
Identification of a New Drug Binding Site in the RNA-Dependent-RNA-Polymerase (RdRp) Domain


Abstract
:1. Introduction

2. Results
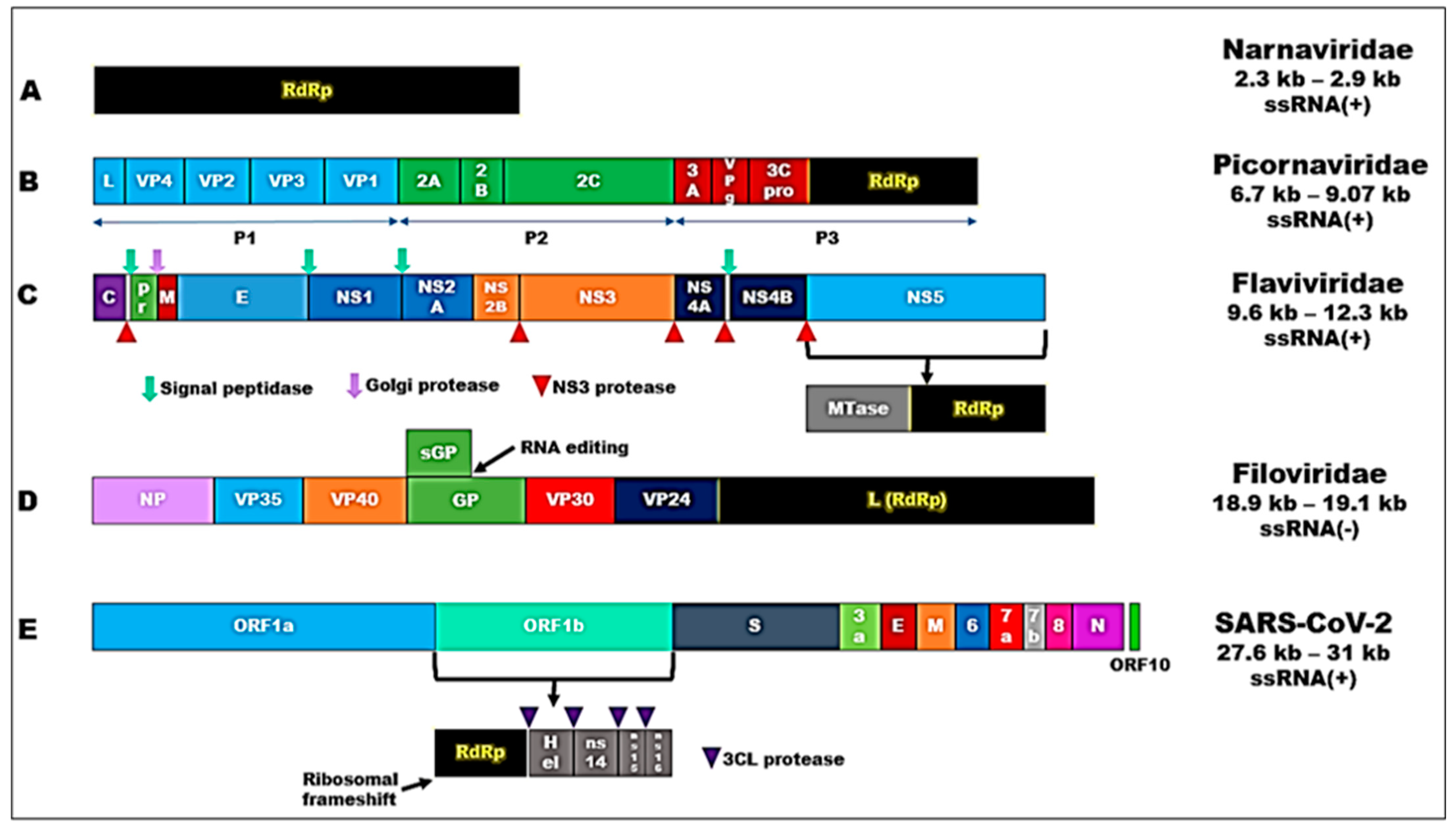
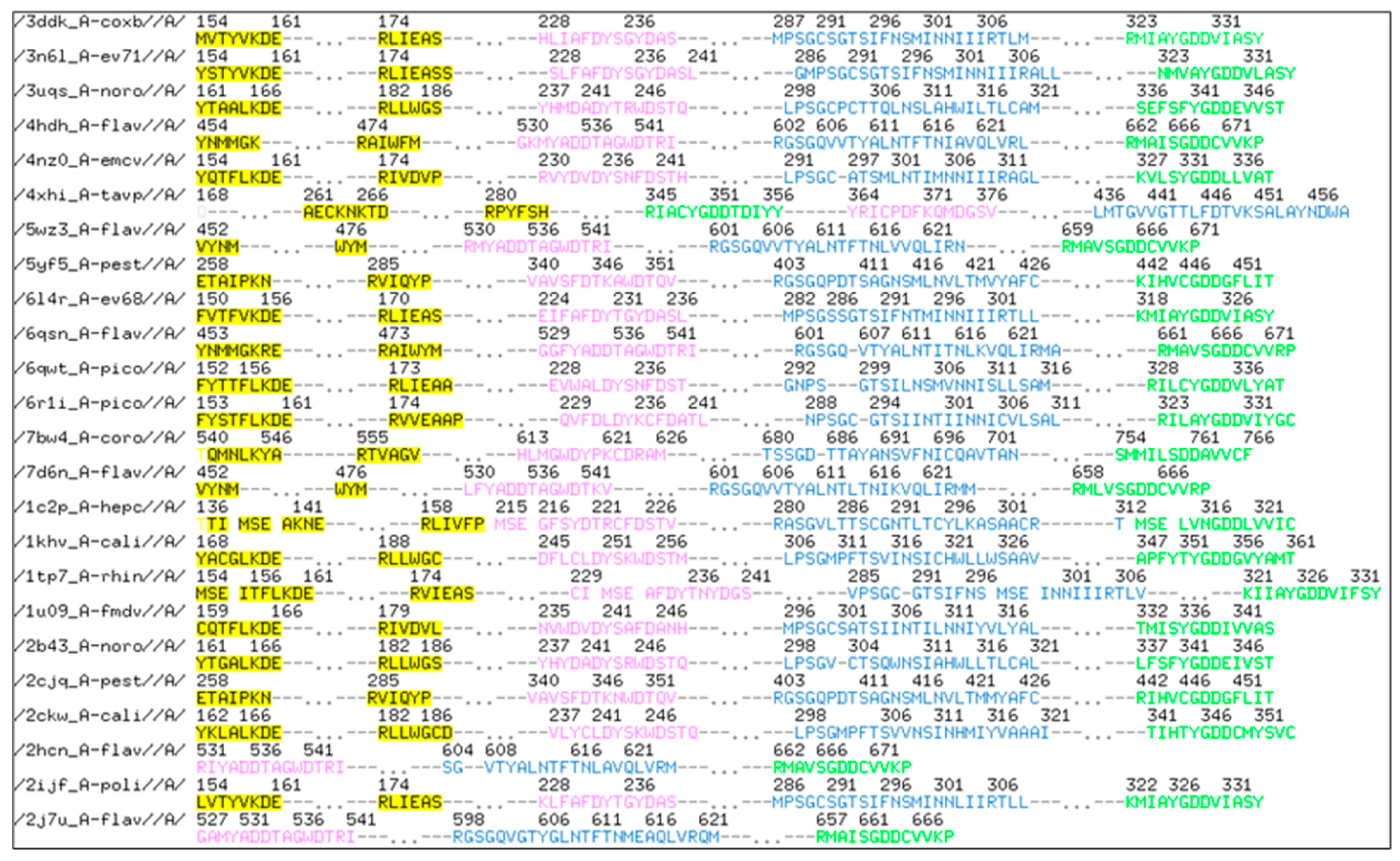
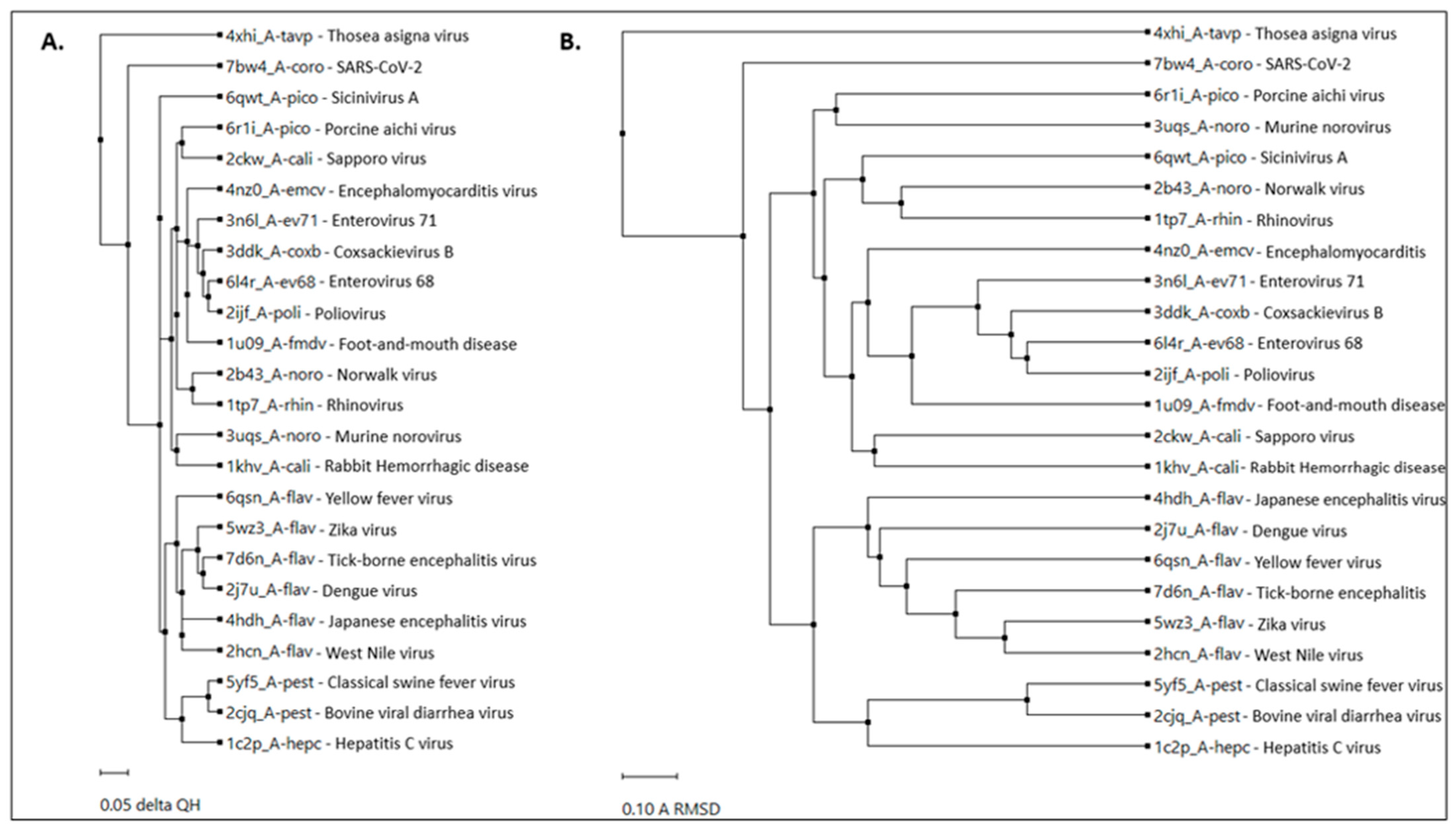
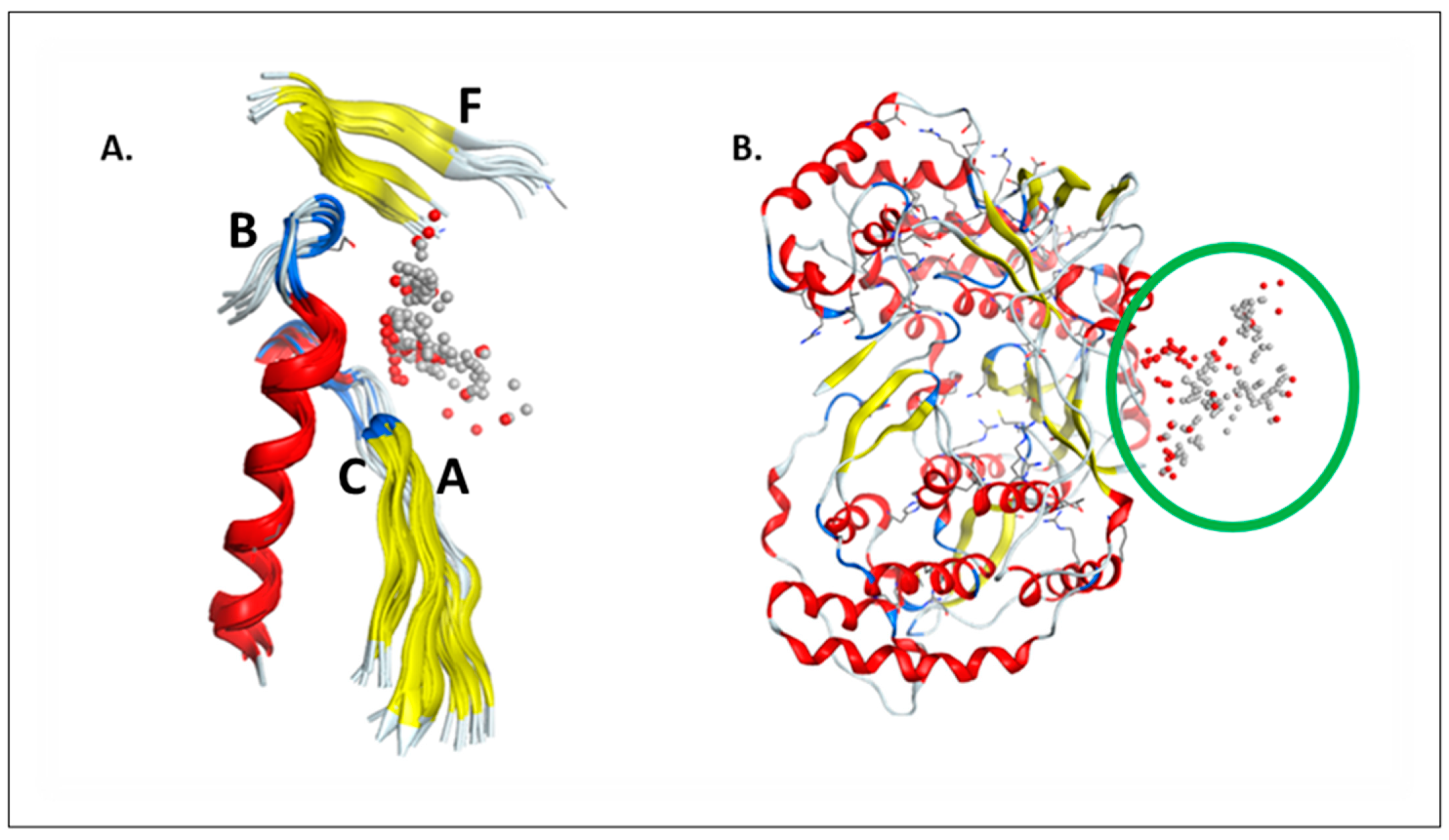
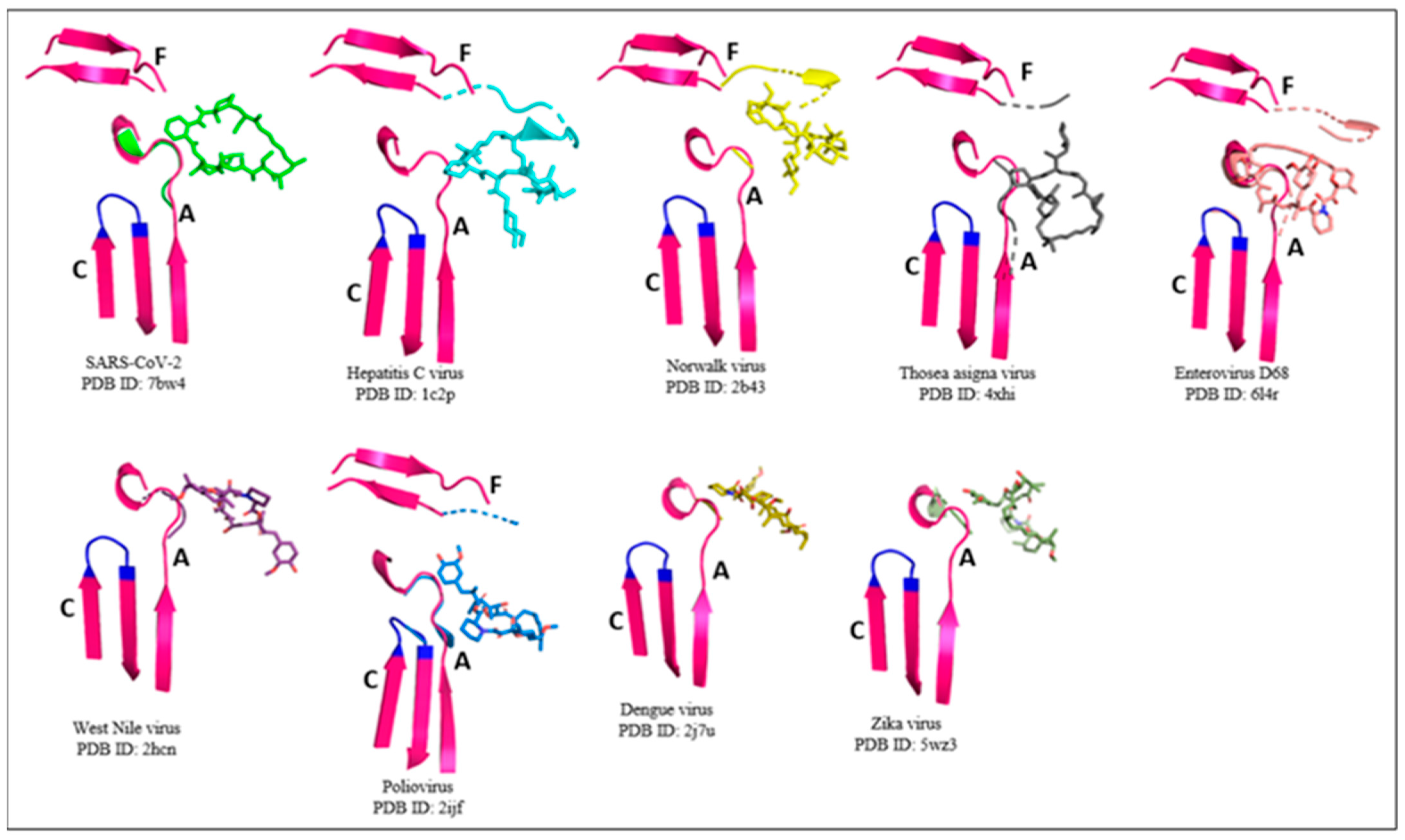
2.1. RdRp Motifs
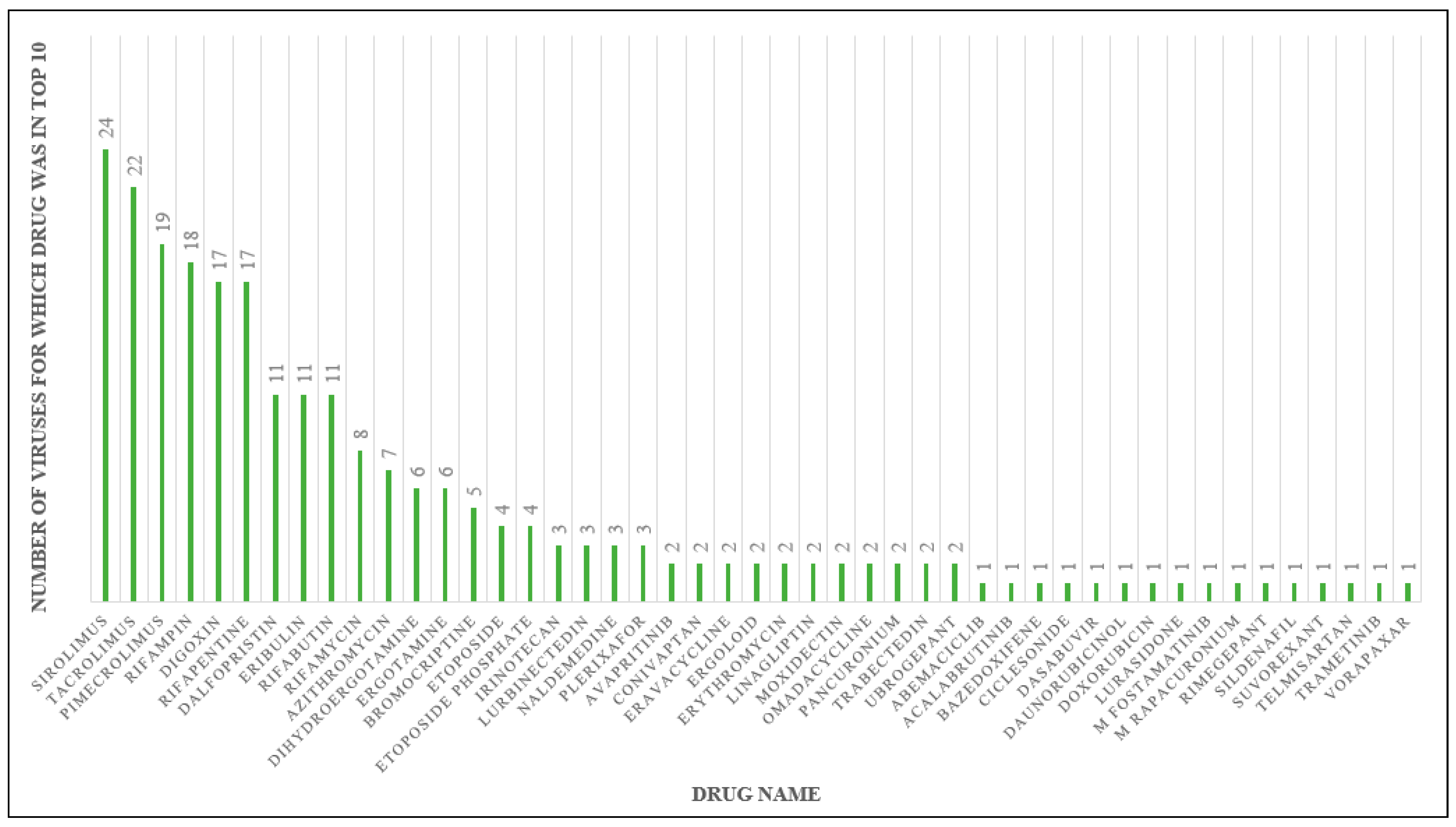
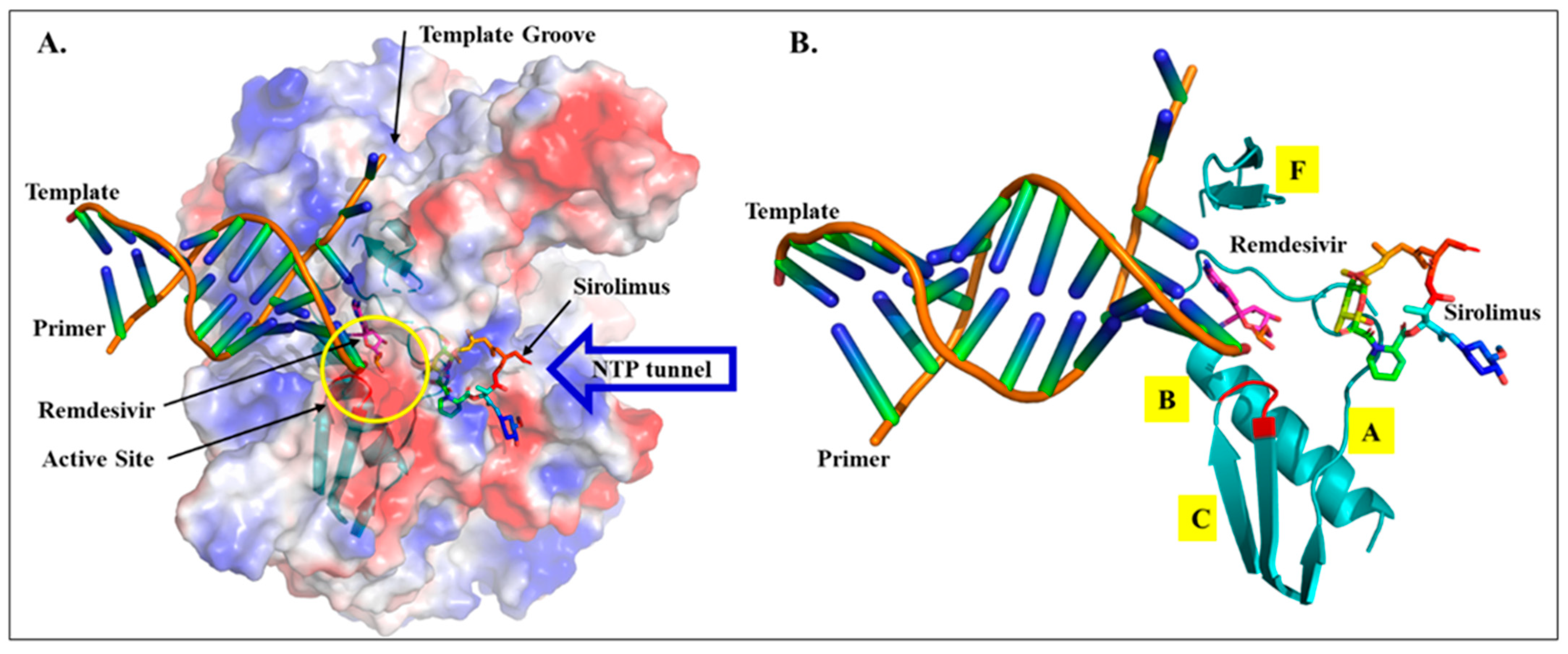
2.2. Drug Docking
2.3. Sirolimus Binding Site
3. Discussion
4. Conclusions
5. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Q.; Allot, A.; Lu, Z. LitCovid: An open database of COVID-19 literature. Nucleic Acids Res. 2021, 49, D1534–D1540. [Google Scholar] [CrossRef] [PubMed]
- Yan, V.C.; Muller, F.L. Why Remdesivir Failed: Preclinical Assumptions Overestimate the Clinical Efficacy of Remdesivir for COVID-19 and Ebola. Antimicrob. Agents Chemother. 2021, 65, e0111721. [Google Scholar] [CrossRef] [PubMed]
- Olender, S.A.; Perez, K.K.; Go, A.S.; Balani, B.; Price-Haywood, E.G.; Shah, N.S.; Wang, S.; Walunas, T.L.; Swaminathan, S.; Slim, J.; et al. Remdesivir for Severe Coronavirus Disease 2019 (COVID-19) Versus a Cohort Receiving Standard of Care. Clin. Infect. Dis. 2021, 73, e4166–e4174. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H. Viral polymerases. Adv. Exp. Med. Biol. 2012, 726, 267–304. [Google Scholar] [CrossRef]
- Jia, H.; Gong, P. A Structure-Function Diversity Survey of the RNA-Dependent RNA Polymerases From the Positive-Strand RNA Viruses. Front. Microbiol. 2019, 10, 1945. [Google Scholar] [CrossRef]
- Hillman, B.I.; Cai, G. The family narnaviridae: Simplest of RNA viruses. Adv. Virus Res. 2013, 86, 149–176. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucia-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol. Mol. Biol. Rev. 2014, 78, 278–303. [Google Scholar] [CrossRef]
- Peersen, O.B. A Comprehensive Superposition of Viral Polymerase Structures. Viruses 2019, 11, 745. [Google Scholar] [CrossRef]
- Lai, M.M.; Liao, C.L.; Lin, Y.J.; Zhang, X. Coronavirus: How a large RNA viral genome is replicated and transcribed. Infect. Agents Dis. 1994, 3, 98–105. [Google Scholar] [PubMed]
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Pringle, F.M.; Zeddam, J.L.; Luke, B.T.; Cameron, C.E.; Kalmakoff, J.; Hanzlik, T.N.; Gordon, K.H.; Ward, V.K. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J. Mol. Biol. 2002, 324, 47–62. [Google Scholar] [CrossRef]
- Bruenn, J.A. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003, 31, 1821–1829. [Google Scholar] [CrossRef]
- te Velthuis, A.J. Common and unique features of viral RNA-dependent polymerases. Cell Mol. Life Sci. 2014, 71, 4403–4420. [Google Scholar] [CrossRef] [PubMed]
- Cerny, J.; Cerna Bolfikova, B.; Valdes, J.J.; Grubhoffer, L.; Ruzek, D. Evolution of tertiary structure of viral RNA dependent polymerases. PLoS ONE 2014, 9, e96070. [Google Scholar] [CrossRef]
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. Computationally repurposed drugs and natural products against RNA dependent RNA polymerase as potential COVID-19 therapies. Mol. Biomed. 2021, 2, 28. [Google Scholar] [CrossRef]
- Pokhrel, R.; Chapagain, P.; Siltberg-Liberles, J. Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. J. Med. Microbiol. 2020, 69, 864–873. [Google Scholar] [CrossRef]
- Elshahawi, H.; Syed Hassan, S.; Balasubramaniam, V. Importance of Zika Virus NS5 Protein for Viral Replication. Pathogens 2019, 8, 169. [Google Scholar] [CrossRef]
- Valle, C.; Martin, B.; Debart, F.; Vasseur, J.J.; Imbert, I.; Canard, B.; Coutard, B.; Decroly, E. The C-Terminal Domain of the Sudan Ebolavirus L Protein Is Essential for RNA Binding and Methylation. J. Virol. 2020, 94, e00520-20. [Google Scholar] [CrossRef]
- Subissi, L.; Imbert, I.; Ferron, F.; Collet, A.; Coutard, B.; Decroly, E.; Canard, B. SARS-CoV ORF1b-encoded nonstructural proteins 12–16: Replicative enzymes as antiviral targets. Antivir. Res. 2014, 101, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Sexton, N.R.; Denison, M.R. Thinking Outside the Triangle: Replication Fidelity of the Largest RNA Viruses. Annu. Rev. Virol. 2014, 1, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef]
- Ashenberg, O.; Gong, L.I.; Bloom, J.D. Mutational effects on stability are largely conserved during protein evolution. Proc. Natl. Acad. Sci. USA 2013, 110, 21071–21076. [Google Scholar] [CrossRef]
- de Farias, S.T.; Dos Santos Junior, A.P.; Rego, T.G.; Jose, M.V. Origin and Evolution of RNA-Dependent RNA Polymerase. Front. Genet. 2017, 8, 125. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Roberts, E.; Eargle, J.; Wright, D.; Luthey-Schulten, Z. MultiSeq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinform. 2006, 7, 382. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Eltahla, A.A.; Luciani, F.; White, P.A.; Lloyd, A.R.; Bull, R.A. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses 2015, 7, 5206–5224. [Google Scholar] [CrossRef]
- Dwivedy, A.; Mariadasse, R.; Ahmad, M.; Chakraborty, S.; Kar, D.; Tiwari, S.; Bhattacharyya, S.; Sonar, S.; Mani, S.; Tailor, P.; et al. Characterization of the NiRAN domain from RNA-dependent RNA polymerase provides insights into a potential therapeutic target against SARS-CoV-2. PLoS Comput. Biol. 2021, 17, e1009384. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.F.; Perry, J.K.; Olinares, P.D.B.; Lee, H.W.; Chen, J.; Appleby, T.C.; Feng, J.Y.; Bilello, J.P.; Ng, H.; Sotiris, J.; et al. Structural basis for substrate selection by the SARS-CoV-2 replicase. Nature 2023, 614, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Unchwaniwala, N.; Rebolledo-Viveros, A.; Pennington, J.; Horswill, M.; Broadberry, R.; Myers, J.; den Boon, J.A.; Grant, T.; Ahlquist, P. Nodavirus RNA replication crown architecture reveals proto-crown precursor and viral protein A conformational switching. Proc. Natl. Acad. Sci. USA 2023, 120, e2217412120. [Google Scholar] [CrossRef]
- Zhang, K.; Law, Y.S.; Law, M.C.Y.; Tan, Y.B.; Wirawan, M.; Luo, D. Structural insights into viral RNA capping and plasma membrane targeting by Chikungunya virus nonstructural protein 1. Cell Host Microbe 2021, 29, 757–764.e3. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Limpens, R.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grunewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Silva, R.; Ribeiro, J.S.; da Silva, G.P.D.; da Costa, L.J.; Travassos, L.H. Autophagy Modulators in Coronavirus Diseases: A Double Strike in Viral Burden and Inflammation. Front. Cell Infect. Microbiol. 2022, 12, 845368. [Google Scholar] [CrossRef]
- Carneiro, L.A.; Travassos, L.H. Autophagy and viral diseases transmitted by Aedes aegypti and Aedes albopictus. Microbes Infect. 2016, 18, 169–171. [Google Scholar] [CrossRef]
- Bowman, L.J.; Brueckner, A.J.; Doligalski, C.T. The Role of mTOR Inhibitors in the Management of Viral Infections: A Review of Current Literature. Transplantation 2018, 102, S50–S59. [Google Scholar] [CrossRef]
- Joubert, P.E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef]
- Karsulovic, C.; Lopez, M.; Tempio, F.; Guerrero, J.; Goecke, A. mTORC inhibitor Sirolimus deprograms monocytes in “cytokine storm” in SARS-CoV2 secondary hemophagocytic lymphohistiocytosis-like syndrome. Clin. Immunol. 2020, 218, 108539. [Google Scholar] [CrossRef] [PubMed]
- Mahdian, S.; Zarrabi, M.; Panahi, Y.; Dabbagh, S. Repurposing FDA-approved drugs to fight COVID-19 using in silico methods: Targeting SARS-CoV-2 RdRp enzyme and host cell receptors (ACE2, CD147) through virtual screening and molecular dynamic simulations. Inform. Med. Unlocked 2021, 23, 100541. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Roy, A.; Roy, L.; Chattopadhyay, S.; Chatterjee, S. Immune response and possible therapeutics in COVID-19. RSC Adv. 2020, 11, 960–977. [Google Scholar] [CrossRef]
- Patocka, J.; Kuca, K.; Oleksak, P.; Nepovimova, E.; Valis, M.; Novotny, M.; Klimova, B. Rapamycin: Drug Repurposing in SARS-CoV-2 Infection. Pharmaceuticals 2021, 14, 217. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Byrareddy, S.N. Rapamycin as a potential repurpose drug candidate for the treatment of COVID-19. Chem. Biol. Interact. 2020, 331, 109282. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; Nicoli, F.; Borgatti, M.; Finotti, A.; Gambari, R. Possible effects of sirolimus treatment on the longterm efficacy of COVID19 vaccination in patients with betathalassemia: A theoretical perspective. Int. J. Mol. Med. 2022, 49, 33. [Google Scholar] [CrossRef]
- Cho, J.; Lee, Y.J.; Kim, J.H.; Kim, S.I.; Kim, S.S.; Choi, B.S.; Choi, J.H. Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci. Rep. 2020, 10, 16200. [Google Scholar] [CrossRef]
- Min, J.S.; Kwon, S.; Jin, Y.H. SARS-CoV-2 RdRp Inhibitors Selected from a Cell-Based SARS-CoV-2 RdRp Activity Assay System. Biomedicines 2021, 9, 996. [Google Scholar] [CrossRef]
- Akamatsu, N.; Sugawara, Y.; Kokudo, N. Asunaprevir (BMS-650032) for the treatment of hepatitis C virus. Expert. Rev. Anti Infect. Ther. 2015, 13, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yokosuka, O.; Omata, M. Faldaprevir for the treatment of hepatitis C. Int. J. Mol. Sci. 2015, 16, 4985–4996. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Buonomo, A.R.; Zappulo, E.; Minei, G.; Morisco, F.; Borrelli, F.; Coppola, N.; Borgia, G. Asunaprevir, a protease inhibitor for the treatment of hepatitis C infection. Ther. Clin. Risk Manag. 2014, 10, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.S.; Nguyen, L.P.; Lee, G.H.; Lee, S.G.; Lyoo, K.S.; Kim, B.; Hwang, S.B. Asunaprevir, a Potent Hepatitis C Virus Protease Inhibitor, Blocks SARS-CoV-2 Propagation. Mol. Cells 2021, 44, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, A.P.; Weissmann, F.; Zeng, J.; Posse, V.; Milligan, J.C.; Canal, B.; Ulferts, R.; Wu, M.; Drury, L.S.; Howell, M.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp12/7/8 RNA-dependent RNA polymerase. Biochem. J. 2021, 478, 2425–2443. [Google Scholar] [CrossRef]
- Yin, W.; Luan, X.; Li, Z.; Zhou, Z.; Wang, Q.; Gao, M.; Wang, X.; Zhou, F.; Shi, J.; You, E.; et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 2021, 28, 319–325. [Google Scholar] [CrossRef]
- Jacome, R.; Campillo-Balderas, J.A.; Becerra, A.; Lazcano, A. Structural Analysis of Monomeric RNA-Dependent Polymerases Revisited. J. Mol. Evol. 2022, 90, 283–295. [Google Scholar] [CrossRef]
- Jacome, R.; Becerra, A.; Ponce de Leon, S.; Lazcano, A. Structural Analysis of Monomeric RNA-Dependent Polymerases: Evolutionary and Therapeutic Implications. PLoS ONE 2015, 10, e0139001. [Google Scholar] [CrossRef]
- Finnegan, D.J. Retrotransposons. Curr. Biol. 2012, 22, R432–R437. [Google Scholar] [CrossRef]
- Zhao, C.; Pyle, A.M. Structural Insights into the Mechanism of Group II Intron Splicing. Trends Biochem. Sci. 2017, 42, 470–482. [Google Scholar] [CrossRef]
- Galej, W.P.; Oubridge, C.; Newman, A.J.; Nagai, K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature 2013, 493, 638–643. [Google Scholar] [CrossRef]
- Novikova, O.; Belfort, M. Mobile Group II Introns as Ancestral Eukaryotic Elements. Trends Genet. 2017, 33, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [Google Scholar] [CrossRef] [PubMed]
- Picarazzi, F.; Vicenti, I.; Saladini, F.; Zazzi, M.; Mori, M. Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge. Molecules 2020, 25, 5695. [Google Scholar] [CrossRef] [PubMed]
- Shehzadi, K.; Saba, A.; Yu, M.; Liang, J. Structure-Based Drug Design of RdRp Inhibitors against SARS-CoV-2. Top. Curr. Chem. 2023, 381, 22. [Google Scholar] [CrossRef] [PubMed]
- Stephen, P.; Lin, S.X. RNA-dependent RNA polymerase: Addressing Zika outbreak by a phylogeny-based drug target study. Chem. Biol. Drug Des. 2018, 91, 322–327. [Google Scholar] [CrossRef]
- Xu, H.T.; Colby-Germinario, S.P.; Hassounah, S.A.; Fogarty, C.; Osman, N.; Palanisamy, N.; Han, Y.; Oliveira, M.; Quan, Y.; Wainberg, M.A. Evaluation of Sofosbuvir (beta-D-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine) as an inhibitor of Dengue virus replication. Sci. Rep. 2017, 7, 6345. [Google Scholar] [CrossRef]
- Yap, T.L.; Xu, T.; Chen, Y.L.; Malet, H.; Egloff, M.P.; Canard, B.; Vasudevan, S.G.; Lescar, J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007, 81, 4753–4765. [Google Scholar] [CrossRef]
- Laurila, M.R.; Makeyev, E.V.; Bamford, D.H. Bacteriophage phi 6 RNA-dependent RNA polymerase: Molecular details of initiating nucleic acid synthesis without primer. J. Biol. Chem. 2002, 277, 17117–17124. [Google Scholar] [CrossRef]
- Gytz, H.; Mohr, D.; Seweryn, P.; Yoshimura, Y.; Kutlubaeva, Z.; Dolman, F.; Chelchessa, B.; Chetverin, A.B.; Mulder, F.A.; Brodersen, D.E.; et al. Structural basis for RNA-genome recognition during bacteriophage Qbeta replication. Nucleic Acids Res. 2015, 43, 10893–10906. [Google Scholar] [CrossRef]
- Kesy, J.; Patil, K.M.; Kumar, S.R.; Shu, Z.; Yong, H.Y.; Zimmermann, L.; Ong, A.A.L.; Toh, D.K.; Krishna, M.S.; Yang, L.; et al. A Short Chemically Modified dsRNA-Binding PNA (dbPNA) Inhibits Influenza Viral Replication by Targeting Viral RNA Panhandle Structure. Bioconjug. Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE). 2019. Available online: https://www.chemcomp.com/Products.htm (accessed on 28 June 2023).
- Douguet, D. Data Sets Representative of the Structures and Experimental Properties of FDA-Approved Drugs. ACS Med. Chem. Lett. 2018, 9, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Picornaviridae | Flaviviridae | Caliciviridae | Coronaviridae | Permutotetraviridae |
|---|---|---|---|---|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gana, A.S.; Baraniuk, J.N. Identification of a New Drug Binding Site in the RNA-Dependent-RNA-Polymerase (RdRp) Domain. BioMedInformatics 2023, 3, 885-907. https://doi.org/10.3390/biomedinformatics3040055
Gana AS, Baraniuk JN. Identification of a New Drug Binding Site in the RNA-Dependent-RNA-Polymerase (RdRp) Domain. BioMedInformatics. 2023; 3(4):885-907. https://doi.org/10.3390/biomedinformatics3040055
Chicago/Turabian StyleGana, Aparna S., and James N. Baraniuk. 2023. "Identification of a New Drug Binding Site in the RNA-Dependent-RNA-Polymerase (RdRp) Domain" BioMedInformatics 3, no. 4: 885-907. https://doi.org/10.3390/biomedinformatics3040055