1. Introduction
Thermochemical and spectroscopic investigation of N-methyl Acetamide (NMA) and other substituted amides are considered as model compounds for the peptide bonds in proteins; understanding their thermochemistry can provide information about the secondary structure of proteins in the gas phase as well as inferences in solution and helpful information toward understanding kinetics. There are no studies that we are aware of that have targeted these molecules’ thermochemical properties and bond energies.
There are several reasons for interest in the structure and chemistry of these amide systems. These include: (i) clear understanding of the NMA structure is considered as the basis for understanding the geometric constraints imposed by the peptide linkages that determine, at least partly, the protein structure; (ii) detailed understanding of NMA spectroscopic features is assumed as the fundamental basis for spectroscopic methods to monitor protein structure and dynamics [
1]. Both of these properties are of interest for future applications, only if both structures and spectroscopic properties of NMA are observed in the natural environment of the biological system(s).
There are many infrared (I.R.), and Raman experiments that have focused on the spectral region spanned by the three amide bands of NMA, particularly in the easily detectable I.R. amide I regime that overlaps with the C.O. stretch. In water (aq), the C.O. stretch responds to water molecules’ presence by forming hydrogen bonds, and the resulting frequency shift can be used to assess the dynamics of protein–solvent interactions [
2,
3,
4,
5,
6,
7,
8,
9]. Similarly, the amide II and amide III bands, which overlap with the N.H. in-plane wagging motion, can be used to describe the interaction between C.O. and H.N., which are part of a protein backbone. The amide hydrogen can also form a hydrogen bond with the solvent (H
2O∙HN), and the corresponding frequency shift provides further information on protein behavior in an aqueous solution [
10].
The structural stability of acetohydrazide CH
3–CO–NH–NH
2 was investigated by DFT-B3LYP and ab initio MP2 calculations with a 6-311+G ** basis set. The C–N rotational barrier in the molecule was calculated to be 26 kcal mol
−1, which suggested the planar sp2 nature of the nitrogen atom of the central N.H. moiety with the two-fold barrier. The N atom of the terminal NH
2 group was predicted to prefer the pyramidal sp3 structure with an inversion barrier of 7–8 kcal mol
−1. The molecule was predicted to have a
trans–syn (N–H bond is
trans with respect to C=O bond and the NH
2 moiety is
syn to C–N bond) conformation as the lowest energy structure. The vibrational frequencies were computed at the B3LYP/6-311+G ** level of theory and normal coordinate calculations were carried out for the
trans–syn acetohydrazide. Complete vibrational assignments were made based on normal coordinate analyses and experimental infrared and Raman data [
11].
The study of amide C–N rotation barriers is important because amide C–N bonds make up protein backbones. The preferred amide conformations play an important role in enzyme structure and the barrier to the rotation affects the rigidity of the structure. The rigidity of an enzyme’s structure can affect its selectivity in binding substrates. Ab initio calculations by Jasien et al. [
12] have been used to determine the gas-phase rotational barrier about the C-N bond in acetamide. Their results indicate that the inclusion of polarization functions in the basis sets leads to a substantial decrease (ca. 5 kcal mol
−1) in the calculated barrier height at the H.F.–SCF level. Electron correlation effects decrease the barrier by less than 1 kcal mol
−1, while the addition of zero-point energy corrections changed the barrier height only slightly. Based upon the current (
DZ + d/SCF) calculations, the 0 K rotational barrier for acetamide is predicted to be 12.5 kcal mol
−1. An investigation of the photolysis of acetamide was performed using light in the 250 Å regions of the spectrum, where the goal was to break down the molecule into CH
3 and CONH
2 radicals. The authors reported this was probably accompanied by a second process yielding CH
3CN and H
2O. Methyl radicals were observed to react with the parent acetamide and with the CONH
2 radical to give methane as a product and to recombine yielding ethane. The CONH
2 radicals were reported to decompose both spontaneously and thermally to give C.O. and NH
2 radicals. The subsequent reaction of the NH
2 radicals with Acetamide gives ammonia. In a separate experiment with acetone as a photo methyl radical source, the activation energy for the abstraction of hydrogen by methyl radical was found to be 9.2 kcal mol
−1 [
13].
The importance of reliable and accessible thermochemical data (enthalpies of formation, entropies, and heat capacities) is universally accepted among scientists and engineers. This work provides thermochemical data for acetic acid hydrazide, acetamide, and N-methyl acetamide and their radicals corresponding to the loss of hydrogen atoms through the use of computational chemistry.
2. Computational Methods
Density functional theory and composite calculations via series of isodesmic reactions: the structure and thermochemical parameters of CH
3CONHNH
2, CH
3CONH
2, and CH
3CONHCH
3 are based on the density functional and composite ab initio levels using Gaussian 98 [
14] and Gaussian 09 [
15]. Computation levels include B3LYP/6-31G(d,p), B3LYP/6-31G(2d,2p). These methods combine the three-parameter Becke exchange functional B3 [
16], with the Lee–Yang–Parr correlation functional, LYP, [
17], and are used here with the 6-31G(d,p) basis set. B3LYP/6-31G(d,p) is chosen because it is computational, economical, and, thus, possibly applicable to larger molecules [
18]. Energies are further refined using the procedures of the complete basis method developed by Petersson and co-workers, CBS-QB3 [
19]. The CBS-QB3 method is utilized for improved energies and serves to check the DFT calculations. CBS models, a series of calculations made on a defined geometry, and a complete basis set model chemistry including corrections for basis set truncation errors. These methods show accuracy in structure and energy that requires convergence in basis set size and the degree of correlation [
18].
Standard enthalpies of formation for stable species are calculated using the total energies at B3LYP/6-31G(d,p), B3LYP/6-31G(2d,2p), and CBS-QB3 levels with work reactions that are isodesmic in most cases. Isodesmic reactions conserve the number and type of bonds on both sides of an equation. The use of a work reaction with similar bonding on both sides of an equation results in a cancellation of calculation error and improves the accuracy for energy analysis [
20]. The reported enthalpy values can be compared with the known enthalpies of several molecules in the system to serve as a calibration on the thermochemistry.
Contributions to S°298 and Cp°(T) of each species are calculated using the “SMCPS” (Statistical Mechanics for Heat Capacity and Entropy Cp and S) program, which incorporates the frequencies, moments of inertia, mass, symmetry, number of optical isomers, from the Gaussian calculation. It also incorporates frequency corrections. Contributions from hindered internal rotors to S°298 and Cp(T) are determined using the “VIBIR” program. The hindered rotor corrections to the S°298 and Cp(T) are obtained by adding the S and Cp values, respectively, obtained by employing the VIBIR program to those obtained from SMCPS.
3. Results and Discussion
Optimized, lowest energy structures of the parent molecules—acetohydrazide, acetamide, and
N-methyl acetamide are shown in
Figure 1.
The torsional potentials of CH3–C=ONHNH2 and CH3–C=ONHCH3 (vide infra) show that corresponding anti- and syn-isomers respective to H8N7---C5O6 dihedral angles (see Figure 3). The anti-acetohydrazide has a near −4.86 kcal mol−1 lower energy than for the syn configuration, with a 20.7 kcal mol−1 barrier to the internal rotation converting the two-isomer configuration.
In contrast, the syn-N-methyl acetamide has a near −2.50 kcal mol−1 lower energy than for the anti-configuration, with a 19.3 kcal mol−1 barrier. The internal rotation energies for these syn–anti isomerizations are high, typically greater than 13 kcal mol−1 (see below). This isomerization does not occur at standard temperature, and the isomers should be considered as different molecules in their thermochemistry and probably in their reactions.
The optimized geometries at the B3LYP/6-31G (d,p) density functional calculation level for CH
3–C=ONHNH
2, CH
3–C=ONH
2, and CH
3–C=ONHCH
3 are presented in the
Supplementary Materials [SM]. The Geometric Parameters (See Section SM0,
Tables S1–S3 of the SM) are listed.
3.1. Enthalpies of Formation of the Parent Molecules
Enthalpies of formation (ΔH
f°298) of the parent molecules have been determined using corresponding ΔHrxn (298) from the enthalpy of reaction in the isodesmic work reactions and calculated enthalpies of each species. The standard enthalpies of formation of the reference molecules at 298.15 K and the calculated ΔHrxn°298 are used to calculate the Δ
fHrxn°298 of the target molecule; the enthalpies are summarized in
Table 1.
The work reactions and the enthalpies obtained from three isodesmic reactions for the parent molecules are listed in
Table 2. Comparing the values of the enthalpies of the parent molecules calculated by two DFT and the CBS-QB3 methods shows that the values obtained by the DFT methods method are in close agreement with those obtained by CBS-QB3 calculations. We recommend the values obtained by the CBS-QB3 because it is a composite method and is known to have higher accuracy. The agreement of the DFT values suggests that the use of B3LYP calculations with the 6-31G(d,p) and 6-31G(2d,2p) basis sets coupled with work reactions results in the cancelation of error and provides reasonable results for these amide systems.
The recommended enthalpies of formation for the CH3–C=ONHNH2, CH3–C=ONH2, and CH3–C=ONHCH3 molecules obtained in this study are: −27.08 kcal mol−1, −57.40 kcal mol−1, and −56.48 kcal mol−1 by the CBS-QB3 calculation method.
Moments of Inertia (See Section SM1,
Table S4 of the SM), vibrational frequencies (
Table S5 of the SM), and Mulliken Atomic Charges(Section SM2,
Tables S6–S8 of the SM) of CH
3–C=ONHNH
2, CH
3–C=ONH
2, and CH
3–C=ONHCH
3 and their radicals are calculated and presented.
3.2. Radicals Corresponding to the Loss of a Hydrogen Atom
Optimized, lowest energy structures of the radicals derived from the target parent molecules are illustrated in
Figure 2.
The radicals from all three parent molecules show anti-structures relative to the carbonyl group for the low energy conformation where the radical sites are on the carbons and the nitrogen, not adjacent to the carbonyl. When the radical site is on the nitrogen atom adjacent to the carbonyl, all three radicals show that the syn conformer is the lowest energy structure.
3.3. Heats of Formation, Bond Energies, and Relative Stability of the Radicals Derived from the Target Parent Molecules
Four isodesmic reactions for each radical and the calculated standard enthalpies are listed in
Table 3. The data illustrate excellent agreement across the different levels of calculations and through the different reaction analyses.
The enthalpies of formation of the parent molecules averaged over three work reactions for molecules CH3–C=ONHNH2, CH3–C=ONH2 and CH3–C=ONHCH3 were evaluated as −28.1, −57.29, and −56.53 kcal mol−1, respectively (values are average of B3LYP/6-31g(d,p), B3LYP/6-31g(2d,2p) and CBS-QB3 levels).
3.3.1. Enthalpy of Formation—Radicals
The four work reactions and calculated ΔH
f°298 for use in determining the enthalpy of formation for each radical are shown in
Table 3.
The recommended enthalpies of formation, in kcal mol−1, from the CBS-QB3 level calculations, averaged over four work reactions are:
- (i)
Radicals from CH3–C=ONHNH2:
- (a)
C•H2–C=ONHNH2 (19.27),
- (b)
CH3–C=ON•NH2 (−2.07),
- (c)
CH3–C=ONHN•H (1.60).
- (ii)
Radicals from CH3–C=ONH2:
C•H2–C=ONH2 (−9.67),
CH3–C=ON•H (2.11).
- (iii)
Radicals from CH3–C=ONHCH3
C•H2−C=ONHCH3 (−9.12),
CH3–C=ON•CH3 (−4.43),
CH3–C=ONHC•H2 (−15.39).
3.3.2. Bond Energies
Bond energies for the formation of radicals by loss of H atom reported at 298.15 K were calculated from the standard ΔH
f°298 values of the parent molecules and of the radicals, obtained at CBS-QB3 level.
ΔH
f°298 of 52.1 kcal mol
−1 was used for H atom enthalpy.
The bond dissociation enthalpies of the radicals calculated at three different levels of theory are listed in
Table 3. The largest difference in R–H bond energy for a given radical, considering the two DFT and the CBS-QB3 calculation methods and the four isodesmic reactions of each radical, was less than 1.5 kcal mol
−1.
The bond dissociation energies for the C–H bonds in the methyl group adjacent to the carbonyl in CH3–C=ONHNH2 (99.51), CH3–C(=O)NH2 (99.72), and CH3–C=ONHCH3 (99.51) kcal mol−1 are in parentheses. These compare with the typical bond energy on a primary methyl site of a normal hydrocarbon of 101 kcal mol−1, and are ca. 2 kcal mol−1 lower. In contrast, they are ca. 3 kcal mol−1 higher than a typical primary methyl C–H bond on a ketone, which is 96 kcal mol−1. The C–H bond energy on the methyl group bonded to the amine to form the CH3–C=ONHC•H2 is 93.24 kcal mol−1.
The N–H bond strengths for nitrogen atom adjacent to the carbonyl groups in CH3–C=ONHNH2 (78.17), CH3–C(=O)NH2 (111.50), and CH3–C=ONHCH3 (104.19) kcal mol−1 are in the parenthesis. For comparison, the N–H bond in ammonia is (108), the CH3NH–H bond in methylamine is (102.4), and the NH2NH–H bond in hydrazine is (82) kcal mol−1.
The C–H bond strength in the CH3 group bonded to an amine in CH3–C=ONHCH2--H is 93.37 kcal mol−1 at the CBS-QB3 level. This compares with the C–H bond energy of H–CH2–NH2 (~94.4) in CH3NH2. This C–H bond in CH3–C=ONHC•H2 is 1 kcal mol−1 lower than on the methyl groups bonded to C=O.
The N–H bond of the N.H. group in CH3–C=O–N•–NH2 is the weakest in this molecule at 78.17 kcal mol−1, where the C–H bond in C•H2–C=ONHNH2 is 99.51 kcal mol−1, which is significantly higher. The N–H bond energy for acetohydrazide (CH3–C=ONHNH•) is markedly different at only 81.84 kcal mol−1. In the N–H bond cleavage of CH3–C=ON•NH2, the electrons from the radical site re-arrange to form a second double bond, from nitrogen to carbon. The N–H bond energy for CH3–C=ON•CH3 is 104.20 kcal mol−1 which is much higher than the CH3–C=ON•NH2.
The weakening of the N–H bonds in this hydrazide is essentially independent of the nature of the β-substituent (H, RCO, CO
2Et, or PhSO
2) and the stabilizing effect on the radical is brought about entirely by the three-electron on N–NH
2 moiety [
27].
3.4. Internal Rotor Potentials
Energy profiles for internal rotations about the C–C=O, O=C–N, N–C, and N–N bonds in the acetohydrazide, acetamide, and N-methyl acetamide were calculated to determine the lowest energy configurations, energies of the rotational conformers, and to identify the interconversion barriers between isomers. Torsional potentials were used to evaluate contributions to the entropy and heat capacity values when there were low barriers (less than 3.5 kcal mol−1) and internal rotation occurred.
The total energies as a function of the corresponding dihedral angles were computed at the B3LYP/6-31G(d,p) level of theory by scanning the torsion angles between 0° and 360° in steps of 15°, while all remaining coordinates were fully optimized. All potentials were rescanned when a lower energy conformer, relative to the initial low-energy conformer was found. The total energy of the corresponding most stable molecular conformer was arbitrarily set to zero and used as a reference point to plot the potential barriers. The resulting potential energy barriers for internal rotations in the stable nonradical and radical molecules are shown in
Figure 3,
Figure 4,
Figure 5,
Figure 6,
Figure 7,
Figure 8 and
Figure 9. Dihedral angles obtained for the optimized lowest energy structures are shown in parentheses.
The CH
3–C=O-NH-NH
2 rotor is illustrated in
Figure 3 for both the CH
3–C=O–N(–H)—NH
2 (H10–N9—N7–H8) and CH
3–C(=O)—NH–NH
2 (H8–N7—C5–O6) systems with barriers at 7.82 and 20.66 kcal mol
−1, respectively. The CH
3–C(=O)—NH–CH
3 (H8–N7—C5–O6) rotor shows a two-fold symmetry with a barrier at 21.7 kcal mol
−1. One reason for the high barriers for the rotation about the carbonyl (C=O)–NH bond involves the repulsive interaction of the carbonyl π bond with the NH lone pair.
The study of amide C–N rotation barriers is important for the evaluation of their reactivity, for input data in biochemical structure calculations other than finding the lowest energy conformer because the amide C–N bonds make up protein backbones. The amide conformations play an important role in enzyme structure and the barrier to the rotation affects the rigidity of that structure. The rigidity of an enzyme’s structure can affect its selectivity in binding substrates.
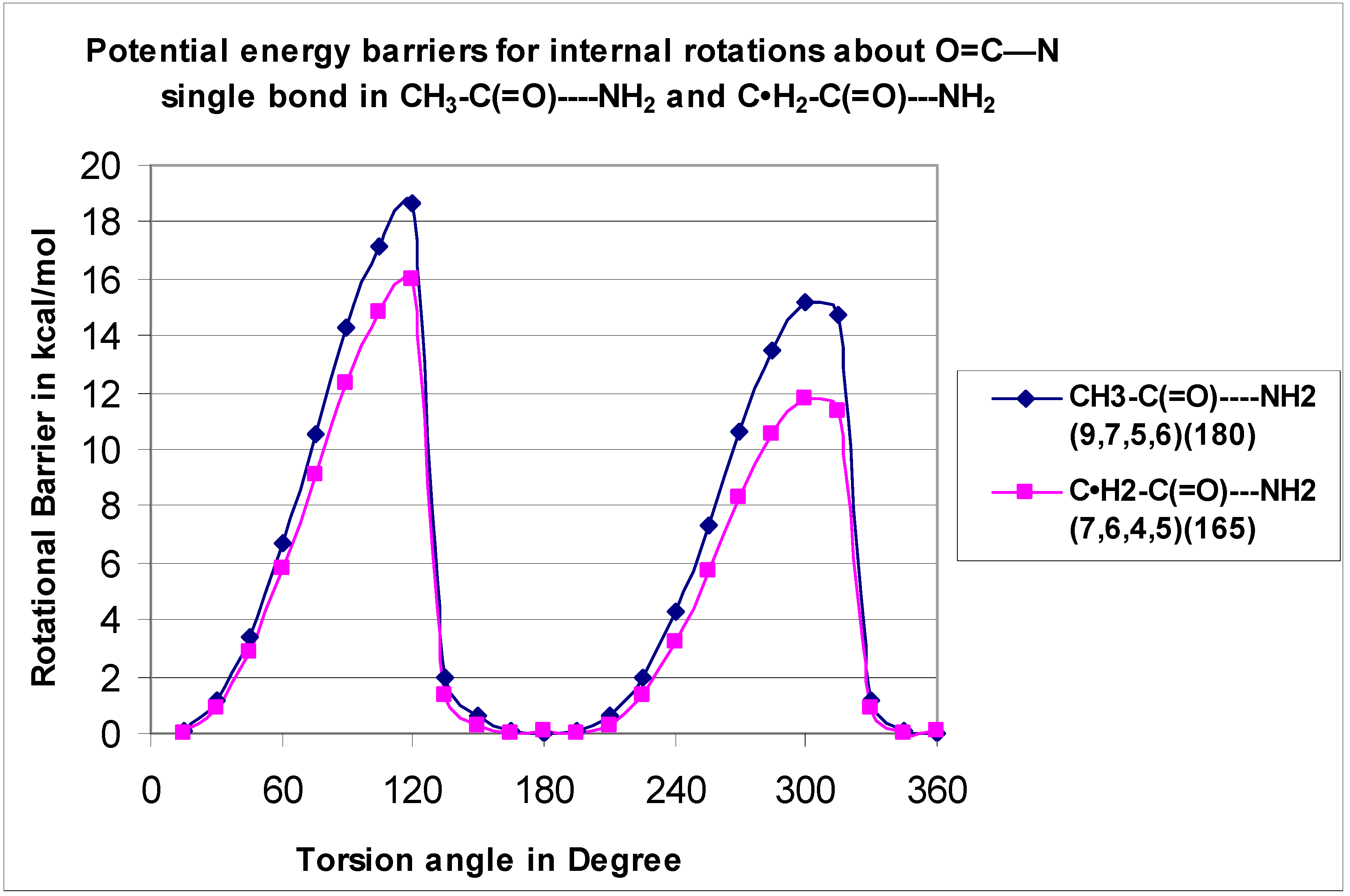
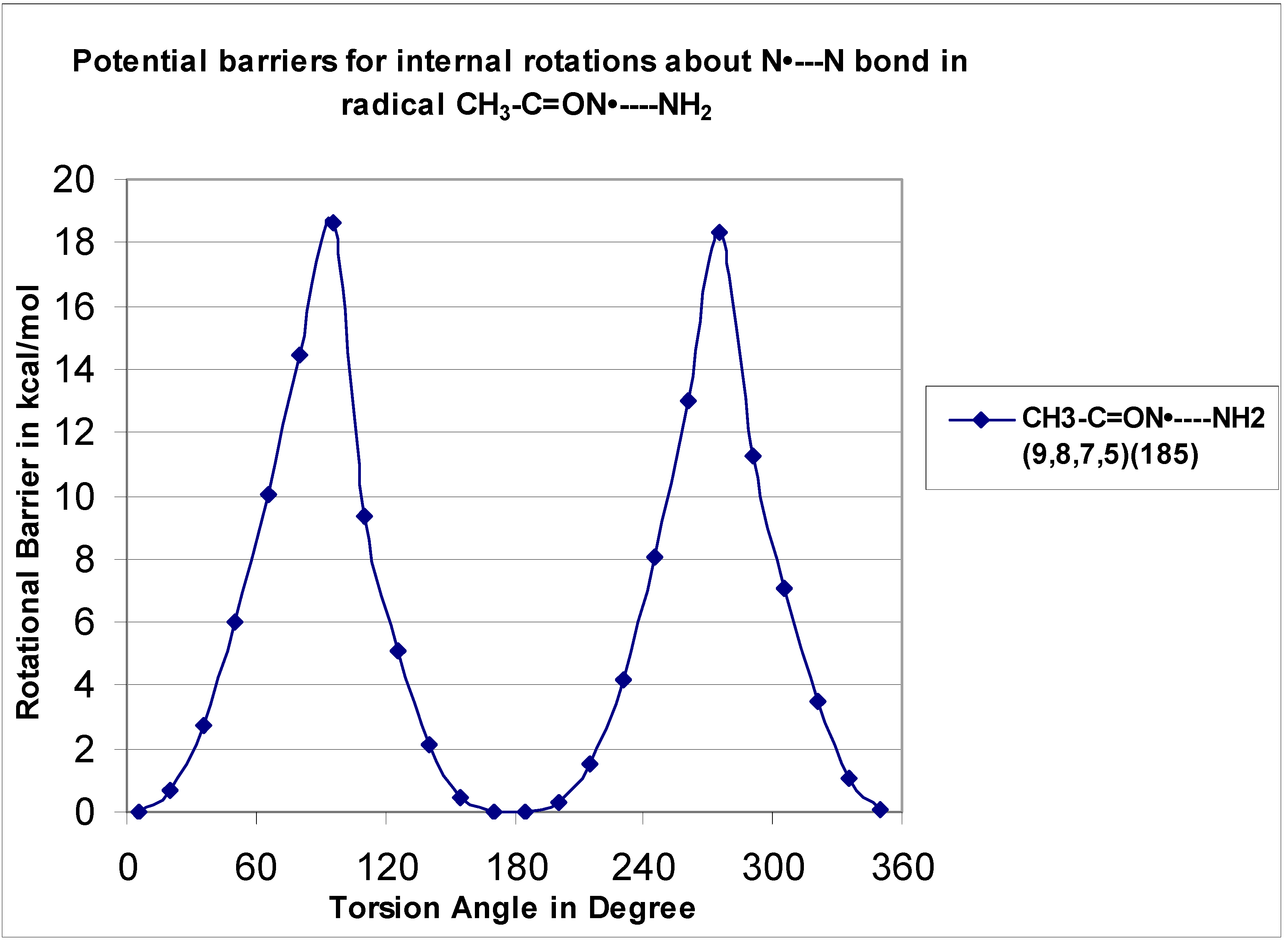
The C–N rotor for the radical C•H
2–C(=O)—NH
2 (H
7–N
6---C
4–O
5 is 15.97 kcal mol
−1) and the value of its parent CH
3–C(=O)—NH
2 (H
8–N
7—C
5–O
6 is 18.66 kcal mol
−1). According to Jasien et al. [
12], based upon the current (
DZ + d/SCF) calculations, the 0 K rotational barrier for acetamide between the C–N bond is predicted to be 12.5 kcal mol
−1.
The two protons attached to N in acetamide are inequivalent at low temperatures but become averaged by C–N rotation at higher temperatures. Typical bond rotation barriers for amides in solution are experimentally determined by NMR to be between 17 and 22 kcal/mol. However, the acetamide enolate, [CH2CONH2] has a much lower barrier; according to NMR experiments, the enolate has free rotation at all accessible temperatures [
28]. The HNCO
anti/
trans/
Z configuration is significantly more probable in proteins than the
syn/
cis/
E geometry. The ratio is 95:5 or even higher [
29].
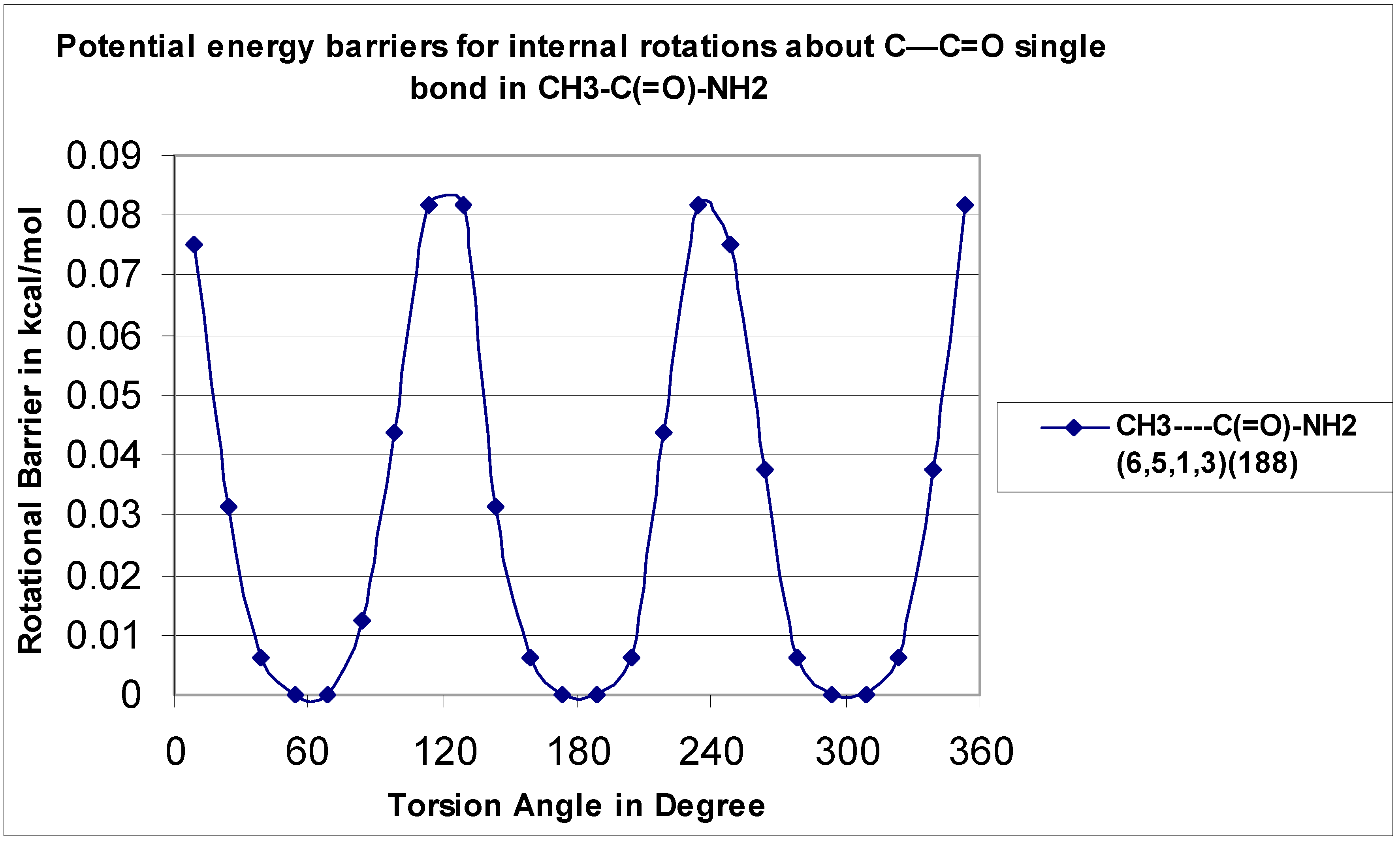
The CH
3–C=O rotor for the CH
3—C(=O)–NH
2 (O
6–C
5—C
1–H
2) has a small barrier at 0.08 kcal mol
−1 and shows three-fold symmetry The data are in reasonable agreement with the previous study conducted by J.R. Bailey [
30], which also reported a low, calculated barrier monosubstituted
N-methyl acetamide to be 0.21 kcal mol
−1.
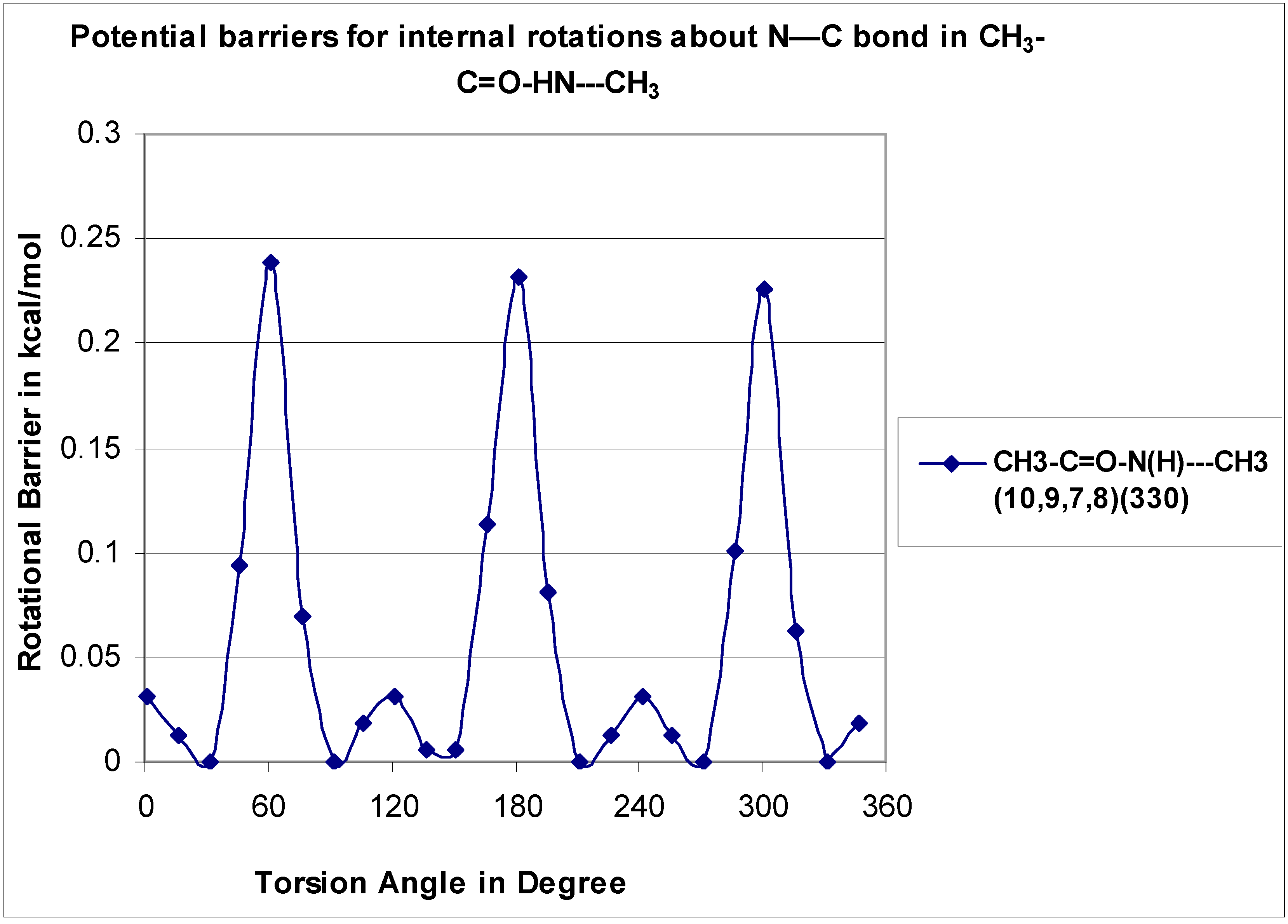
The CH
3–C=O–HN—CH
3 rotor is illustrated in
Figure 6 which shows a three-fold symmetry with a barrier at 0.24 kcal mol
−1.
The C(=O)NH
2 rotor is illustrated in
Figure 8 for both the C•H
2–C(=O)—NH
2 (H7–N6—C4–O5) and C•H2---C(=O)NH
2 (O5–C4—C1–H2) systems with a great difference in bariers at 16.0 and 6.6 kcal mol
−1, respectively, but with the same two-fold symmetry.
The CH
3–C(=O)N•—CH
3 (H9–C8—N
7–C5) and CH
3–C(=O)N—C•H
2 (H10–C9—N7–H8) rotors are illustrated in
Figure 9 and there is a significant difference in the internal rotor barriers. CH
3–C(=O)N•—CH
3 shows extremely low three-fold symmetry at 0.7 kcal mol
−1, and CH
3–C(=O)N—C•H
2 shows two-fold symmetry at 8.7 kcal mol
−1 barrier. The low barrier in CH
3–C(=O)N•—CH
3 is a result of the overlap between the carbonyl bond and the unpaired electron of the N atom, which reduces the interaction of the methyl H atoms with the N nitrogen π orbitals. The higher barrier for CH
3–C(=O)N—C•H
2 results from overlap (resonance) between the radical and the nitrogen π bonds.
3.5. Entropy and Heat Capacity
The entropy and heat capacity data for the parent molecules and their radicals as a function of temperature were determined from the optimized structures, moments of inertia, vibrational frequencies, symmetries, the known mass of the molecules, and internal rotor contributions when barriers were low. The calculations use standard formulas from statistical mechanics for the contributions of translation, external rotation, and vibrations [
31,
32]. Contributions to the entropy and the heat capacity from translation, vibrations, and external rotation were calculated using the SMCPS program. This program utilizes the rigid-harmonic oscillator approximation from the optimized structures obtained at B3LYP/6-31G(d,p) level. The number of optical isomers and the spin degeneracy of unpaired electrons is also incorporated for the calculation of S°298.
Contributions from hindered internal rotors to S°298 and
Cp(
T) are determined using the VIBIR program. This program utilizes the method of Pitzer and Gwinn [
33,
34], the potential barriers, folds, and moments of inertia from the internal rotor analysis. The moments of inertia were calculated. The rotors with a barrier value greater than 3.5 kcal mol
−1 were treated as torsion vibrations. The internal rotor data were combined with the S(T) and Cp(T) data from frequencies, mass, moments of inertia, symmetry, and electronic degeneracy in the our statistical mechanics program SMCPS [
35] and are presented in
Table 4 for the parent molecules,
Table 5 for radicals from acetic acid hydrazide,
Table 6 for radicals from acetamide, and
Table 7 for radicals from
N-methyl acetamide.
Entropy and heat capacity contributions of the parent molecules and radicals using VIBIR have been calculated at temperatures ranging from 1–5000 K.
4. Summary
Thermochemical properties are presented for acetic acid hydrazide, Acetamide, N-methyl acetamide, and radicals that result from the loss of H atoms from the carbon and the nitrogen atoms. Standard enthalpies from all the work reactions and each of the calculation methods are in reasonably good agreement, suggesting that the B3LYP DFT calculations, in conjunction with the work reactions used here, are acceptable methods for larger hydrazide and amides. C–H bond energy values for the radicals C•H2–C=ONHNH2, C•H2–C=ONH2 and C•H2–C=ONHCH3 from the B3LYP/6-31G(d,p), B3LYP/6-31G(2d,2p) and CBS-QB3 levels of calculation were 99.50, 99.40, 99.61 and 100.22, 99.43, 99.52 and 99.54, 99.42, 99.56 kcal mol−1 respectively. The N–H bond in the acetohydrazide was weak, at 78.17 kcal mol−1, but strong in N-methyl acetamide at 104.20 kcal mol−1. The HN–H bond energies for the formation of the radicals CH3–C=ONHN•H and CH3–C=ON•H from the parent molecules were also similar across the different B3LYP basis sets and CBS-QB3 level of calculations (CH3–C=ONHN•H = 81.93, 81.45, 82.13; CH3-C=ON•H = 111.74, 110.76, 111.99 kcal mol−1) respectively.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}