
PAX 2 Mutation in an Indian Family with Renal Coloboma Syndrome
 , , ,
, , ,
Abstract
:1. Introduction
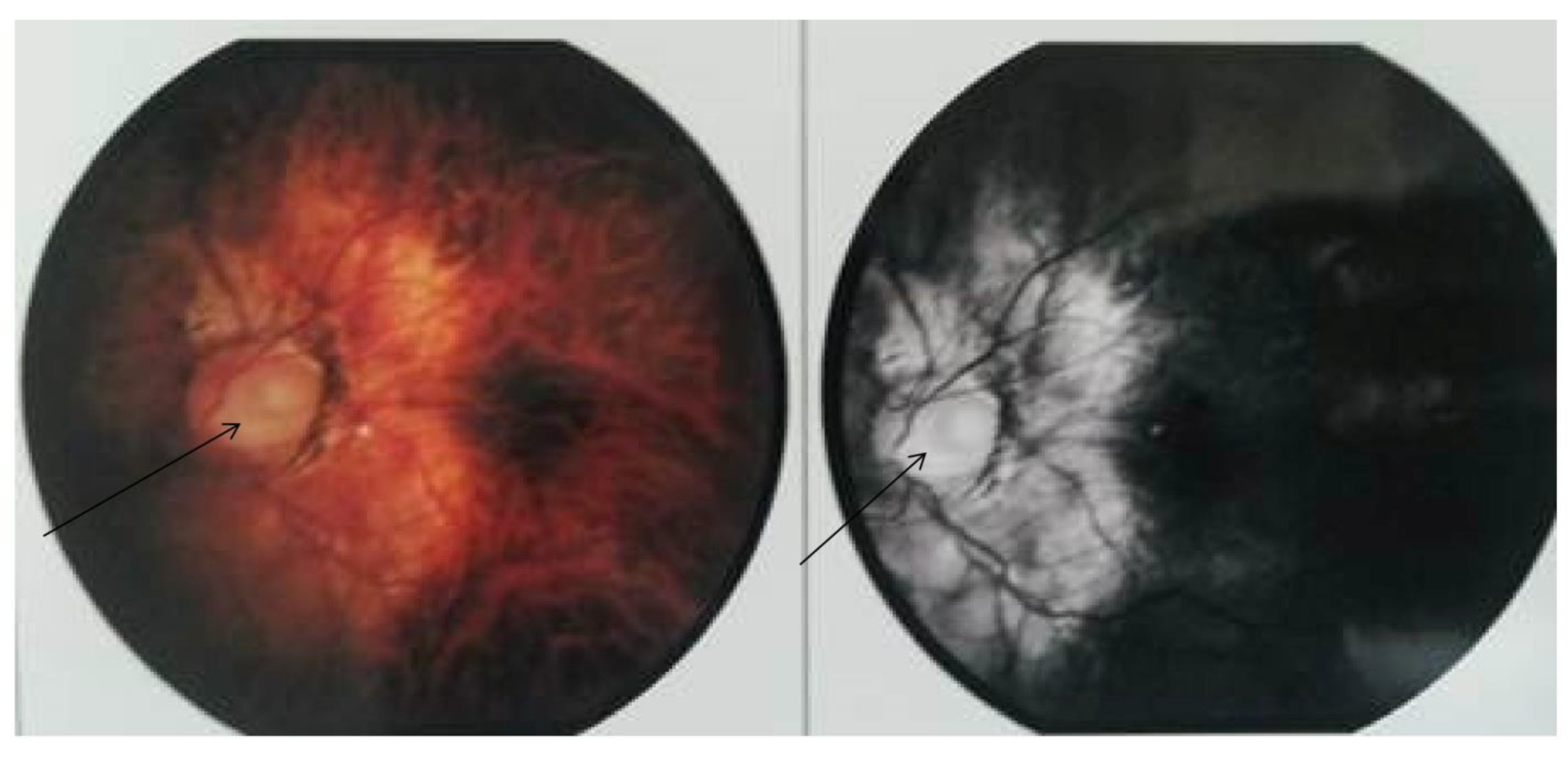
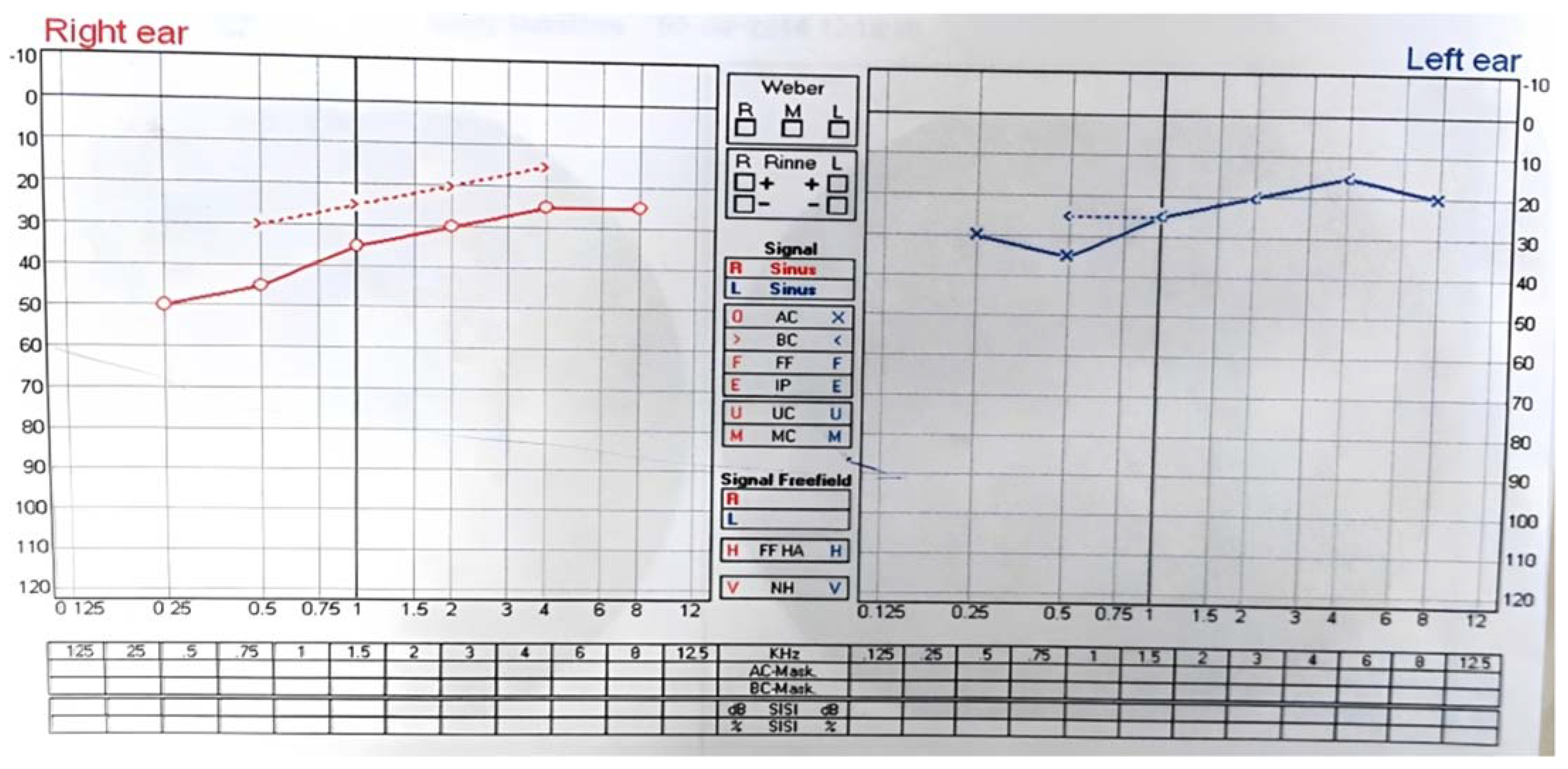
2. Case Report/Case Presentation
3. Materials and Methods
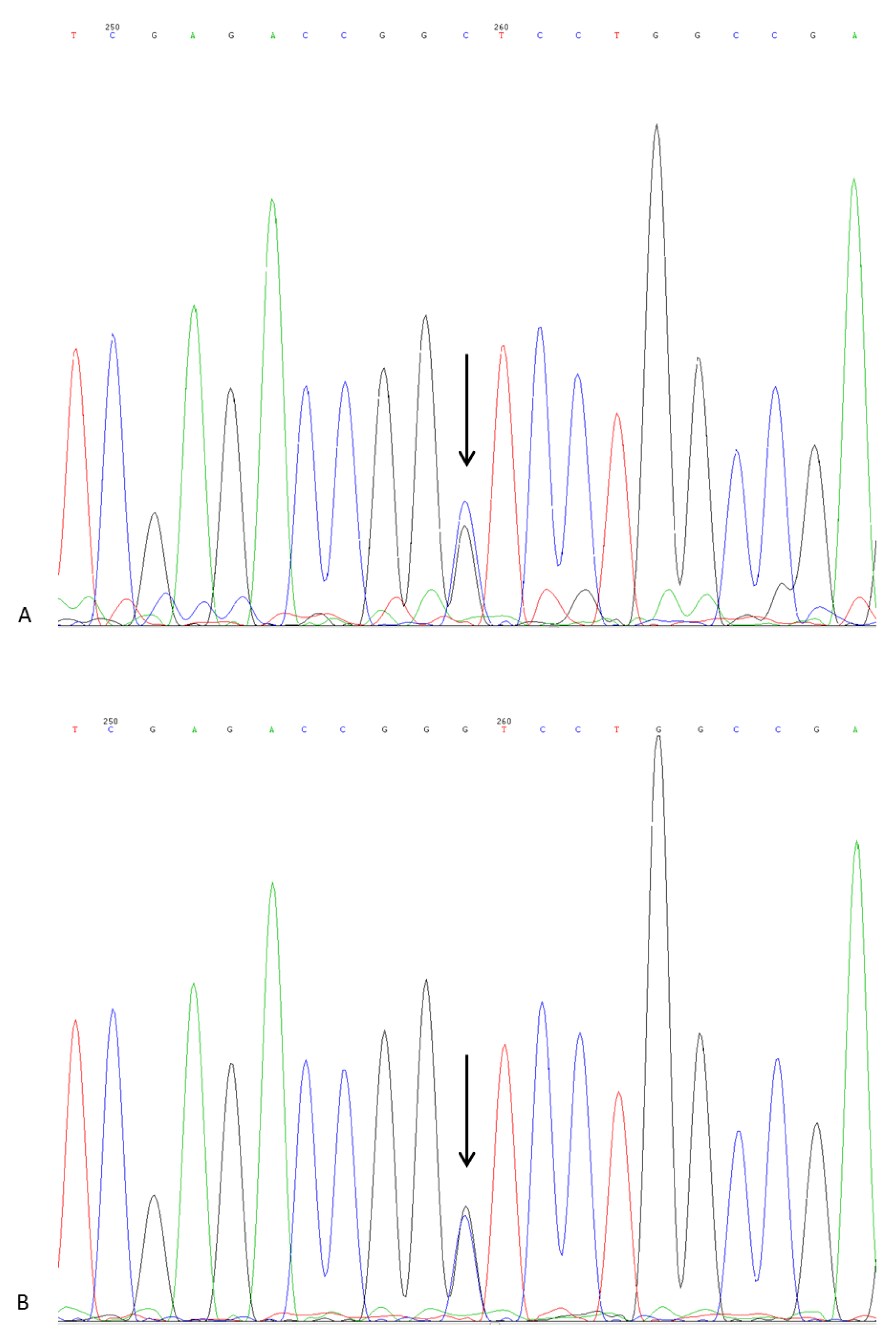
DNA Sequencing
4. Results and Discussion
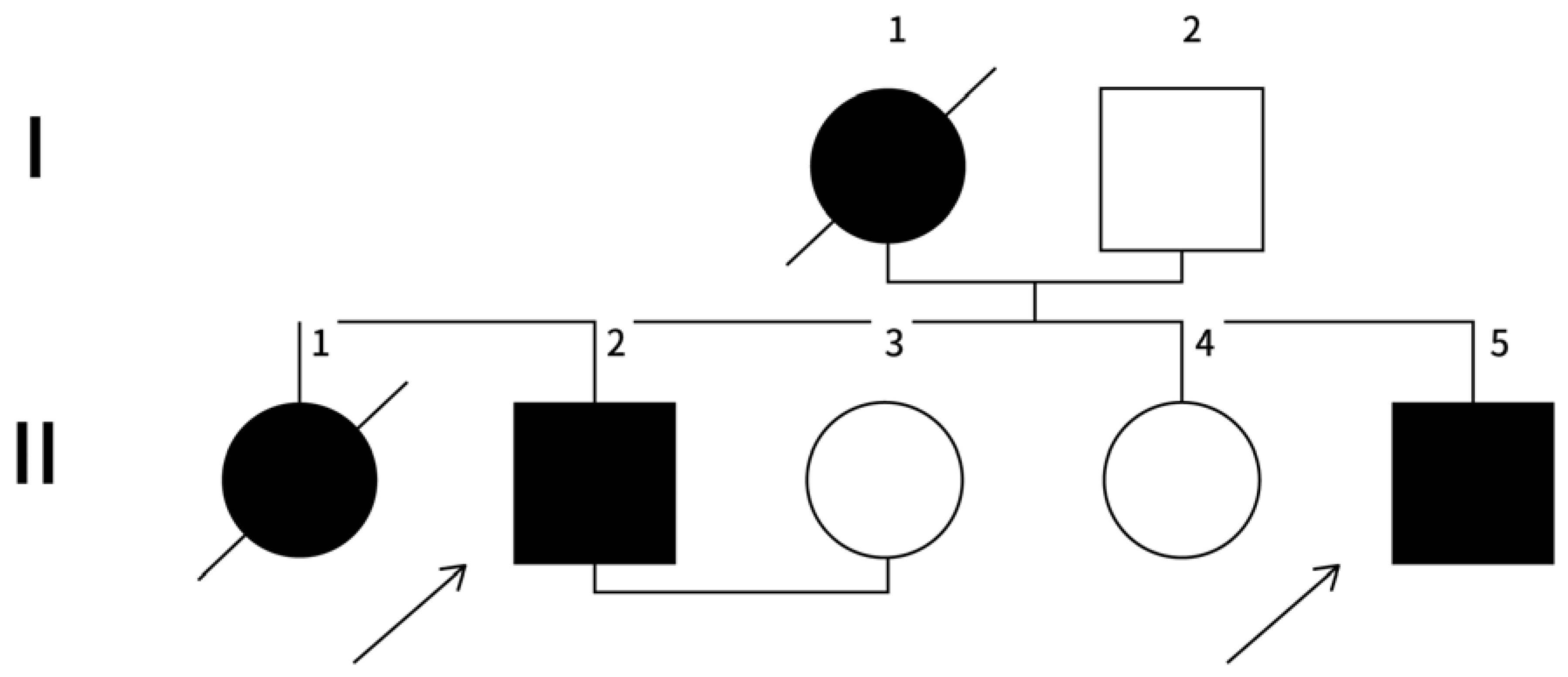
Family History
5. Genetic Analysis and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanyanusin, P. Papillorenal Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Sanyanusin, P.; Schimmenti, L.A.; McNoe, L.A.; Ward, T.A.; Pierpont, M.E.; Sullivan, M.J.; Dobyns, W.B.; Eccles, M.R. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat. Genet. 1995, 9, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Iijima, K.; Shirakawa, T.; Kitagawa, K.; Satomura, K.; Nakamura, H.; Yoshikawa, N. Papillorenal syndrome: The frequency of clinical symptoms and PAX2 mutations in Japanese pediatric patients. Pediatr. Nephrol. 2005, 20, 1723–1728. [Google Scholar]
- Schedl, A.; Hastie, N.D. Genetics of kidney development. Nat. Genet. 2000, 24, 210–211. [Google Scholar]
- Singh, A.D.; De Potter, P.; Fijal, B.A.; Shields, C.L.; Shields, J.A.; Elston, R.C. Lifetime prevalence of uveal melanoma in white patients with oculo(dermal) melanocytosis. Ophthalmology 1998, 105, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Winyard, P.J.; Risdon, R.A.; Sams, V.R.; Dressler, G.R.; Woolf, A.S. The PAX2 transcription factor is expressed in cystic and hyperproliferative dysplastic epithelia in human kidney malformations. J. Clin. Investig. 1996, 98, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Favor, J.; Peters, H.; Hermann, T. Molecular characterization of Pax2 mutants suggest that neural crest cells contribute to the development of the urogenital system. Dev. Dyn. 1996, 206, 169–179. [Google Scholar]
- Sanyanusin, P.; McNoe, L.A.; Sullivan, M.J.; Weaver, R.G.; Eccles, M.R. Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum. Mol. Genet. 1995, 4, 2183–2184. [Google Scholar] [CrossRef] [PubMed]
- Eccles, M.R.; Schimmenti, L.A. Renal-coloboma syndrome: A multi-system developmental disorder caused by PAX2 mutations. Clin. Genet. 1999, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weaver, R.G., Jr.; Torczynski, E.; Mao, J.I. Renal-coloboma syndrome: A report of 2 cases and review of the literature. Am. J. Kidney Dis. 1988, 12, 406–410. [Google Scholar]
- Weaver, R.G., Jr.; Hopper, K.; Torczynski, E.; Scharre, J. Familial renal-coloboma syndrome: Report of a second family. Am. J. Kidney Dis. 1995, 26, 498–506. [Google Scholar]
- Lakshmanaiah, L.; Rangarajan, T.; Chaitanya Ramana Reddy, V. Renal Coloboma Syndrome: A Novel PAX2 Mutation Involving An Indian Family. Indian J. Nephrol. 2016, 26, 327–331. [Google Scholar]
- Yovagood, S.; Vuppala, S.; Jalali, S. Phenotypic Spectrum and PAX2 Mutation Analysis in a Second Indian Family with Renal Coloboma Syndrome. Indian J. Nephrol. 2018, 28, 310–313. [Google Scholar]
- Ammayappan, S.K.; Rajagopalan, A.; Arunachalam, J.; Prasath, A.; Durai, R. A case of renal coloboma syndrome. J. Nephrol. 2023, 36, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. ACMG Laboratory Quality Assurance Committee. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safieh, L.; Vithana, E.N.; Chan, W.M. Genetics of primary inherited optic neuropathies. Asia-Pac. J. Ophthalmol. 2015, 4, 318–325. [Google Scholar]
- Schimmenti, L.A.; Cunliffe, H.E.; McNoe, L.A.; Ward, T.A.; French, M.C.; Shim, H.H.; Zhang, Y.H.; Proesmans, W.; Leys, A. Further delineation of renal-coloboma syndrome in patients with extreme variability of phenotype and identical PAX2 mutations. Am. J. Hum. Genet. 1997, 60, 869–878. [Google Scholar] [PubMed]
- Fletcher, J.; Hu, M.; Berman, Y.; Collins, F.; Grigg, J.; McIver, M.; Jüppner, H.; Alexander, S.I. Multicystic dysplastic kidney and variable phenotype in a family with a novel deletion mutation of PAX2. J. Am. Soc. Nephrol. 2005, 16, 2754–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.; Sanna-Cherchi, S.; Warady, B.A.; Furth, S.L.; Kaskel, F.J.; Gharavi, A.G. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr. Nephrol. 2011, 26, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′ to 3′) | Exon/Intron Boundary | Genetic Analyzer Used |
|---|---|---|---|
| PaxF1 | CCCTCCCTTTTCTCCTCAAG | Intron 1/Exon 2 | 3500xl |
| PaxR1 | GGTGACAGAAGGCAGGAGAG | Intron 1/Exon 2 | 3500xl |
| PaxF2 | CTTCTCAAGCTCGGGAACAT | Intron 2/Exon 3 | 3500xl |
| PaxR2 | GCTGGACTTTTAGCCACGTC | Intron 2/Exon 3 | 3500xl |
| PaxF3 | CACCCTCAGGAAGTCAGCTC | Intron 3/Exon 4 | 3500xl |
| PaxR3 | CTCTCTGCCTCACAGGTTCC | Intron 3/Exon 4 | 3500xl |
| PaxF4 | GACACAGGGAGCAGATGGAT | Exon 4/Intron 4 | 3500xl |
| PaxR4 | GCTCAGGTGAGAGGAGTTGG | Exon 4/Intron 4 | 3500xl |
| PaxF5 | GGCTCCTCATCCCTCCTTAT | Intron 4/Exon 5 | 3500xl |
| PaxR5 | GGACCTGGGCTTTGCTACTA | Intron 4/Exon 5 | 3500xl |
| PaxF6 | GGCCCAAGACTAGGGAGACT | Exon 5/Intron 5 | 3500xl |
| PaxR6 | GAGCCAACTCCTCCTTTTCC | Exon 5/Intron 5 | 3500xl |
| PaxF7 | CTCTTTGCCCTGCACTGTTC | Intron 5/Exon 6 | 3500xl |
| PaxR7 | TGGCTATGCATGTGGTGTTT | Intron 5/Exon 6 | 3500xl |
| PaxF8 | ACTACCCGCACAAAACTTGC | Exon 6/Intron 6 | 3500xl |
| PaxR8 | CAGGAAGCACCCTGGTTTTA | Exon 6/Intron 6 | 3500xl |
| PaxF9 | TTTCCTCTCCGTGCAGTACC | Intron 6/Exon 7 | 3500xl |
| PaxR9 | GAGGGGCCATGACATACAGT | Intron 6/Exon 7 | 3500xl |
| PaxF10 | ATGCCTCCTAGAACCGGAGT | Exon 7/Intron 7 | 3500xl |
| PaxR10 | CTGCACTAACAAGCCTGTCC | Exon 7/Intron 7 | 3500xl |
| PaxF11 | CAGGGGAGGTCTTTTCTGTG | Intron 7/Exon 8 | 3500xl |
| PaxR11 | GGGGTGATGTGAAGGGTTG | Intron 7/Exon 8 | 3500xl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Digvijay, K.; Virzi, G.M.; Pomarè Montin, D.; da Luz, L.G.; Taramsari, M.M.; Gupta, A.; Malik, M.; Gupta, A.; Bhargava, V.; Verma, M.; et al. PAX 2 Mutation in an Indian Family with Renal Coloboma Syndrome. Kidney Dial. 2023, 3, 255-264. https://doi.org/10.3390/kidneydial3030023
Digvijay K, Virzi GM, Pomarè Montin D, da Luz LG, Taramsari MM, Gupta A, Malik M, Gupta A, Bhargava V, Verma M, et al. PAX 2 Mutation in an Indian Family with Renal Coloboma Syndrome. Kidney and Dialysis. 2023; 3(3):255-264. https://doi.org/10.3390/kidneydial3030023
Chicago/Turabian StyleDigvijay, Kumar, Grazia Maria Virzi, Diego Pomarè Montin, Lucas Gobetti da Luz, Maryam Momeni Taramsari, Ashwani Gupta, Manish Malik, Anurag Gupta, Vinant Bhargava, Meenakshi Verma, and et al. 2023. "PAX 2 Mutation in an Indian Family with Renal Coloboma Syndrome" Kidney and Dialysis 3, no. 3: 255-264. https://doi.org/10.3390/kidneydial3030023