
β-Lactams and Ureas as Cross Inhibitors of Prokaryotic Systems
Abstract
:1. Introduction
- General Chemistry and Structure
- Lactams—Cyclic Amides
- Bioisosters of Amides—Ureas
2. Microbe-Specific Targeting by β-Lactams and Ureas
2.1. Gram-Positive Staphylococcus aureus and Enterococcus faecalis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorg. | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
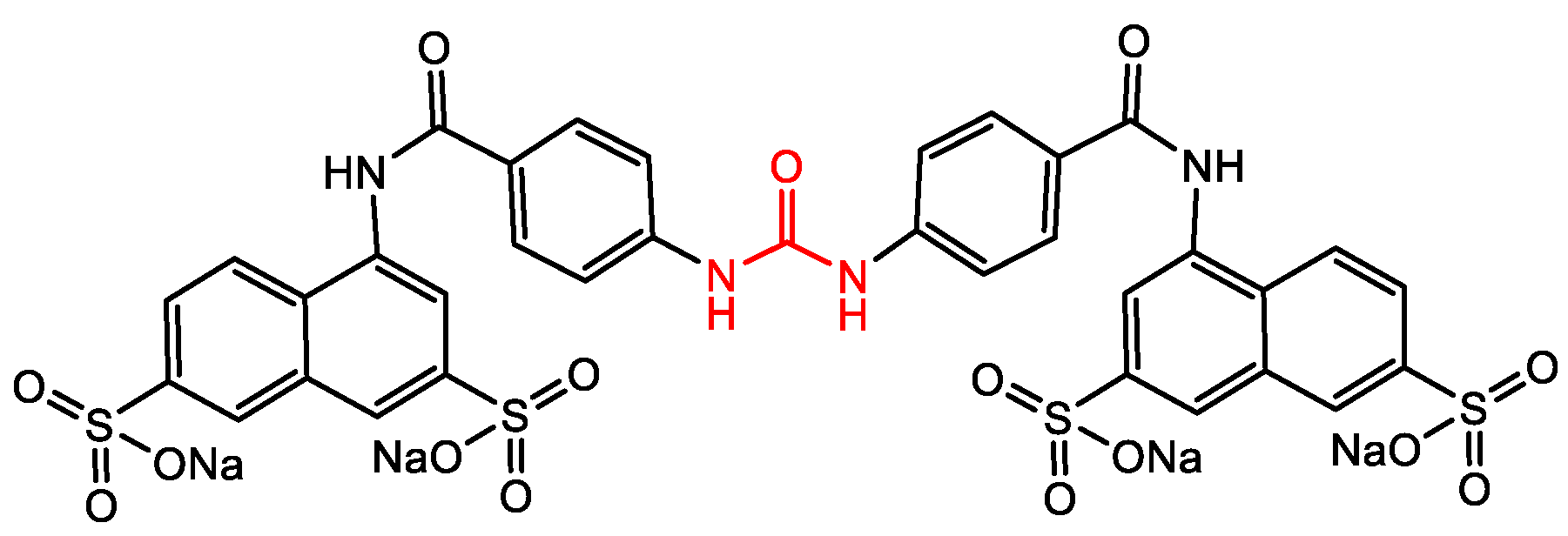
| S. aureus |  Cephazolin (first-generation cephalosporin) [22]  Ceftaroline (first-generation cephalosporin) [23]  Cefiderocol (approved for medical use in the United States in November 2019, and in the European Union in April 2020) [10,11,12]  1 [24] MIC = 1 mg/mL |  Triclocarban (TCC) [26,27,28,29]  2 [25,30] S. aureus (ATCC, MIC50 = 0.5 µg/mL) as well as MRSA (MIC50 = 0.05 µg/mL)  PK150 [25] MIC 0.3 µM for MSSA, MIC 0.03 µM for MRSA.  PQ 401 [31,32] |
| MRSA/ Biofilm |  3 [33,34]  4 [33,34] MIC values for 3 and 4 between 2.0 and 4.0 µg/mL as compared with vancomycin (from 0.5 to 1 µg/mL). Molecular target: cell wall synthesis | |
| S. aureus |  5 [35] MIC = 250 mg/mL Compound 5 was evaluated as an antibacterial and antifungal agent against Staphylococcus aureus (ATCC 25923), Escherichia coli (ATCC 25922), Bacillus subtilis (ATCC 8236F800), and Candida albicans (ATCC 885-653), showing a moderate antimicrobial activity.  6 [36] IC50 ranging from 0.054 to 4.24 μM | |
| E. faecalis |  Cefepime—fourth-generation cephalosporin [39]  Meropenem [40] |  7 [38]  8 [38] |
2.2. Gram-Negative Multi-Drug Resistant Bacteria: Proteus mirabilis, Klebsiella pneumoniae, and Acinetobacter baumannii
| Microorg. | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| P. mirabilis |  Cefepime [39] fourth-generation cephalosporin  Meropenem [39] |  10 [42]  11 [42] |
| K. pneumoniae |  Cefepime [40] fourth-generation cephalosporin  Meropenem [40]  Aztreonam [41] |  7 [38]  8 [38] |
| A. baumannii |  Aztreonam [41]  9 [41] Aztreonam with siderophore mimetic |  12 [43] |
2.3. Mycobacterium tuberculosis (Mtb)
| Microorg. | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| Mtb |  Meropenem [44,45]  13 [46,47]  14 [46,47] (TBBL-0000316)  15 [48] R = o-F, MIC 1.5 µg/mL (M. tuberculosis pathogenic strain H37Rv) |  16 [50] MIC 6.25 µg/mL (M. tuberculosis pathogenic strain H37Rv)  17 [50] MIC 3.125 µg/mL (M. tuberculosis pathogenic strain H37Rv)  18 [31] MIC 6.0 µg/mL (M. tuberculosis pathogenic strain H37Rv)  19 [31] MIC 5.2 µg/mL (M. tuberculosis pathogenic strain H37Rv) |
3. β-Lactams and Ureas as Antiviral Agents
3.1. Herpesviridae (DNA viruses): Human Cytomegalovirus (HCMV)
| Virus | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| HCMV |  20 [49]  21 [52]  22 [52] The most active compound of the series.  23 [53]   24 [54,55] 25 [55] EC50 0.6−7.4 μM |  26 [56] (R1 = Ar, R2 = H); 27 (R1 = Ar, R2 = H) (EC50 = 1.2–13.4 μM)  A939572 [57,58,59,60,61,62] IC50 < 4 nM (mSCD1) |
3.2. Flaviviridae, Picornaviridae, Retroviridae, and Coronaviridae (Single-Stranded Positive Sense RNA Viruses): DENV, ZKV, WNV, HCV, HRV, HIV, and SARS-CoV-2
| Virus | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
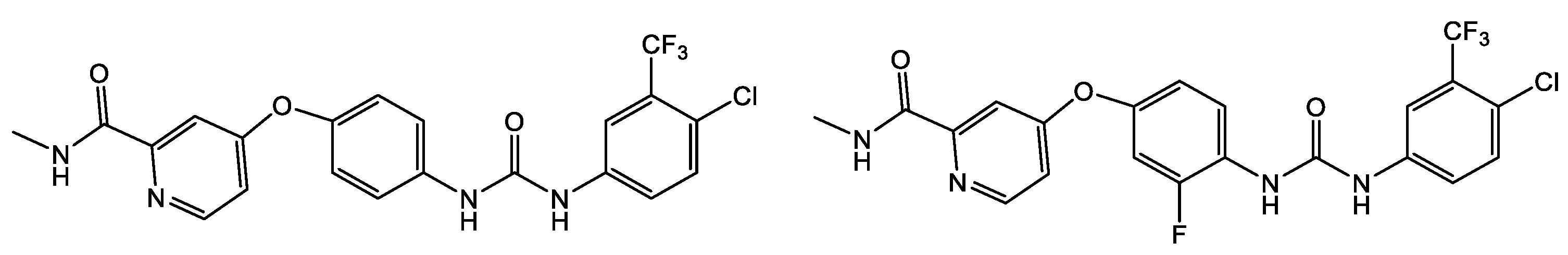
| Hepatitis C virus Flaviviridae |  28 [63] |  38 [57,58,59,60,61,62]  A939572 [59,60,61]  Boceprevir (SCH 503034) [69,70,71,72] (Ki = 14 nM, EC90 = 0.35 mM) |
| Dengue virus Flaviviridae |  A939572 [59,60,61]  39 [73] IC50 = 91 mM  40 [73] IC50 = 110 mM  41 [74] IC50 = 26 mM  42 [74] IC50 = 24 mM | |
| West Nile virus Flaviviridae |  28 [63] |  39 [73] IC50 = 51 mM  40 [73] IC50 = 71 mM |
| Zika virus Flaviviridae |  41 [74] IC50 = 28 mM  42 IC50 = 19 mM [74]  43 IC50 = 35 mM [75]  44 [75] ZIKV MTase IC50 = 23–48 mM ZIKV EC50 = 1.67–25 mM  ASN 25 [76,77]  Suramin [78] | |
| Rhinovirus Picornaviridae |  45 [79,80,81,82]  46 [79,80,81,82]  47 [83] | |
| HIV Retroviridae |  BSS-730A [64] |  Ritonavir [84]  38 [85,86,87,88] |
| Coronaviruses SARS-CoV-2 Coronaviridae |  34 and 35 [89,90]  36 and 37 [89,90] |  38 [85,86,87,88] |
3.3. Pneumoviridae and Orthomixoviridae (Single-Stranded Negative-Sense RNA Viruses): RSV and IV
| Virus | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| RSV Pneumoviridae |  29 [67]  30 [67]  31 [67]  32 [67] |  47 [83] |
| Influenza virus Orthomixoviridae |  33c [68]  33t [68] |  Regorafenib [78]  Sorafenib [78]  47 [83] |
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCMV | Human Cytomegalovirus |
| DENV | Dengue virus |
| ZKV | Zika virus |
| WNV | West Nile virus |
| HCV | Hepatitis C virus |
| HIV | Human Immunodeficiency virus |
| HRV | Human Rhinovirus |
| RSV | Respiratory Syncytial virus |
| IV | Influenza virus |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus-2 |
References
- Fisher, J.F.; Mobashery, S. Chapter 3: The β-Lactam (Azetidin-2-one) as a Privileged Ring in Medicinal Chemistry. In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Bräse, S., Ed.; Royal Chemical Society of Chemictry, RSC Drug Discovery, 2015; Available online: https://books.rsc.org/books/edited-volume/529/Privileged-Scaffolds-in-Medicinal-Chemistry-Design (accessed on 21 March 2023).
- Konaklieva, M.I.; Plotkin, B.J. The relationship between inhibitors of eukaryotic and prokaryotic serine proteases. Mini Rev. Med. Chem. 2004, 4, 721–739. [Google Scholar] [CrossRef]
- Kluge, A.F.; Petter, R.S. Acylating drugs: Redesigning natural covalent inhibitors. Curr. Opin. Chem. Biol. 2010, 14, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Burnett, D.A. β-Lactam cholesterol absorption inhibitors. Curr. Med. Chem. 2004, 11, 1873–1887. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Calvo, M.; Lisnock, J.-M.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Revell, K.D.; Heldreth, B.; Long, T.E.; Jang, S.; Turos, E. N-thiolated β-lactams: Studies on the mode of action and identification of a primary cellular target in Staphylococcus aureus. Bioorg. Med. Chem. 2007, 15, 2453–2467. [Google Scholar] [CrossRef]
- Konaklieva, M.I. Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects. Antibiotics 2014, 3, 128–142. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. β-Lactams against the Fortress of the Gram-Positive Staphylococcus aureus Bacterium. Chem. Rev. 2021, 121, 3412–3463. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. β-Lactams from the Ocean. Mar. Drugs 2023, 21, 86. [Google Scholar] [CrossRef]
- WHO: More New Antibiotics Needed. Available online: https://www.idse.net/Bacterial-Infections/Article/03-23/WHO-More-New-Antibiotics-Needed/69730 (accessed on 21 March 2023).
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.K.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A.; et al. Cefiderocol: A siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-negative bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin. Infect. Dis. 2019, 69, S538–S543. [Google Scholar] [CrossRef]
- Kumar, L.; Brenner, N.; Brice, J.; Klein-Seetharaman, J.; Sarkar, S.K. Cephalosporins Interfere with Quorum Sensing and Improve the Ability of Caenorhabditis elegans to Survive Pseudomonas aeruginosa Infection. Front. Microbiol. 2021, 12, 598498. [Google Scholar] [CrossRef] [PubMed]
- Mondeme, M.; Carnoy, C.; Sirard, J.-C.; Faveeuw, C. Treatment of Bacterial Infections with β-Lactams: Cooperation with Innate Immunity. Infect. Immun. 2023, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Carmona, A.V.; Towari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M. Urea Derivatives in Modern Drug Discovery and Medicinal Chemistry. J. Med. Chem. 2020, 63, 2751–2788. [Google Scholar] [CrossRef]
- Ronchetti, R.; Moroni, G.; Carotti, A.; Gioiello, A.; Camaioni, E. Recent advances in urea- and thiourea-containing compounds: Focus on innovative approaches in medicinal chemistry and organic synthesis. RSC Med. Chem. 2021, 12, 1046. [Google Scholar] [CrossRef]
- Adibekian, A.; Martin, B.R.; Wang, C.; Hsu, K.-L.; A Bachovchin, D.; Niessen, S.; Hoover, H.; Cravatt, B.F. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat. Chem. Biol. 2011, 7, 469–478. [Google Scholar] [CrossRef]
- Chang, J.W.; Nomura, D.K.; Cravatt, B.F. A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem. Biol. 2011, 18, 476–484. [Google Scholar] [CrossRef]
- Hsu, K.L.; Tsuboi, K.; Adibekian, A.; Pugh, H.; Masuda, K.; Cravatt, B.F. DAGLbeta inhibition perturbs a lipid network involved in macrophage infammatory responses. Nat. Chem. Biol. 2012, 8, 999–1007. [Google Scholar] [CrossRef]
- Remsberg, J.R.; Suciu, R.M.; Zambetti, N.A.; Hanigan, T.W.; Firestone, A.J.; Inguva, A.; Long, A.; Ngo, N.; Lum, K.M.; Henry, C.L.; et al. ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat. Chem. Biol. 2021, 17, 856–864. [Google Scholar] [CrossRef]
- Li, J.; Echevarria, K.L.; Traugott, K.A. β-Lactam therapy for methicillin-susceptible Staphylococcus aureus bacteremia: A comparative review of cefazolin versus antistaphylococcal penicillins. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 346–360. [Google Scholar] [CrossRef]
- Dingle, T.C.; Gamage, D.; Gomez-Villegas, S.; Hanson, B.M.; Reyes, J.; Abbott, A.; Burnham, C.-A.D.; Bard, J.D.; Fritz, S.; Miller, W.R.; et al. Prevalence and Characterization of the Cefazolin Inoculum Effect in North American Methicillin-Susceptible Staphylococcus aureus Isolates. J. Clin. Microbiol. 2022, 60. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.K.; Ghosh, S.; Bhat, H.R.; Naesens, L.; Singh, U.P. Synthesis and biological evaluation of substituted phenyl azetidine-2-one sulphonyl derivatives as potential antimicrobial and antiviral agents. Bioorg. Chem. 2020, 104, 104320. [Google Scholar] [CrossRef]
- Le, P.; Kunold, E.; Macsics, R.; Rox, K.; Jennings, M.C.; Ugur, I.; Reinecke, M.; Chaves-Moreno, D.; Hackl, M.W.; Fetzer, C.; et al. Repurposing human kinase inhibitors to create an antibiotic active against drug-resistant Staphylococcus aureus, persisters and biofilms. Nat. Chem. 2020, 12, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Macsics, R.; Hackl, M.W.; Fetzer, C.; Mostert, D.; Bender, J.; Layer, F.; Sieber, S.A. Comparative Target Analysis of Chlorinated Biphenyl Antimicrobials Highlights MenG as Molecular Target of Triclocarban. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- FDA Issues Final Rule on Safety and Effectiveness of Antibacterial Soaps. Available online: https://www.fda.gov/news-events/press-announcements/fda-issues-final-rule-safety-and-effectiveness-antibacterial-soaps (accessed on 12 February 2023).
- Wang, Y.; Teng, Y.; Wang, D.; Han, K.; Wang, H.; Kang, L. The fate of triclocarban in activated sludge and its influence on biological wastewater treatment system. J. Environ. Manag. 2020, 276, 111237. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Pellegrino, M.; Aquaro, S.; Franchini, C.; Sinicropi, M.S. Diarylureas: Repositioning from Antitumor to Antimicrobials or Multi-Target Agents against New Pandemics. Antibiotics 2021, 10, 92. [Google Scholar] [CrossRef]
- Pujol, E.; Blanco-Cabra, N.; Julián, E.; Leiva, R.; Torrents, E.; Vázquez, S. Pentafluorosulfanyl-containing triclocarban analogs with potent antimicrobial activity. Molecules 2018, 23, 2853. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Iacopetta, D.; Sinicropi, M.S.; Franchini, C. Diarylureas as antitumor agents. Appl. Sci. 2021, 11, 374. [Google Scholar] [CrossRef]
- Kim, W.; Zou, G.; Pan, W.; Fricke, N.; Faizi, H.A.; Kim, S.M.; Khader, R.; Li, S.; Lee, K.; Escorba, L.; et al. The neutrally charged diarylurea compound PQ401 kills antibiotic-resistant and antibiotic-tolerant Staphylococcus aureus. Mbio 2020, 11, 603. [Google Scholar] [CrossRef]
- Eissa, I.H.; Mohammad, H.; Qassem, O.A.; Younis, W.; Abdelghany, T.M.; Elshafeey, A.; Moustafa, M.M.A.R.; Seleem, M.N.; Mayhoub, A.S. Diphenylurea derivatives for combating methicillin- and vancomycin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2017, 130, 73–85. [Google Scholar] [CrossRef]
- Mohammad, H.; Younis, W.; Ezzat, H.G.; Peters, C.E.; AbdelKhalek, A.; Cooper, B.; Pogliano, K.; Pogliano, J.; Mayhoub, A.S.; Seleem, M.N. Bacteriological profiling of diphenylureas as a novel class of antibiotics against methicillin-resistant Staphylococcus aureus. PLoS ONE 2017, 12, e0182821. [Google Scholar] [CrossRef] [PubMed]
- Bratenko, M.K.; Barus, M.M.; Rotar, D.V.; Vovk, M.V. Polyfunctional Pyrazoles. 9*. Synthesis of 1-Alkyl(Aryl)-3-[4-(Hydroxymethyl)-1H-Pyrazol-3-Yl]ureas. Chem. Heterocyc. Compd. 2014, 50, 1252–1258. [Google Scholar] [CrossRef]
- Kane, J.L.; Hirth, B.H.; Liang, B.; Gourlie, B.B.; Nahill, S.; Barsomian, G. Ureas of 5-aminopyrazole and 2-aminothiazole inhibit growth of gram-positive bacteria. Bioorg. Med. Chem. Lett. 2003, 13, 4463–4466. [Google Scholar] [CrossRef] [PubMed]
- Brullo, C.; Rapetti, F.; Bruno, O. Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry. Molecules 2020, 25, 3457. [Google Scholar] [CrossRef]
- Gezegen, H.; Hepokur, C.; Tutar, U.; Ceylan, M. Synthesis and Biological Evaluation of Novel 1-(4-(Hydroxy(1-oxo-1,3-dihydro-2H-inden-2-ylidene)methyl)phenyl)-3-phenylurea Derivatives. Chem. Biodivers. 2017, 14, e1700223. [Google Scholar] [CrossRef]
- Jamil, R.T.; Foris, L.A.; Snowden, J. Proteus Mirabilis Infections. Available online: https://www.ncbi.nlm.nih.gov/books/NBK442017/ (accessed on 30 March 2023).
- Ashurst, J.V.; Dawson, A. Klebsiella Pneumoniae. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519004/ (accessed on 30 March 2023).
- Liu, R.; Miller, P.A.; Miller, M.J. Conjugation of Aztreonam, a Synthetic Monocyclic β-Lactam Antibiotic, to a Siderophore Mimetic Significantly Expands Activity Against Gram-Negative Bacteria. ACS Infect. Dis. 2021, 7, 2979–2986. [Google Scholar] [CrossRef] [PubMed]
- Sarveswari, S.; Vijayakumar, V. A Facile Synthesis of diaylureas and their antimicrobial evaluation. Chiang Mai J. Sci. 2018, 45, 997–1007. [Google Scholar]
- Patil, M.; Noonikara-Poyil, A.; Joshi, S.D.; Patil, S.A.; Patil, S.A.; Bugarin, A. New Urea Derivatives as Potential Antimicrobial Agents: Synthesis, Biological Evaluation, and Molecular Docking Studies. Antibiotics 2019, 8, 178. [Google Scholar] [CrossRef]
- Diacon, A.H.; van der Merwe, L.; Barnard, M.; von Groote-Bidlingmaier, F.; Lange, C.; García-Basteiro, A.L.; Sevene, E.; Ballell, L.; Barros-Aguirre, D. β-Lactams against Tuberculosis--New Trick for an Old Dog? N. Engl. J. Med. 2016, 375, 393–394. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, S.P.; Zuur, M.A.; Anthony, R.; Wilffert, B.; van Altena, R.; Akkerman, O.W.; de Lange, W.C.M.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.C. Evaluation of Carbapenems for Treatment of Multi- and Extensively Drug-Resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e01489-18. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; Zhang, J.; Quezada, L.L.; Roberts, J.; Ling, Y.; Wood, M.; Shinwari, W.; Goullieux, L.; Roubert, C.; Fraisse, L.; et al. Identification of β-Lactams Active against Mycobacterium tuberculosis by a Consortium of Pharmaceutical Companies and Academic Institutions. ACS Infect. Dis. 2022, 8, 557–573. [Google Scholar] [CrossRef]
- Lopez Quezada, L.; Li, K.; McDonald, S.L.; Nguyen, Q.; Perkowski, A.J.; Pharr, C.W.; Gold, B.; Roberts, J.; McAulay, K.; Saito, K.; et al. Dual-Pharmacophore Pyrithione-Containing Cephalosporins Kill Both Replicating and Nonreplicating Mycobacterium tuberculosis. ACS Infect. Dis. 2019, 5, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Kuskovsky, R.; Lloyd, D.; Arora, K.; Plotkin, B.; Green, J.; Boshoff, H.; Barry, C.; Deschamps, J.; Konaklieva, M.I. C4-Phenylthio β-lactams: Effect of the Chirality of the β-Lactam Ring on Antimicrobial Activity. Bioorg. Med. Chem. 2019, 27, 115050. [Google Scholar] [CrossRef]
- Déziel, R.; Malenfant, E. Inhibition of human cytomegalovirus protease No with monocyclic β-lactams. Bioorg. Med. Chem. Lett. 1998, 8, 1437–1442. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V. Chattapadhayaya, Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Velappan, A.B.; Raja, M.R.C.; Datta, D.; Tsai, Y.T.; Halloum, I.; Wan, B.; Kremer, L.; Gramajo, H.; Franzblau, S.G.; Mahapatra, S.K.; et al. Attenuation of Mycobacterium species through direct and macrophage mediated pathway by unsymmetrical diaryl urea. Eur. J. Med. Chem. 2017, 125, 825–841. [Google Scholar] [CrossRef]
- Yoakim, C.; Ogilvie, W.W.; Cameron, D.R.; Chabot, C.; Grand-Maître, C.; Guse, I.; Haché, B.; Kawai, S.; Naud, J.; O’Meara, J.A.; et al. Potent β-Lactam Inhibitors of Human Cytomegalovirus Protease. Antivir. Chem. Chemother. 1998, 9, 379–387. [Google Scholar] [CrossRef]
- Yoakim, C.; Ogilvie, W.W.; Cameron, D.R.; Chabot, C.; Guse, I.; Haché, B.; Naud, J.; O’Meara, J.A.; Plante, R.; Déziel, R. β-Lactam Derivatives as Inhibitors of Human Cytomegalovirus Protease. J. Med. Chem. 1998, 41, 2882–2891. [Google Scholar] [CrossRef]
- Gerona-Navarro, G.; Pérez de Vega, M.J.; García-López, M.T.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E.; González-Muñiz, R. Synthesis and anti-HCMV activity of 1-acyl-β-lactams and 1-acylazetidines derived from phenylalanine. Bioorg. Med. Chem. Lett. 2004, 14, 2253–2256. [Google Scholar] [CrossRef]
- Gerona-Navarro, G.; Pérez de Vega, M.J.; García-López, M.T.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J.; González-Muñiz, R. From 1-Acyl-β-lactam Human Cytomegalovirus Protease Inhibitors to 1-Benzyloxycarbonylazetidines with Improved Antiviral Activity. A Straightforward Approach to Convert Covalent to Noncovalent Inhibitors. J. Med. Chem. 2005, 48, 2612–2621. [Google Scholar] [CrossRef]
- Džimbeg, G.; Zorc, B.; Kralj, M.; Ester, K.; Pavelić, K.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E.; Mintas, M. The novel primaquine derivatives of N-alkyl, cycloalkyl or aryl urea: Synthesis, cytostatic and antiviral activity evaluations. Eur. J. Med. Chem. 2008, 43, 1180–1187. [Google Scholar] [CrossRef]
- Xin, Z.; Zhao, H.; Serby, M.D.; Liu, B.; Liu, M.; Szczepankiewicz, B.G.; Nelson, L.T.; Smith, H.T.; Suhar, T.S.; Janis, R.S.; et al. Discovery of piperidine-aryl urea-based stearoyl-CoA desaturase 1 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4298–4302. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Krauss, M.R.; Gray-Keller, M.P.; Brownlie, A.; Miyazaki, M.; Kastelein, J.J.; Lusis, A.J.; Stalenhoef, A.F.; Stoehr, J.P.; Hayden, M.R.; et al. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J. Lipid Res. 2002, 43, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, R.C.; Steel, J.J.; Pujari, V.; Rovnak, J.; Crick, D.C.; Perera, R. Stearoly-CoA desaturase 1 differentiates early and advanced dengue virus infections and determines virus particle infectivity. PLoS Pathog. 2018, 14, e1007261. [Google Scholar] [CrossRef]
- Uto, Y. Recent progress in the discovery and development of stearoyl CoA desaturase inhibitors. Chem. Phys. Lipids 2016, 197, 3–12. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Moquin-Beaudry, G.; Mangahas, C.L.; Pratesi, F.; Aubin, M.; Aumont, A.; Joppé, S.E.; Légiot, A.; Vachon, A.; Plourde, M.; et al. Stearoyl-CoA Desaturase inhibition reverses immune, synaptic and cognitive impairments in an Alzheimer’s disease mouse model. Nat. Commun. 2002, 13, 2061. [Google Scholar] [CrossRef]
- Dražić, T.; Kopf, S.; Corridan, J.; Leuthold, M.M.; Bertoša, B.D.; Klein, C.D. Peptide-β-lactam Inhibitors of Dengue and West Nile Virus NS2B-NS3 Protease Display Two Distinct Binding Modes. J. Med. Chem. 2020, 63, 140–156. [Google Scholar] [CrossRef]
- Bártolo, I.; Santos, S.S.; Fontinha, D.; Machado, M.; Francisco, D.; Sepodes, B.; Rocha, J.; Mota-Filipe, H.; Pinto, R.; Figueira, M.E.; et al. Spiro-β-lactam BSS-730A Displays Potent Activity against HIV and Plasmodium. ACS Infect. Dis. 2021, 7, 421–434. [Google Scholar] [CrossRef]
- Dömling, A.; Starnecker, M.; Ugi, I. The β-lactam-nucleoside chimera. Angew. Chem. Int. Ed. 1995, 34, 2238–2239. [Google Scholar] [CrossRef]
- Dang, Q.; Zhang, Z.; Bai, Y.; Sun, R.; Yin, J.; Chen, T.; Bogen, S.; Girijavallabhan, V.; Olsen, D.B.; Meinke, P.T. Syntheses of nucleosides with a 1′,2′-β-lactam moiety as potential inhibitors of hepatitis C virus NS5B polymerase. Tetrahedron Lett. 2014, 55, 5576–5579. [Google Scholar] [CrossRef]
- D’hooghe, M.; Mollet, K.; De Vreese, R.; Jonckers, T.H.M.; Dams, G.; De Kimpe, N. Design, Synthesis, and Antiviral Evaluation of Purine-β-lactam and Purine-aminopropanol Hybrids. J. Med. Chem. 2012, 55, 5637–5641. [Google Scholar] [CrossRef]
- Głowacka, I.E.; Grabkowska-Drużyc, M.; Andrei, G.; Schols, D.; Snoeck, R.; Witek, K.; Podlewska, S.; Handzlik, J.; Piotrowska, D.G. Novel N-Substituted 3-Aryl-4-(diethoxyphosphoryl)azetidin-2-ones as Antibiotic Enhancers and Antiviral Agents in Search for a Successful Treatment of Complex Infections. Int. J. Mol. Sci. 2021, 22, 8032. [Google Scholar] [CrossRef]
- Oerlemans, R.; Ruiz-Moreno, A.J.; Cong, Y.; Dinesh Kumar, N.; Velasco-Velazquez, M.A.; Neochoritis, C.G.; Smith, J.; Reggiori, F.; Groves, M.R.; Dömling, A. Repurposing the HCV NS3-4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 2020, 12, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Li, H.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Arnould, M.A.; Jonsson, C.B.; Surendranathan, S.; Parvathareddy, J.; Blakeley, M.P.; et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022, 13, 2268. [Google Scholar] [CrossRef] [PubMed]
- Benmansour, F.; Trist, I.; Coutard, B.; Decroly, E.; Querat, G.; Brancale, A.; Barral, K. Discovery of novel dengue virus NS5 methyltransferase non-nucleoside inhibitors by fragment-based drug design. Eur. J. Med. Chem. 2017, 125, 865–880. [Google Scholar] [CrossRef]
- Hernandez, J.; Hoffer, L.; Coutard, B.; Querat, G.; Roche, P.; Morelli, X.; Decroly, E.; Barral, K. Optimization of a fragment linking hit toward Dengue and Zika virus NS5 methyltransferases inhibitors. Eur. J. Med. Chem. 2019, 161, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Spizzichino, S.; Mattedi, G.; Lauder, K.; Valle, C.; Aouadi, W.; Canard, B.; Decroly, E.; Kaptein, S.J.F.; Neyts, J.; Graham, C.; et al. Design, Synthesis and Discovery of N,N′-Carbazoyl-aryl-urea Inhibitors of Zika NS5 Methyltransferase and Virus Replication. ChemMedChem 2020, 15, 385–390. [Google Scholar] [CrossRef]
- Micewicz, E.D.; Khachatoorian, R.; French, S.W.; Ruchala, P. Identification of novel small-molecule inhibitors of Zika virus infection. Bioorg. Med. Chem. Lett. 2018, 28, 452–458. [Google Scholar] [CrossRef]
- Khachatoorian, R.; Micewicz, E.D.; Micewicz, A.; French, S.W.; Ruchala, P. Optimization of 1, 3-disubstituted urea-based inhibitors of Zika virus infection. Bioorg. Med. Chem. Lett. 2019, 29, 126626. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, M.; Chi, X.; Liu, X.; Zhou, J.; Lin, T.; Yang, W. High-Throughput Screening Identifies Mixed-Lineage Kinase 3 as a Key Host Regulatory Factor in Zika Virus Infection. J. Virol. 2019, 93, e00758-19. [Google Scholar] [CrossRef]
- Voss, S.; Nitsche, C. Inhibitors of the Zika virus protease NS2B-NS3. Bioorg. Med. Chem. Lett. 2020, 30, 126965. [Google Scholar] [CrossRef]
- Lesch, M.; Luckner, M.; Meyer, M.; Weege, F.; Gravenstein, I.; Raftery, M.; Sieben, C.; Martin-Sancho, L.; Imai-Matsushima, A.; Welke, R.-W.; et al. RNAi-based small molecule repositioning reveals clinically approved urea-based kinase inhibitors as broadly active antivirals. PLoS Pathog. 2019, 15, e1007601. [Google Scholar] [CrossRef] [PubMed]
- Longshaw, A.I.; Fordyce, E.A.F.; Onions, S.T.; King-Underwood, J.; Venable, J.D. Preparation of Aromatic Heterocyclic Compounds as p38 MAP Kinase Inhibitors with Antiinflammatory Activity. WO Patent 2,015,121,444, 20 August 2015. [Google Scholar]
- King-Underwood, J.; Murray, P.J.; Williams, J.G.; Buck, I.; Onions, S.T. Preparation of Pyrazolyl Naphthyl Ureas as p38 MAP Kinase Inhibitors. WO Patent 2,011,124,930, 13 October 2011. [Google Scholar]
- King-Underwood, J.; Ito, K.; Strong, P.; Rapeport, W.G.; Charron, C.E.; Murray, P.J.; Williams, J.G.; Onions, S.T. 1-(5-Pyrazolyl)-3-(1-naphthyl)ureas as Enzyme Inhibitors and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Inflammatory Disorders. WO Patent 2,011,124,923, 13 October 2011. [Google Scholar]
- Ito, K.; Strong, P.; Rapeport, W.G.; Murray, P.J.; King-Underwood, J.; Williams, J.G.; Onions, S.T.; Joly, K.; Charron, C.E. Preparation of Pyrazolyl Ureas as p38 MAP Kinase Inhibitors. WO Patent 2,010,067,130, 21 April 2011. [Google Scholar]
- Murray, P.J.; Onions, S.T.; Williams, J.G.; Joly, K. Preparation of Pyrido[2,3-b]pyrazine Compounds for Use in Drug Formulations for Treating Inflammation, Respiratory Disorders, and Viral Infections. WO2012119690A1, 22 December 2011. Available online: https://www.ncbi.nlm.nih.gov/books/NBK544312 (accessed on 3 April 2023).
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Nevers, Q.; Gelin, M.; Bessin, Y.; Brillet, R.; Cala, O.; Douguet, D.; Bourguet, W.; et al. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat. Commun. 2016, 7, 12777. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Gallay, P.A. The role of immunophilins in viral infection. Biochim. Biophys. Acta 2014, 1850, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Feinman, S.V.; Jablkowski, M.; Horban, A.; Kryczka, W.; Pawlowska, M.; Heathcote, J.E.; Mazzella, G.; Vandelli, C.; Nicolas-Métral, V.; et al. The cyclophilin inhibitor Debio-025 combined with PEG-IFN alpha-2a significantly reduces viral load in treatment-naïve hepatitis C patients. Hepatology 2009, 49, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Flisiak, R.; Sarin, S.K.; Rasenack, J.; Piratvisuth, T.; Chuang, W.L.; Peng, C.Y.; Foster, G.R.; Shah, S.; Wedemeyer, H.; et al. Alisporivir plus ribavirin, interferon-free or in combination with peg-interferon, for HCV genotype 2 or 3 infection. Hepatology 2015, 62, 1013–1023. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, P.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Malla, T.R.; Tumber, A.; John, T.; Brewitz, L.; Strain-Damerell, C.; Owen, C.D.; Schofield, C.J. Mass spectrometry reveals potential of β-lactams as SARS-CoV-2 M pro inhibitors. Chem. Commun. 2021, 57, 1430–1433. [Google Scholar] [CrossRef]
- Malla, T.R.; Brewitz, L.; Muntean, D.-G.; Aslam, H.; Owen, C.D.; Salah, E.; Tumber, A.; Lukacik, P.; Strain-Damerell, C.; Mikolajek, H.; et al. Penicillin Derivatives Inhibit the SARS-CoV-2 Main Protease by Reaction with Its Nucleophilic Cysteine. J. Med. Chem. 2022, 65, 7682–7696. [Google Scholar] [CrossRef]
- Niphakis, M.J.; Cravatt, B.F. Enzyme inhibitor discovery by activity-based protein profling. Annu. Rev. Biochem. 2014, 83, 341–377. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konaklieva, M.I.; Plotkin, B.J. β-Lactams and Ureas as Cross Inhibitors of Prokaryotic Systems. Appl. Microbiol. 2023, 3, 605-628. https://doi.org/10.3390/applmicrobiol3030043
Konaklieva MI, Plotkin BJ. β-Lactams and Ureas as Cross Inhibitors of Prokaryotic Systems. Applied Microbiology. 2023; 3(3):605-628. https://doi.org/10.3390/applmicrobiol3030043
Chicago/Turabian StyleKonaklieva, Monika I., and Balbina J. Plotkin. 2023. "β-Lactams and Ureas as Cross Inhibitors of Prokaryotic Systems" Applied Microbiology 3, no. 3: 605-628. https://doi.org/10.3390/applmicrobiol3030043