
Vascular Calcification in Diabetic Kidney Disease
{kind=link}
Abstract
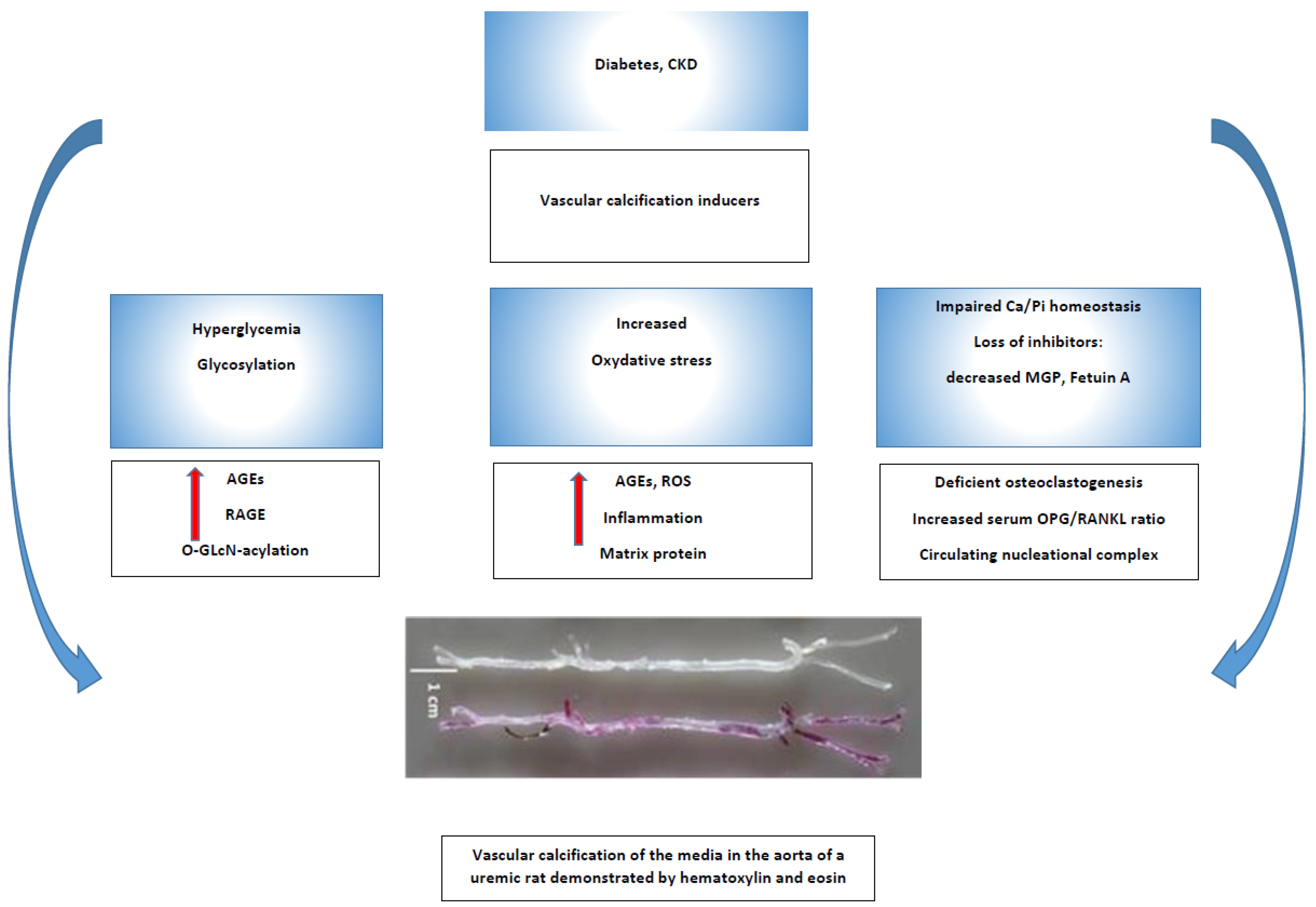
:1. Introduction
2. Vascular Calcification—General Overview
3. Vascular Calcification Risk Factors in Patients with Diabetes
3.1. Advanced Glycosylated End Products (AGEs) and Hyperglycemia
3.2. Hypertension and Diabetes
3.3. Diabetes, Lipid Metabolism Disorder, and VC
3.4. Insulin Resistance, Diabetes, and VC
3.5. Obesity and VC
3.6. Chronic Kidney Disease-Bone Disorder (CKD-MBD), microRNAs, Calciprotein Particles, and Diabetes
4. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimmet, P.Z.; Magliano, D.J.; Herman, W.H.; Shaw, J.E. Diabetes: A 21st century challenge. Lancet Diabetes Endocrinol. 2014, 2, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; Pignone, M.P.; The ADA/AHA Primary Prevention Consensus Panel. Primary Prevention of Cardiovascular Diseases in People with Diabetes Mellitus: A Scientific Statement from the American Heart Association and the American Diabetes Association. Diabetes Care 2007, 30, e58. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhao, X.; Wu, H. Arterial Stiffness: A Focus on Vascular Calcification and Its Link to Bone Mineralization. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef] [PubMed]
- Rabia, K.; Khoo, E.M. Peripheral arterial disease in people with diabetes. Diabetes Care 2003, 26, 3333–3341. [Google Scholar]
- De Angelis, M.; Scrucca, L.; Leandri, M.; Mincigrucci, S.; Bistoni, S.; Bovi, M.; Calabrese, G.; Pippi, R.; Parretti, D.; Grilli, P.; et al. Prevalence of carotid stenosis in type 2 diabetic patients asymptomatic for cerebrovascular disease. Diabetes Nutr. Metab. 2003, 16, 48–55. [Google Scholar]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate Regulation of Vascular Smooth Muscle Cell Calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Schantl, A.E.; Verhulst, A.; Neven, E.; Behets, G.J.; D’Haese, P.C.; Maillard, M.; Mordasini, D.; Phan, O.; Burnier, M.; Spaggiari, D.; et al. Inhibition of vascular calcification by inositol phosphates derivatized with ethylene glycol oligomers. Nat. Commun. 2020, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Moe, S.M.; Duan, D.; Doehle, B.P.; O’Neill, K.D.; Chen, N.X. Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. 2003, 63, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Berezovska, O.; Yildirim, G.; Budell, W.; Yagerman, S.; Pidhaynyy, B.; Bastien, C.; van der Meulen, M.; Dowd, T. Osteocalcin affects bone mineral and mechanical properties in female mice. Bone 2019, 128, 115031. [Google Scholar] [CrossRef]
- Bolton, K.; Segal, D.; McMillan, J.; Jowett, J.; Heilbronn, L.; Abberton, K.; Zimmet, P.; Chisholm, D.; Collier, G.; Walder, K. Decorin is a secreted protein associated with obesity and type 2 diabetes. Int. J. Obes. 2008, 32, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wright, W.; Bernlohr, D.A.; Cushman, S.W.; Chen, X. Alterations of the classic pathway of complement in adipose tissue of obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1433–E1440. [Google Scholar] [CrossRef]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Fukami, K.; Matsui, T. Evaluation of tissue accumulation levels of advanced glycation end products by skin autofluorescence: A novel marker of vascular complications in high-risk patients for cardiovascular disease. Int. J. Cardiol. 2015, 185, 263–268. [Google Scholar] [CrossRef]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced Glycation End Products Induce Calcification of Vascular Smooth Muscle Cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef]
- Baxevanis, A.D.; Bryant, S.H.; Landsman, D. Homology model building of the HMG-1 box structural domain. Nucleic Acids Res. 1995, 23, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Lavery, K.; Swain, P.; Falb, D.; Alaoui-Ismaili, M.H. BMP-2/4 and BMP-6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow-derived mesenchymal stem cells. J. Biol. Chem. 2008, 28, 20948–20958. [Google Scholar] [CrossRef] [Green Version]
- Tóbon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Wei, G.S.; Coady, S.A.; Goff, D.C., Jr.; Brancati, F.L.; Levy, D.; Selvin, E.; Vasan, R.S.; Fox, C.S. Blood Pressure and the Risk of Developing Diabetes in African Americans and Whites: ARIC, CARDIA, and the Framingham Heart Study. Diabetes Care 2011, 34, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Kawamori, R.; Daida, H.; Tanaka, Y.; Miyauchi, K.; Kitagawa, A.; Hayashi, D.; Kishimoto, J.; Ikeda, S.; Imai, Y.; Yamazaki, T. Amlodipine versus angiotensin II receptor blocker; control of blood pressure evaluation trial in diabetics (ADVANCED-J). BMC Cardiovasc. Disord. 2006, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Bendix, E.F.; Johansen, E.; Ringgaard, T.; Wolder, M.; Starup-Linde, J. Diabetes and Abdominal Aortic Calcification—A Systematic Review. Curr. Osteoporos. Rep. 2018, 16, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.; Zhao, Y.; Ibrahimi, P.; Olivecrona, G.; Henein, M. Diabetes and Hypertension Consistently Predict the Presence and Extent of Coronary Artery Calcification in Symptomatic Patients: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2016, 17, 1481. [Google Scholar] [CrossRef] [PubMed]
- Khurrami, L.; Møller, J.E.; Lindholt, J.S.; Urbonaviciene, G.; Steffensen, F.H.; Lambrechtsen, J.; Karon, M.; Frost, L.; Busk, M.; Egstrup, K.; et al. Cross-sectional study of aortic valve calcification and cardiovascular risk factors in older Danish men. Heart 2021, 107, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.-Y.; Shan, B.-S.; Jiang, Y.-N. The protective effects of renin–angiotensin system componts on vascular calcification. J. Hum. Hypertens. 2020, 35, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; López-Ongil, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL Increases Vascular Smooth Muscle Cell Calcification Through a RANK-BMP4–Dependent Pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, S.B.; McGraw, A.P.; Jaffe, I.Z.; Sowers, J.R. Mineralocorticoid receptor-mediated vascular insulin resistance: An early contributor to diabetes-related vascular disease? Diabetes 2013, 62, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Hwang, M.-H.; Yoo, J.-K.; Luttrell, M.; Kim, H.-K.; Meade, T.H.; English, M.; Segal, M.S.; Christou, D.D. Mineralocorticoid receptors modulate vascular endothelial function in human obesity. Clin. Sci. 2013, 125, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Voelkl, J.; Alesutan, I.; Leibrock, C.B.; Quintanilla-Martinez, L.; Kuhn, V.; Feger, M.; Mia, S.; Ahmed, M.S.; Rosenblatt, K.P.; Kuro-o, M.; et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J. Clin. Investig. 2013, 123, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Feng, W.; Su, X.; Luo, D.; Li, Z.; Zhou, Y.; Zhu, Y.; Zhang, M.; Chen, J.; Liu, B.; et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J. Clin. Investig. 2022, 132, e150051. [Google Scholar] [CrossRef]
- Okui, T.; Iwashita, M.; Rogers, M.A.; Halu, A.; Atkins, S.K.; Kuraoka, S.; Abdelhamid, I.; Higashi, H.; Ramsaroop, A.; Aikawa, M.; et al. CROT (Carnitine O-Octanoyltransferase) Is a Novel Contributing Factor in Vascular Calcification via Promoting Fatty Acid Metabolism and Mitochondrial Dysfunction. Arter. Thromb. Vasc. Biol. 2021, 41, 755–768. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Gautam, S.; Banerjee, M. The macrophage Ox-LDL receptor, CD36 and its association with type II diabetes mellitus. Mol. Genet. Metab. 2011, 102, 389–398. [Google Scholar] [CrossRef]
- Farzaneh-Far, A.; Proudfoot, D.; Shanahan, C.; Weissberg, P.L. Vascular and valvar calcification: Recent advances. Heart 2001, 85, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Davies, J.; Skepper, J.; Weissberg, P.; Shanahan, C. Acetylated Low-Density Lipoprotein Stimulates Human Vascular Smooth Muscle Cell Calcification by Promoting Osteoblastic Differentiation and Inhibiting Phagocytosis. Circulation 2002, 106, 3044–3050. [Google Scholar] [CrossRef] [Green Version]
- Vergeer, M.; Holleboom, A.G.; Kastelein, J.J.P.; Kuivenhoven, J. The HDL hypothesis: Does high-density lipoprotein protect from atherosclerosis? J. Lipid Res. 2010, 51, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Tsao, C.W.; Preis, S.R.; Peloso, G.M.; Hwang, S.-J.; Kathiresan, S.; Fox, C.S.; Cupples, L.A.; Hoffmann, U.; O’Donnell, C.J. Relations of Long-Term and Contemporary Lipid Levels and Lipid Genetic Risk Scores with Coronary Artery Calcium in the Framingham Heart Study. J. Am. Coll. Cardiol. 2012, 60, 2364–2371. [Google Scholar] [CrossRef]
- Parhami, F.; Tintut, Y.; Ballard, A.; Fogelman, A.M.; Demer, L.L. Leptin enhances the calcification of vascular cells: Artery wall as a target of leptin. Circ. Res. 2001, 88, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Ebisawa, T.; Tada, K.; Kitajima, I.; Tojo, K.; Sampath, T.K.; Kawabata, M.; Miyazono, K.; Imamura, T. Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J. Cell Sci. 1999, 112, 3519–3527. [Google Scholar] [CrossRef]
- Hruska, K.; Mathew, S.; Lund, R.; Fang, Y.; Sugatani, T. Cardiovascular risk factors in chronic kidney disease: Does phosphate qualify? Kidney Int. 2011, 79121, S9–S13. [Google Scholar] [CrossRef] [Green Version]
- Parhami, F.; Basseri, B.; Hwang, J.; Tintut, Y.; Demer, L.L. High-density lipoprotein regulates calcification of vascular cells. Circ. Res. 2002, 91, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Varma, B.; Ogunmoroti, O.; Ndumele, C.E.; Zhao, D.; Szklo, M.; Sweeney, T.; Allison, M.A.; Budoff, M.J.; Subramanya, V.; Bertoni, A.G.; et al. Higher leptin levels are associated with coronary artery calcium progression: The multi-ethnic study of atherosclerosis (MESA). Diabetes Epidemiol. Manag. 2021, 6, 100047. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, J.C.; Ng, K.F.; Aung, H.H.; Wilson, D.W. Role of triglyceride-rich lipoproteins in diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kaze, A.D.; Santhanam, P.; Musani, S.K.; Ahima, R.; Echouffo-Tcheugui, J.B. Metabolic Dyslipidemia and Cardiovascular Outcomes in Type 2 Diabetes Mellitus: Findings From the Look AHEAD Study. J. Am. Heart Assoc. 2021, 10, e016947. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-L.; Shao, J.-S.; Behrmann, A.; Krchma, K.; Towler, D.A. Abstract 222: Dkk1 and Msx2-Wnt7b Signaling Reciprocally Regulate the Endothelial-Mesenchymal Transition in Aortic Endothelial Cells. Arter. Thromb. Vasc. Biol. 2013, 33, 1679–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Sun, Y.-T.; Sun, W.; Xu, T.-H.; Ren, C.; Fan, X.; Sun, L.; Liu, L.-L.; Feng, J.-M.; Ma, J.-F.; et al. High Phosphorus Level Leads to Aortic Calcification via β-Catenin in Chronic Kidney Disease. Am. J. Nephrol. 2015, 41, 28–36. [Google Scholar] [CrossRef]
- Guo, C.; Yang, R.-J.; Jang, K.; Zhou, X.-L.; Liu, Y.-Z. Protective Effects of Pretreatment with Quercetin Against Lipopolysaccharide-Induced Apoptosis and the Inhibition of Osteoblast Differentiation via the MAPK and Wnt/β-Catenin Pathways in MC3T3-E1 Cells. Cell. Physiol. Biochem. 2017, 43, 1547–1561. [Google Scholar] [CrossRef]
- Schauer, I.E.; Snell-Bergeon, J.K.; Bergman, B.C.; Maahs, D.M.; Kretowski, A.; Eckel, R.H.; Rewers, M. Insulin resistance, defective insulin-mediated fatty acid suppression, and coronary artery calcification in subjects with and without type 1 diabetes: The CACTI study. Diabetes 2011, 60, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Dabelea, D.; Kinney, G.; Snell-Bergeon, J.K.; Hokanson, J.E.; Eckel, R.H.; Ehrlich, J.; Garg, S.; Hamman, R.F.; Rewers, M. Effect of type 1 diabetes on the gender difference in coronary artery calcification: A role for insulin resistance? The Coronary Artery Calcification in Type 1 Diabetes (CACTI) Study. Diabetes 2003, 52, 2833–2839. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, L.A.; Poot, M.; Gerrity, R.G.; Bornfeldt, K.E. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: Lack of direct growth-promoting effects of high glucose levels. Diabetes 2001, 50, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Ikeda, K.; Ito, T.; Tamaki, K.; Nara, Y.; Yamori, Y. Protective effect of inducible type nitric oxide synthase against intracellular oxidative stress caused by advanced glycation end-products in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. J. Hypertens. 2000, 18, 1071–1079. [Google Scholar] [CrossRef]
- Hirafuji, M.; Tsunoda, M.; Machida, T.; Hamaue, N.; Endo, T.; Miyamoto, A.; Minami, M. Reduced expressions of inducible nitric oxide synthase and cyclooxygenase-2 in vascular smooth muscle cells of stroke-prone spontaneously hypertensive rats. Life Sci. 2002, 70, 917–926. [Google Scholar] [CrossRef]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Paschaliori, C.; Galiatsatos, N.; Tsioufis, K.; Tousoulis, D. Inflammation in Coronary Microvascular Dysfunction. Int. J. Mol. Sci. 2021, 22, 13471. [Google Scholar] [CrossRef]
- Iguchi, T.; Hasegawa, T.; Otsuka, K.; Matsumoto, K.; Yamazaki, T.; Nishimura, S.; Nakata, S.; Ehara, S.; Kataoka, T.; Shimada, K.; et al. Insulin resistance is associated with coronary plaque vulnerability: Insight from optical coherence tomography analysis. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Tavintharan, S.; Pek, L.T.S.; Liu, J.; Ng, X.W.; Yeoh, L.Y.; Chi, L.S.; Fang, S.C. Osteoprotegerin is independently associated with metabolic syndrome and microvascular complications in type 2 diabetes mellitus. Diab. Vasc. Dis. Res. 2014, 11, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Ávila, M.; Mora, C.; Zavala, M.; Prado, M.D.C.; Paniagua, R. Osteoprotegerin Is the Strongest Predictor for Progression of Arterial Calcification in Peritoneal Dialysis Patients. Am. J. Nephrol. 2017, 46, 39–46. [Google Scholar] [CrossRef]
- Ávila, M.; Prado, M.d.; Romero, R.; Córdova, R.; Rigo, M.d.; Trejo, M.; Mora, C.; Paniagua, R.; for The Mexican Nephrology Collaborative Study Group. Osteoprotegerin Is a Better Predictor for Cardiovascular and All-Cause Mortality than Vascular Calcifications in a Multicenter Cohort of Patients on Peritoneal Dialysis. Biomolecules 2022, 12, 551. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, A.S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef] [Green Version]
- Yaturu, S.; Rains, J.; Jain, S.K. Relationship of elevated osteoprotegerin with insulin resistance, CRP, and TNF-α levels in men with type 2 diabetes. Cytokine 2008, 44, 168–171. [Google Scholar] [CrossRef]
- Knudsen, S.T.; Foss, C.H.; Poulsen, P.L.; Andersen, N.H.; Mogensen, C.E.; Rasmussen, L.M. Increased plasma concentrations of osteoprotegerin in type 2 diabetic patients with microvascular complications. Eur. J. Endocrinol. 2003, 149, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ Links BMP2 and TGFβ1 Pathways in Vascular Smooth Muscle Cells, Regulating Cell Proliferation and Glucose Metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marulanda, J.; Gao, C.; Roman, H.; Henderson, J.E.; Murshed, M. Prevention of arterial calcification corrects the low bone mass phenotype in MGP-deficient mice. Bone 2013, 57, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Lefebvre, A.T.; Horuzsko, A. Kupffer Cell Metabolism and Function. J. Enzymol. Metab. 2015, 1, 101. [Google Scholar] [PubMed]
- Barchetta, I.; Ceccarelli, V.; Cimini, F.A.; Bertoccini, L.; Fraioli, A.; Alessandri, C.; Lenzi, A.; Baroni, M.G.; Cavallo, M.G. Impaired bone matrix glycoprotein pattern is associated with increased cardio-metabolic risk profile in patients with type 2 diabetes mellitus. J. Endocrinol. Investig. 2019, 42, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. state-level prevalence of adult obesity and severe obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef]
- Vasim, I.; Majeed, C.N.; DeBoer, M.D. Intermittent Fasting and Metabolic Health. Nutrients 2022, 14, 631. [Google Scholar] [CrossRef]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Greif, M.; Becker, A.; von Ziegler, F.; Lebherz, C.; Lehrke, M.; Broedl, U.C.; Tittus, J.; Parhofer, K.; Becker, C.; Reiser, M.; et al. Pericardial Adipose Tissue Determined by Dual Source CT Is a Risk Factor for Coronary Atherosclerosis. Arter. Thromb. Vasc. Biol. 2009, 29, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Noblet, J.N.; Goodwill, A.G.; Sassoon, D.J.; Kiel, A.M.; Tune, J.D. Leptin augments coronary vasoconstriction and smooth muscle proliferation via a Rho-kinase-dependent pathway. Basic Res. Cardiol. 2016, 111, 25. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.K.; Witzmann, F.A.; McKenney, M.L.; Lai, X.; Berwick, Z.C.; Moberly, S.P.; Alloosh, M.; Sturek, M.; Tune, J.D. Perivascular adipose tissue potentiates contraction of coronary vascular smooth muscle: Influence of obesity. Circulation 2013, 128, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Yerramasu, A.; Dey, D.; Venuraju, S.; Anand, D.V.; Atwal, S.; Corder, R.; Berman, D.S.; Lahiri, A. Increased volume of epicardial fat is an independent risk factor for accelerated progression of sub-clinical coronary atherosclerosis. Atherosclerosis 2011, 220, 223–230. [Google Scholar] [CrossRef]
- Shibasaki, I.; Nishikimi, T.; Mochizuki, Y.; Yamada, Y.; Yoshitatsu, M.; Inoue, Y.; Kuwata, T.; Ogawa, H.; Tsuchiya, G.; Ishimitsu, T.; et al. Greater expression of inflammatory cytokines, adrenomedullin, and natriuretic peptide receptor-C in epicardial adipose tissue in coronary artery disease. Regul. Pept. 2010, 165, 210–217. [Google Scholar] [CrossRef]
- Kerr, J.D.; Holden, R.M.; Morton, A.R.; Nolan, R.L.; Hopman, W.M.; Pruss, C.M.; Garland, J.S. Associations of epicardial fat with coronary calcification, insulin resistance, inflammation, and fibroblast growth factor-23 in stage 3–5 chronic kidney disease. BMC Nephrol. 2013, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Cannata-Andía, J.B.; Martín-Carro, B.; Martín-Vírgala, J.; Rodríguez-Carrio, J.; Bande-Fernández, J.J.; Alonso-Montes, C.; Carrillo-López, N. Chronic Kidney Disease—Mineral and Bone Disorders: Pathogenesis and Management. Calcif. Tissue Res. 2020, 108, 410–422. [Google Scholar] [CrossRef]
- Kruzel-Davila, E.; Wasser, W.G.; Aviram, S.; Skorecki, K. APOL1 nephropathy: From gene to mechanisms of kidney injury. Nephrol. Dial. Transplant. 2015, 31, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.G.; Bomback, A.S.; Radhakrishnan, J.; Herlitz, L.C.; Stokes, M.B.; Markowitz, G.S.; D’Agati, V.D. The Modern Spectrum of Renal Biopsy Findings in Patients with Diabetes. Clin. J. Am. Soc. Nephrol. 2013, 8, 1718–1724. [Google Scholar] [CrossRef] [Green Version]
- Anguiano Gómez, L.; Lei, Y.; Devarapu, S.K.; Anders, H.-J. The diabetes pandemic suggests unmet needs for ‘CKD with diabetes’ in addition to ‘diabetic nephropathy’-implications for pre-clinical research and drug testing. Nephrol. Dial Transplant. 2018, 33, 1292–1304. [Google Scholar] [CrossRef] [Green Version]
- Goodman, W.G.; Goldin, J.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-Artery Calcification in Young Adults with End-Stage Renal Disease Who Are Undergoing Dialysis. N. Engl. J. Med. 2000, 342, 1478–1483. [Google Scholar] [CrossRef]
- Tomiyama, C.; Higa, A.; Dalboni, M.A.; Cendoroglo, M.; Draibe, S.A.; Cuppari, L.; Carvalho, A.B.; Neto, E.M.; Canziani, M.E.F. The impact of traditional and non-traditional risk factors on coronary calcification in pre-dialysis patients. Nephrol. Dial. Transplant. 2006, 21, 2464–2471. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.L.; Fairman, R.; Mitchell, M.E.; Carpenter, J.P.; Golden, M.; Khalapyan, T.; Wolfe, M.; Neschis, D.; Milner, R.; Scoll, B.; et al. Bone formation in carotid plaques: A clinicopathological study. Stroke 2002, 33, 1214–1219. [Google Scholar] [CrossRef] [Green Version]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Phan, O.; Burnier, M.; Wuerzner, G.; Nephrology, S.O. Hypertension in Chronic Kidney Disease—Role of Arterial Calcification and Impact on Treatment. Eur. Cardiol. Rev. 2014, 9, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Krasniak, A.; Drozdz, M.; Pasowicz, M.; Chmiel, G.; Michalek, M.; Szumilak, D.; Podolec, P.; Klimeczek, P.; Konieczynska, M.; Wicher-Muniak, E.; et al. Factors involved in vascular calcification and atherosclerosis in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2007, 22, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, S.; Drüeke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-Y.; Chen, L.-R.; Chen, K.-H. Osteoporosis in Patients with Chronic Kidney Diseases: A Systemic Review. Int. J. Mol. Sci. 2020, 21, 6846. [Google Scholar] [CrossRef] [PubMed]
- Perneger, T.V.; Whelton, P.K.; Klag, M.J. Race and end-stage renal disease. Socioeconomic status and access to health care as mediating factors. Arch. Intern. Med. 1995, 155, 1201–1208. [Google Scholar] [CrossRef]
- Phan, O.; Ivanovski, O.; Nguyen-Khoa, T.; Mothu, N.; Angulo, J.; Westenfeld, R.; Ketteler, M.; Meert, N.; Maizel, J.; Nikolov, I.G.; et al. Sevelamer Prevents Uremia-Enhanced Atherosclerosis Progression in Apolipoprotein E–Deficient Mice. Circulation 2005, 112, 2875–2882. [Google Scholar] [CrossRef] [Green Version]
- Bessueille, L.; Magne, D. Inflammation: A culprit for vascular calcification in atherosclerosis and diabetes. Cell. Mol. Life Sci. 2015, 72, 2475–2489. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Norris, K. Lipid Disorders and Their Relevance to Outcomes in Chronic Kidney Disease. Blood Purif. 2011, 31, 189–196. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, M.; Huang, K.; He, Q.; Li, H.; Chang, G. MicroRNA-125b Affects Vascular Smooth Muscle Cell Function by Targeting Serum Response Factor. Cell. Physiol. Biochem. 2018, 46, 1566–1580. [Google Scholar] [CrossRef]
- Casella, S.; Bielli, A.; Mauriello, A.; Orlandi, A. Molecular Pathways Regulating Macrovascular Pathology and Vascular Smooth Muscle Cells Phenotype in Type 2 Diabetes. Int. J. Mol. Sci. 2015, 16, 24353–24368. [Google Scholar] [CrossRef] [Green Version]
- Cavallari, C.; Dellepiane, S.; Fonsato, V.; Medica, D.; Marengo, M.; Migliori, M.; Quercia, A.D.; Pitino, A.; Formica, M.; Panichi, V.; et al. Online Hemodiafiltration Inhibits Inflammation-Related Endothelial Dysfunction and Vascular Calcification of Uremic Patients Modulating miR-223 Expression in Plasma Extracellular Vesicles. J. Immunol. 2019, 202, 2372–2383. [Google Scholar] [CrossRef] [Green Version]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural Basis of Calcification Inhibition by α2-HS Glycoprotein/Fetuin-A. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [Green Version]
- Heiss, A.; Eckert, T.; Aretz, A.; Richtering, W.; van Dorp, W.; Schäfer, C.; Jahnen-Dechent, W. Hierarchical Role of Fetuin-A and Acidic Serum Proteins in the Formation and Stabilization of Calcium Phosphate Particles. J. Biol. Chem. 2008, 283, 14815–14825. [Google Scholar] [CrossRef] [Green Version]
- Wald, J.; Wiese, S.; Eckert, T.; Jahnen-Dechent, W.; Richtering, W.; Heiss, A. Formation and stability kinetics of calcium phosphate–fetuin-A colloidal particles probed by time-resolved dynamic light scattering. Soft Matter 2011, 7, 2869–2874. [Google Scholar] [CrossRef]
- Pasch, A.; Farese, S.; Gräber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-Based Test Measures Overall Propensity for Calcification in Serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752. [Google Scholar] [CrossRef] [Green Version]
- Mencke, R.; van der Vaart, A.; Pasch, A.; Harms, G.; Waanders, F.; Bilo, H.J.G.; van Goor, H.; Hillebrands, J.-L.; van Dijk, P.R. Serum calcification propensity is associated with HbA1c in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2021, 9, e002016. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, P.R.; Hop, H.; Waanders, F.; Mulder, U.J.; Pasch, A.; Hillebrands, J.; van Goor, H.; Bilo, H.J.G. Serum calcification propensity in type 1 diabetes associates with mineral stress. Diabetes Res. Clin. Pract. 2019, 158, 107917. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Fitzpatrick, J.; Monroy-Trujillo, J.M.; Sozio, S.M.; Jaar, B.G.; Estrella, M.M.; Serrano, J.; Anokhina, V.; Miller, B.L.; Melamed, M.L.; et al. Associations of Serum Calciprotein Particle Size and Transformation Time with Arterial Calcification, Arterial Stiffness, and Mortality in Incident Hemodialysis Patients. Am. J. Kidney Dis. 2021, 77, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.D.; Ketteler, M.; Tur, F.; Tur, E.; Isern, B.; Salcedo, C.; Joubert, P.H.; Behets, G.J.; Neven, E.; D’Haese, P.C.; et al. Characterization of SNF472 pharmacokinetics and efficacy in uremic and non-uremic rats models of cardiovascular calcification. PLoS ONE 2018, 13, e0197061. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Bellasi, A.; Bushinsky, D.; Bover, J.; Rodriguez, M.; Ketteler, M.; Sinha, S.; Salcedo, C.; Gillotti, K.; Padgett, C.; et al. Slowing Progression of Cardiovascular Calcification with SNF472 in Patients on Hemodialysis: Results of a Randomized Phase 2b Study. Circulation 2020, 141, 728–739. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, O.; Joki, N. Vascular Calcification in Diabetic Kidney Disease. Kidney Dial. 2022, 2, 595-606. https://doi.org/10.3390/kidneydial2040054
Phan O, Joki N. Vascular Calcification in Diabetic Kidney Disease. Kidney and Dialysis. 2022; 2(4):595-606. https://doi.org/10.3390/kidneydial2040054
Chicago/Turabian StylePhan, Olivier, and Nobuhiko Joki. 2022. "Vascular Calcification in Diabetic Kidney Disease" Kidney and Dialysis 2, no. 4: 595-606. https://doi.org/10.3390/kidneydial2040054