34.36 | Autosomal Recessive | [134,135,136] |
| WT1 | Encodes a zinc finger DNA-binding protein. Mutations associated with Wilms Tumor, Denys–Drash Syndrome with NS, somatic mesothelioma, and other diseases. | Mutations associated with Wilms Tumor, Denys–Drash Syndrome with NS, somatic mesothelioma, and other diseases. | 11p13 | Autosomal Dominant (Wilms Tumor, Denys–Drash Syndrome), Somatic Mutation | [137,138,139,140,141,142] |
| OSGEP | Encodes O-sialoglycoprotein endopeptidase. Mutations lead to Galloway–Mowat syndrome and GBM disorder. | Mutations cause Galloway–Mowat syndrome and GBM disorder. | 14q11.2-12 | Autosomal Recessive | [143,144,145] |
| LAGE3 | Encodes L-antigen family member 3. | Mutations cause X-linked Galloway–Mowat syndrome with NS and primary microcephaly. | Xq28 | X-Linked Recessive | [146,147] |
| WDR73 | Encodes WD repeat-containing protein 73. | Mutations cause Galloway–Mowat syndrome with microcephaly and SRNS. | 15q25.2 | Autosomal Recessive | [148,149,150] |
| TP53RK | Encodes p-53-related protein kinase. | Mutations cause Galloway–Mowat syndrome. | 20q13.2 | Autosomal Recessive | [146,151] |
| NUP93 | Encodes nucleoporin 93 kd, a subunit of the 12 million Da nuclear pore complex. | Mutations cause SRNS. | 16q13 | Autosomal Recessive | [152,153] |
| NUP107 | Encodes nucleoporin 107 kd, a subunit of the 120 million Da nuclear pore complex. | Mutations cause SRNS and ovarian dysgenesis. | 12q15 | Autosomal Recessive | [154,155,156] |
| NUP205 | Encodes nucleoporin 205 kd, a subunit of the 120 million Da nuclear pore complex. | Mutations cause SRNS. | 7q33 | Autosomal Recessive | [153,157] |
| XPO5 | Encodes exportin 5. | Mutations associated with early onset NS. | 6p21.1 | Autosomal Recessive | [153,158] |
| Proximal tubule protein reabsorption mutations | |||||
| CUBN | Encodes cubilin, the intestinal receptor for endocytosis of intrinsic factor, vitamin B12, and a receptor in epithelial apoA-I/HDL metabolism. | Mutations cause Imerslund–Grasbeck syndrome, low molecular weight proteinuria, and megaloblastic anemia. | 10p13 | Autosomal Recessive | [159,160] |
| AMN | Encodes amnion-associated transmembrane protein. Along with cubilin, it forms a receptor complex called “cubam.” | Mutations cause Imerslund–Grasbeck syndrome with B12 deficiency and proteinuria. | 14q32 | Autosomal Recessive | [161,162,163] |
| LRP2 | Encodes low-density lipoprotein receptor-related protein 2 or megalin. | Mutations cause Donnai–Barrow syndrome with facial anomalies, ocular alterations, sensorineural hearing loss, and NS. | 2q31.1 | Autosomal Recessive | [164,165,166] |
| Other mutations | |||||
| CFH | Encodes complement factor H, a serum glycoprotein that regulates the function of the alternative complement pathway. | Mutations associated with atypical hemolytic uremic syndrome and C3 glomerulopathy. | 1q31.3 | Autosomal Dominant | [167,168,169,170] |
| DGKE | Encodes diacylglycerol kinase epsilon, an intracellular molecule that phosphorylates diacylglycerol (DAG) to phosphatidic acid. | Mutations associated with susceptibility to atypical hemolytic uremic syndrome and NS. | 17q22 | Autosomal Recessive | [171,172] |
| PMM2 | Encodes phosphomannomutase, an enzyme necessary for the synthesis of GDP-mannose. | Mutations cause congenital disorders of glycosylation type I and SRNS. | 16p13.2 | Autosomal Recessive | [173,174,175] |
| PTPRO | Encodes protein-tyrosine phosphatase receptor-type O or glomerular epithelial protein 1. | Mutations cause childhood-onset NS with varying severities. | 12p12.3 | Autosomal Recessive | [176,177] |
| SCARB2 | Encodes scavenger receptor class B, member 2, a lysosomal integral membrane glycoprotein. | Mutations cause progressive myoclonic epilepsy, with or without renal failure and SRNS. | 4q21.1 | Autosomal Recessive | [178,179,180,181] |
| ZMPSTE24 | Encodes zinc metalloproteinase STE24, a protein involved in the metabolism of farnesylated proteins. | Mutations cause mandibuloacral dysplasia type B, increasing the risk for FSGS. | 1p34.2 | Autosomal Recessive | [182,183] |
| ALG1 | Encodes chitobiosyldiphosphodolichol beta-mannosyltransferase, a protein that acts in the glycosylation process. | Mutations cause congenital disorder of glycosylation type Ik, producing congenital NS. | 16p13.3 | Autosomal Recessive | [184,185,186] |
| EMP2 | Encodes epithelial membrane protein 2, which regulates cell membrane composition and plays a role in glomerular filtration. | Mutations cause childhood-onset NS. | 16p13.2 | Autosomal Recessive | [187,188,189] |
| TTC21B | Encodes tetratricopeptide repeat domain-containing protein 21B, involved in ciliary function. | Mutations cause nephronophthisis, FSGS, and short-rib thoracic dysplasia. | 2q24.3 | Autosomal Recessive | [190,191,192,193] |
| SGPL1 | Encodes sphingosine-1-phosphate lyase 1, involved in sphingolipid catabolism. | Mutations cause primary adrenal insufficiency and SRNS. | 10q22.1 | Autosomal Recessive | [194,195] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaz de Castro, P.A.S.; Fujihara Ide, T.; Crespo Torres, F.; Simões e Silva, A.C. The View of Pediatric Nephrotic Syndrome as a Podocytopathy. Kidney Dial. 2023, 3, 346-372. https://doi.org/10.3390/kidneydial3040030
Vaz de Castro PAS, Fujihara Ide T, Crespo Torres F, Simões e Silva AC. The View of Pediatric Nephrotic Syndrome as a Podocytopathy. Kidney and Dialysis. 2023; 3(4):346-372. https://doi.org/10.3390/kidneydial3040030
Chicago/Turabian StyleVaz de Castro, Pedro Alves Soares, Thomas Fujihara Ide, Fernando Crespo Torres, and Ana Cristina Simões e Silva. 2023. "The View of Pediatric Nephrotic Syndrome as a Podocytopathy" Kidney and Dialysis 3, no. 4: 346-372. https://doi.org/10.3390/kidneydial3040030