
The Effect of Activation on the Structure of Biochars Prepared from Wood and from Posidonia Oceanica: A Spectroscopic Study
 , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Experiment
2.1. The Samples
2.2. Elemental Analysis, X-ray Diffraction and Nitrogen Adsorption/Desorption Measurements
2.3. ICP-OES Measurements
2.4. EPR and NMR Measurements
2.5. FT-IR Spectroscopy
3. Results
3.1. XRD Analysis
3.2. Elemental Composition and Pore Size Distribution
3.3. ICP-OES Measurements
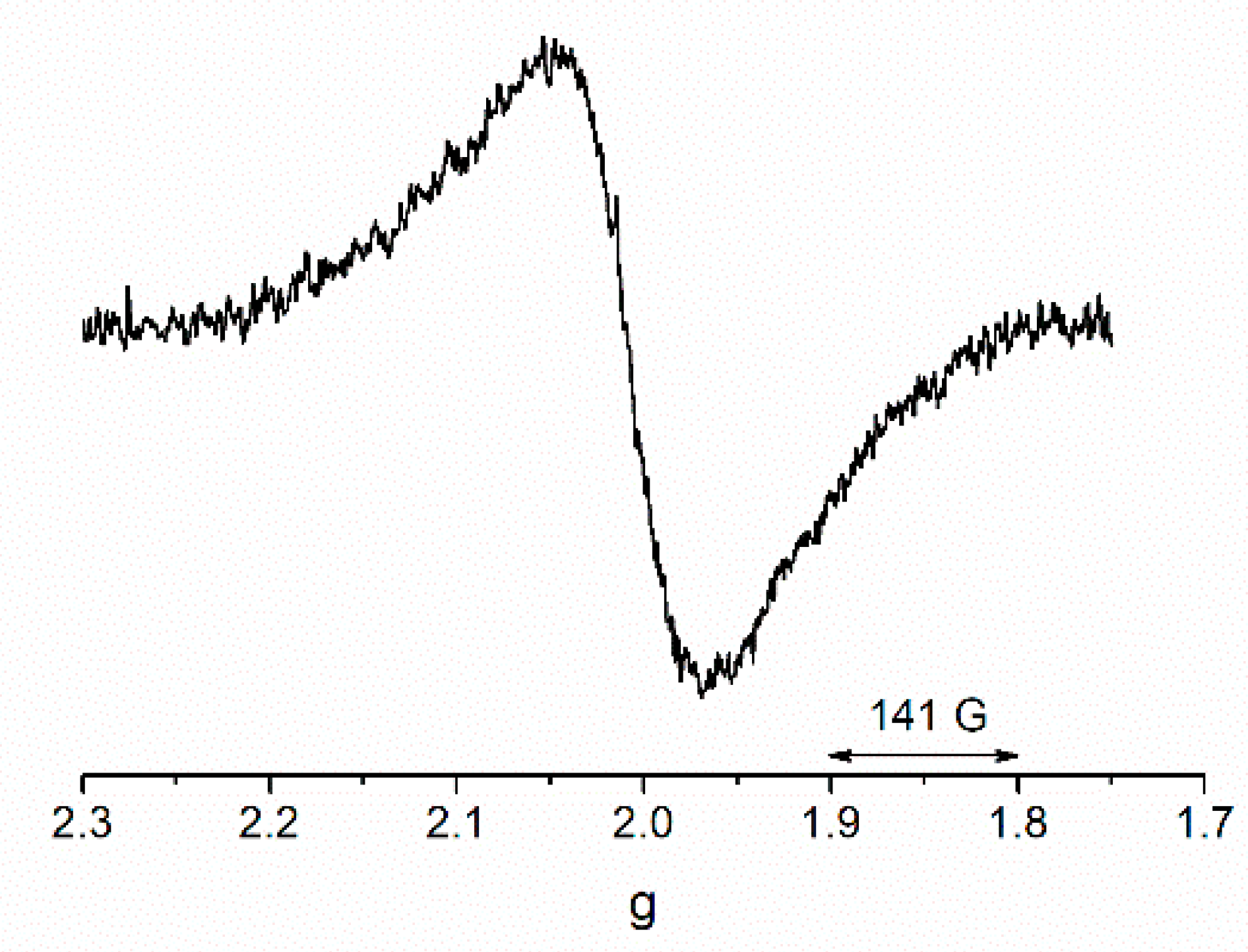
3.4. EPR
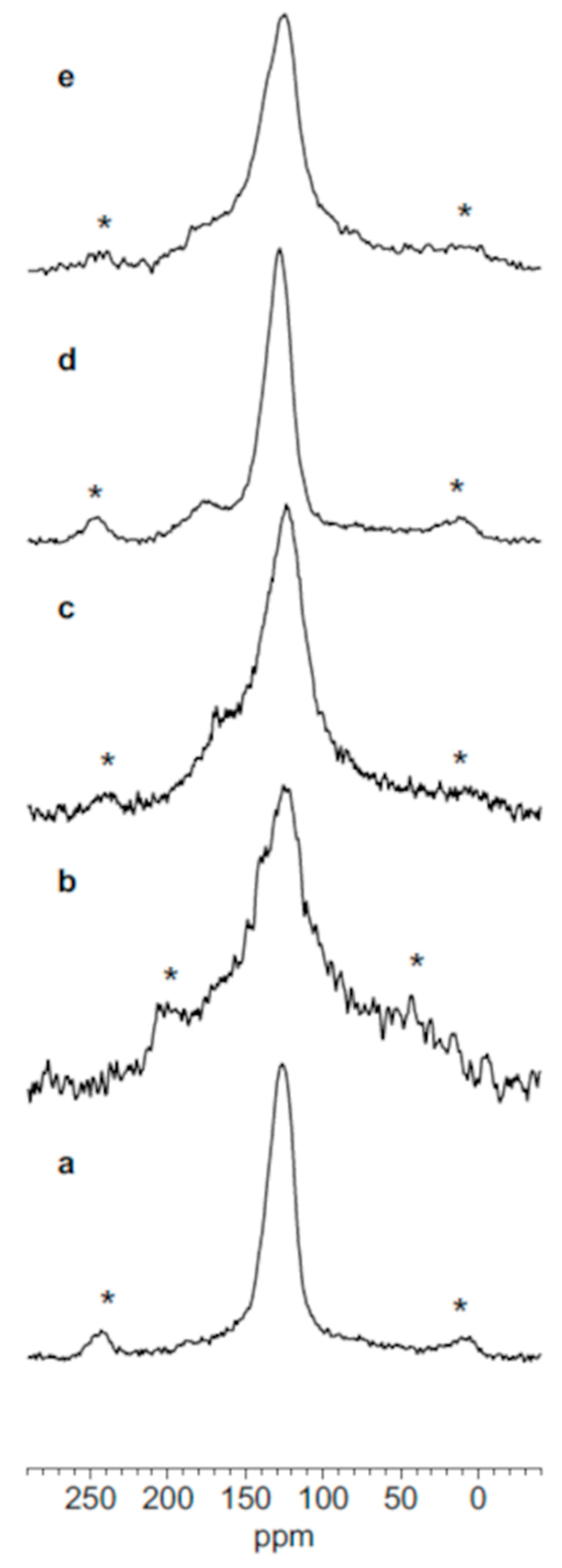
3.5. Solid State NMR
3.6. FT-IR
4. Discussion
4.1. Structure of the Pyrolyzed Biomasses
4.2. Structure of the KOH ACs
4.3. Effect of KOH:Biochar Ratio on the ACs Structures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, W.; Zhang, H.; Duan, X.; Sun, H.; Shao, G.; Wang, S. Porous Carbons: Stucture-Oriented Design and Versatile Applications. Adv. Funct. Mater. 2020, 30, 1909265. [Google Scholar] [CrossRef]
- Mu’azu, N.D.; Jarrah, N.; Zubair, M.; Alagha, O. Removal of Phenolic Compounds fromWater Using Sewage Sludge-Based Activated Carbon Adsorption: A Review. Int. J. Environ. Res. Public Health 2017, 14, 1094. [Google Scholar] [CrossRef]
- Chen, W.; Gong, M.; Li, K.; Xia, M.; Chen, Z.; Xiao, H.; Fang, Y.; Chen, Y.; Yang, H.; Chen, H. Insight into KOH Activation Mechanism During Biomass Pyrolysis: Chemical Reactions Between O-Containing Groups and KOH. Appl. Energy 2020, 278, 115730. [Google Scholar] [CrossRef]
- Leng, L.; Xiong, Q.; Yang, L.; Li, H.; Zhou, Y.; Zhang, W.; Jiang, S.; Li, H.; Huang, H. An Overview on Engineering the Surface Area and Porosity of Biochars. Sci. Total Environ. 2021, 763, 144204. [Google Scholar] [CrossRef]
- Wang, J.; Kaskel, S. KOH Activation of Carbon-Based Materials for Energy Storage. J. Mater. Chem. 2012, 22, 23710. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, J.; Li, D.; Liu, C.; Lu, Y.; Lin, X. Insights into the KOH/KMnO4 Activation Mechanism of Oxygen-Enriched Hierarchical porous Biochar Derived from Biomass Waste by in-Situ Pyrolysis for Methylene Blue Enhanced Adsorption. J. Anal. Appl. Pyrolysis 2021, 158, 1052. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Cazorla-Amorós, D.; Linares-Solano, A. Understanding Chemical Reactions between Carbons and NaOH and KOH: An Insight into the Chemical Activation Mechanism. Carbon 2003, 41, 267–275. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-Based Supercapacitors Produced by Activation of Graphene. Science 2011, 332, 1537–1541. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Azaïs, P.; Cacciaguerra, T.; Cazorla-Amorós, D.; Linares-Solano, A.; Béguin, F. KOH and NaOH Activation Mechanisms of Multiwalled Carbon Nanotubes with Different Structural Organisation. Carbon 2005, 43, 786–795. [Google Scholar] [CrossRef]
- Pedicini, R.; Maisano, S.; Chiodo, V.; Conte, G.; Policicchio, A.; Agostino, R.G. Posidonia Oceanica and Wood Chips Activated Carbon as Interesting Materials for Hydrogen Storage. Int. J. Hydrog. Energy 2020, 45, 14038–14047. [Google Scholar] [CrossRef]
- Grinberg, O.Y.; Williams, B.B.; Ruuge, A.E.; Grinberg, S.A.; Wilcox, D.E.; Swartz, H.M.; Freed, J.H. Oxygen Effects on the EPR Signals from Wood Charcoals: Experimental Results and the Development of a Model. J. Phys. Chem. B 2007, 111, 13316–13324. [Google Scholar] [CrossRef] [PubMed]
- Konchits, A.A.; Shanina, B.D.; Valakh, M.Y.; Yanchuk, I.B.; Yukhymchuk, V.O.; Alexeev, A.D.; Vasilenko, T.A.; Molchanov, A.N.; Kirillov, A.K. Local Structure, Paramagnetic Properties, and Porosity of Natural Coals: Spectroscopic studies. J. Appl. Phys. 2012, 112, 043504. [Google Scholar] [CrossRef]
- Bährle, C.; Custodis, V.; Jeschke, G.; van Bokhoven, J.A.; Vogel, F. In Situ Observation of Radicals and Molecular Products during Lignin Pyrolysis. ChemSusChem 2014, 7, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Trubetskaya, A.; Jensen, P.A.; Jensen, A.D.; Glarborg, P.; Hofmann Larsen, F.; Larsen Andersen, M. Characterization of Free Radicals by Electron Spin Resonance Spectroscopy in Biochars from Pyrolysis at High Heating Rates and at High Temperatures. Biomass Bioenergy 2016, 94, 117–129. [Google Scholar] [CrossRef]
- Bourke, J.; Manley-Harris, M.; Fushimi, C.; Dowaki, K.; Nunoura, T.; Antal, M.J., Jr. Do All Carbonized Charcoals Have the Same Chemical Structure? 2. A Model of the Chemical Structure of Carbonized Charcoal. Ind. Eng. Chem. Res. 2007, 46, 5954–5967. [Google Scholar] [CrossRef]
- Baccile, N.; Falco, C.; Titirici, M.-M. Characterization of Biomass and its Derived Char Using 13C-Solid State Nuclear Magnetic Resonance. Green Chem. 2014, 16, 4839–4869. [Google Scholar] [CrossRef]
- Bardet, M.; Hediger, S.; Gerbaud, G.; Gambarelli, S.; Jacquot, J.F.; Foray, M.F.; Gadelle, A. Investigation with 13C NMR, EPR and Magnetic Susceptibility Measurements of Char Residues Obtained by Pyrolysis of Biomass. Fuel 2007, 86, 1966–1976. [Google Scholar] [CrossRef]
- Knicker, H.; Velasco-Molina, M.; Knicker, M. 2D Solid-State HETCOR 1H-13C NMR Experiments with Variable Cross Polarization Times as a Tool for a Better Understanding of the Chemistry of Cellulose-Based Pyrochars—A Tutorial. Appl. Sci. 2021, 11, 8569. [Google Scholar] [CrossRef]
- Kloss, S.; Zehetner, F.; Dellantonio, A.; Hamid, R.; Ottner, F.; Liedtke, V.; Schwanninger, M.; Gerzabek, M.H.; Soja, G. Characterization of Slow Pyrolysis Biochars: Effects of Feedstocks and Pyrolysis Temperature on Biochar Properties. J. Environ. Qual. 2012, 41, 990–1000. [Google Scholar] [CrossRef]
- Seaton, N.A.; Walton, J.P.R.B.; Quirke, N. A New Analysis Method for the Determination of the Pore Size Distribution of Porous Carbons from Nitrogen Adsorption Measurements. Carbon 1989, 27, 853–861. [Google Scholar] [CrossRef]
- Ustinov, E.A.; Do, D.D. Application of Density Functional Theory to Analysis of Energetic Heterogeneity and Pore Size Distribution of Activated Carbons. Langmuir 2004, 20, 3791–3797. [Google Scholar] [CrossRef] [PubMed]
- Reza, M.T.; Lynam, J.G.; Uddin, M.H.; Coronella, C.J. Hydrothermal Carbonization: Fate of Inorganics. Biomass Bioenergy 2013, 49, 86–94. [Google Scholar] [CrossRef]
- Ambrosetti, R.; Ricci, D. A Fast Time Averaging Data Acquisition-System for the PC-AT bus. Rev. Sci. Instrum. 1991, 62, 2281–2287. [Google Scholar] [CrossRef]
- Cory, D.G.; Ritchey, W.M. Suppression of Signals from the Probe in Bloch Decay Spectra. J. Magn. Reson. 1988, 80, 128–132. [Google Scholar] [CrossRef]
- Mimmo, T.; Panzacchi, P.; Baratieri, M.; Davies, C.A.; Tonon, G. Effect of Pyrolysis Temperature on Miscanthus (Miscanthus × giganteus) Biochar Physical, Chemical and Functional Properties. Biomass Bioenergy 2014, 62, 149–157. [Google Scholar] [CrossRef]
- Chiodo, V.; Zafarana, G.; Maisano, S.; Freni, S.; Urbani, F. Pyrolysis of Different Biomass: Direct Comparison among Posidonia Oceanica, Lacustrine Alga and White-Pine. Fuel 2016, 164, 220–227. [Google Scholar] [CrossRef]
- Borysiak, S.; Doczekalska, B. X-ray Diffraction Study of Pine Wood Treated with NaOH. Fibres Text. East. Eur. 2005, 13, 87–89. [Google Scholar]
- Sun, F.; Wang, L.; Peng, Y.; Gao, J.; Pi, X.; Qu, Z.; Zhao, G.; Qin, Y. Converting Biomass Waste into Microporous Carbon with Simultaneously High Surface Area and Carbon Purity as Advanced Electrochemical Energy Storage Materials. Appl. Surf. Sci. 2018, 436, 486–494. [Google Scholar] [CrossRef]
- Boughriet, A.; Cordier, C.; Deram, L.; Ouddane, B.; Chamley, H.; Wartel, W. Coprecipitation/Accumulation/Distribution of Manganese and Iron, and Electrochemical Characteristics of Mn in Calcareous Seawater. Fresenius’ J. Anal. Chem. 1995, 352, 341–353. [Google Scholar] [CrossRef]
- Lu, D.-Y.; Gao, X.-L.; Chi, Y.; Wang, H.-G.; Yand, X.; Qiao, T. Preliminary Study on the Correlation Between the Trace Mn2+ and the Calcite Polymorph in Gallstones Containing Calcium Carbonate from the Northeast China via Electron Spin Resonance. J. Trace Elem. Med. Biol. 2020, 60, 126494. [Google Scholar] [CrossRef]
- Jeschke, G. Electron Paramagnetic Resonance: Recent Developments and Trends. Curr. Opin. Solid State Mater. Sci. 2003, 7, 181–188. [Google Scholar] [CrossRef]
- Papenguth, H.W.; Kirkpatrick, R.J.; Montez, B.; Sandberg, P.A. 13C MAS NMR Spectroscopy of Inorganic and Biogenic Carbonates. Am. Mineral. 1989, 74, 1152–1158. [Google Scholar]
- Brewer, C.E.; Schmidt-Rohr, K.; Satrio, J.A.; Brown, R.C. Characterization of Biochar from Fast Pyrolysis and Gasification Systems. Environ. Prog. Sustain. Energy 2009, 28, 386–396. [Google Scholar] [CrossRef]
- Cao, X.; Pignatello, J.J.; Li, Y.; Lattao, C.; Chappell, M.A.; Chen, N.; Miller, L.F.; Mao, J. Characterization of Wood Chars Produced at Different Temperatures Using Advanced Solid-State 13C NMR Spectroscopic Techniques. Energy Fuels 2012, 26, 5983–5991. [Google Scholar] [CrossRef]
- Althaus, S.M.; Mao, K.; Kennedy, G.J.; Pruski, M. Solid-State NMR Studies of Fossil Fuels using One- and Two-Dimensional Methods at High Magnetic Field. Energy Fuels 2012, 26, 4405–4412. [Google Scholar] [CrossRef]
- Forse, A.C.; Griffin, J.M.; Presser, V.; Gogotsi, Y.; Grey, C.P. Ring Current Effects: Factors Affecting the NMR Chemical Shift of Molecules Adsorbed on Porous Carbons. J. Phys. Chem. C 2014, 118, 7508–7514. [Google Scholar] [CrossRef]
- Cervini, L.; Lynes, O.D.; Akien, G.R.; Kerridge, A.; Barrow, N.S.; Griffith, J.M. Factors Affecting the Nucleus-independent Chemical Shift in NMR Studies of Microporous Carbon Electrode Materials. Energy Storage Mater. 2019, 21, 335–346. [Google Scholar] [CrossRef]
- Szewczyk, I.; Rokicińska, A.; Michalik, M.; Chen, J.; Jaworski, A.; Aleksis, R.; Pell, A.J.; Hedin, N.; Slabon, A.; Kuśtrowski, P. Electrochemical Denitrification and Oxidative Dehydrogenation of Ethylbenzene over N-doped Mesoporous Carbon: Atomic Level Understanding of Catalytic Activity by 15N NMR Spectroscopy. Chem. Mater. 2020, 32, 7263–7273. [Google Scholar] [CrossRef]
- Le Bozec, G.; Giraudet, S.; Le Polles, L.; Le Cloirec, P. 1H NMR Investigations of Activated Carbon Loaded with Volatile Organic Compounds: Quantification, Mechanisms, and Diffusivity Determination. Langmuir 2017, 33, 1605–1613. [Google Scholar] [CrossRef]
- Chia, C.H.; Gong, B.; Joseph, S.D.; Marjo, C.E.; Munroe, P.; Rich, A.M. Imaging of Mineral-Enriched Biochar by FTIR, Raman and SEM-EDX. Vib. Spectrosc. 2012, 62, 248–257. [Google Scholar] [CrossRef]
- Lian, P.; Zhu, X.; Liang, S.; Li, Z.; Yang, W.; Wang, H. Large Reversible Capacity of High Quality Graphene Sheets as an Anode Material for Lithium-Ion Batteries. Electrochim. Acta 2010, 55, 3909–3914. [Google Scholar] [CrossRef]
- Kalahal, P.B.; Kulkarni, A.S.; Sajjan, A.M.; Khan, T.M.Y.; Anjum Badruddin, I.; Kamangar, S.; Banapurmath, N.R.; Ayachit, N.H.; Naik, M.L.; Marakatti, V.S. Fabrication and Physicochemical Study of B2SA-Grafted Poly (vinyl Alcohol)–Graphene Hybrid Membranes for Dehydration of Bioethanol by Pervaporation. Membranes 2021, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Facchinetto, S.E.; Bortolotto, T.; Neumann, G.E.; Vieira, J.C.B.; de Menezes, B.B.; Giacomelli, C.; Schmidt, V. Synthesis of Submicrometer Calcium Carbonate Particles from Inorganic Salts Using Linear Polymers as Crystallization Modifiers. J. Braz. Chem. Soc. 2017, 28, 547–556. [Google Scholar] [CrossRef]
- Wartel, M.; Skiker, M.; Auger, Y.; Boughriet, A. Interaction of Mn2+ with Carbonates in Seawater: Assessment of the Solubility Product of MnCO3 and Mn Distribution Coefficient between the Liquid Phase and CaCO3 Particles. Mar. Chem. 1990, 29, 99–117. [Google Scholar] [CrossRef]
- Chunlan, L.; Shaoping, X.; Yixiong, G.; Shuqin, L.; Changhou, L. Effect of Pre-Carbonization of Petroleum Cokes on Chemical Activation Process with KOH. Carbon 2005, 43, 2295–2301. [Google Scholar] [CrossRef]
- Zornitta, R.L.; Barcelos, K.M.; Nogueira, F.G.E.; Ruotolo, L.A.M. Understanding the Mechanism of Carbonization and KOH Activation of Polyaniline Leading to Enhanced Electrosorption Performance. Carbon 2020, 156, 346–358. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, Z.; Peng, H.; Qin, Y.; Li, G. Nitrogen- and Oxygen-Containing Hierarchical Porous Carbon Frameworks for High-Performance Supercapacitors. Electrochim. Acta 2014, 134, 471–477. [Google Scholar] [CrossRef]
- Kolanowski, Ł.; Graś, M.; Bartkowiak, M.; Doczekalska, B.; Lota, G. Electrochemical Capacitors Based on Electrodes Made of Lignocellulosic Waste Materials. Waste Biomass Valorization 2020, 11, 3863–3871. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, Y.; Fu, Y.; Zhang, N. Activated Carbons Synthesized from Unaltered and Pelletized Biomass Wastes for Bio-tar Adsorption in Different Phases. Renew. Energy 2020, 146, 1700–1709. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Langmuir Surface Area (m2/g) a | Elemental Composition (wt%) a | Ash (wt%) a | |||
|---|---|---|---|---|---|---|
| C | H | N | O | |||
| wF | 47.1 | 6.3 | 0.3 | 39.8 | 2.0 | |
| pF | 40.3 | 6.0 | 1.1 | 25.7 | 14.0 | |
| wB | 425 | 69.2 | 1.8 | 0.2 | 28.2 | |
| wAC | 2835 | 95.6 | 1.2 | 0.0 | 3.2 | |
| pB | 41 | 58 | 2.8 | 1.8 | 37.5 | |
| pAC_meso | 479 | 80.8 | 2.3 | 1.5 | 15.4 | |
| pAC_micro | 2365 | 92.1 | 2.0 | 0.7 | 5.2 | |
| Sample | Organic Radical Content (Spin/g) |
|---|---|
| wB | 8.7 × 1019 |
| pB | 1.0 × 1016 |
| pAC_meso | 5.3 × 1019 |
| Sample | H/C Molar Ratio | O/C Molar Ratio | Porosity % | XRD | EPR | 13C ssNMR | 1H ssNMR | FT-IR | ||
|---|---|---|---|---|---|---|---|---|---|---|
| micro | meso | macro | ||||||||
| wB | 0.31 | 0.31 | 40.0 | 45.0 | 15.0 | Absence of cellulose and of graphitic planes | Observable signal: absence of interconnected conducting domains; broad signal: meso-microporosity | Absence of cellulose; CP efficiency: presence of small aromatic networks | Porosity accessible to water | Absence of cellulose; presence of C-O groups |
| wAC | 0.15 | 0.03 | 77.0 | 21.2 | 1.8 | Abundant microporous structure; presence of graphitic planes | No signal: presence of interconnected conducting domains | CP inefficiency: presence of large aromatic networks | Water located in bottle-neck pores | Presence of graphene-like structures |
| pB | 0.58 | 0.48 | 0.8 | 65.0 | 34.2 | Absence of cellulose and of graphitic planes; presence of calcite | Observable signal: absence of interconnected conducting domains; narrow signal: macro-mesoporosity; presence of Mn2+ and Fe3+ | Absence of cellulose; presence of calcite | Porosity accessible to water | Absence of cellulose; presence of calcite |
| pAC_meso | 0.34 | 0.14 | 17.1 | 55.0 | 27.9 | Abundant microporous structure; presence of graphitic planes | Observable signal: absence of interconnected conducting domains; narrow signal: macro-mesoporosity | Presence of carboxylic groups; CP efficiency: small aromatic networks | Porosity accessible to water | Presence of carboxylic groups |
| pAC_micro | 0.26 | 0.04 | 70.7 | 23.9 | 5.3 | Abundant microporous structure; presence of graphitic planes | No signal: presence of interconnected conducting domains | CP inefficiency: presence of large aromatic networks | Water located in bottle-neck pores and on the external surface | Presence of graphene-like structures |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizzanelli, S.; Maisano, S.; Pinzino, C.; Manariti, A.; Chiodo, V.; Pitzalis, E.; Forte, C. The Effect of Activation on the Structure of Biochars Prepared from Wood and from Posidonia Oceanica: A Spectroscopic Study. Physchem 2022, 2, 286-304. https://doi.org/10.3390/physchem2030021
Pizzanelli S, Maisano S, Pinzino C, Manariti A, Chiodo V, Pitzalis E, Forte C. The Effect of Activation on the Structure of Biochars Prepared from Wood and from Posidonia Oceanica: A Spectroscopic Study. Physchem. 2022; 2(3):286-304. https://doi.org/10.3390/physchem2030021
Chicago/Turabian StylePizzanelli, Silvia, Susanna Maisano, Calogero Pinzino, Antonella Manariti, Vitaliano Chiodo, Emanuela Pitzalis, and Claudia Forte. 2022. "The Effect of Activation on the Structure of Biochars Prepared from Wood and from Posidonia Oceanica: A Spectroscopic Study" Physchem 2, no. 3: 286-304. https://doi.org/10.3390/physchem2030021