
Formation of Metallic Ag on AgBr by Femtosecond Laser Irradiation





Abstract
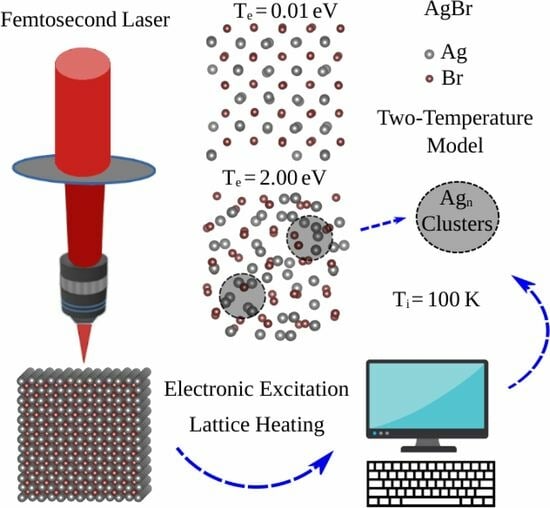
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Theoretical Approach and Computational Details
3. Results
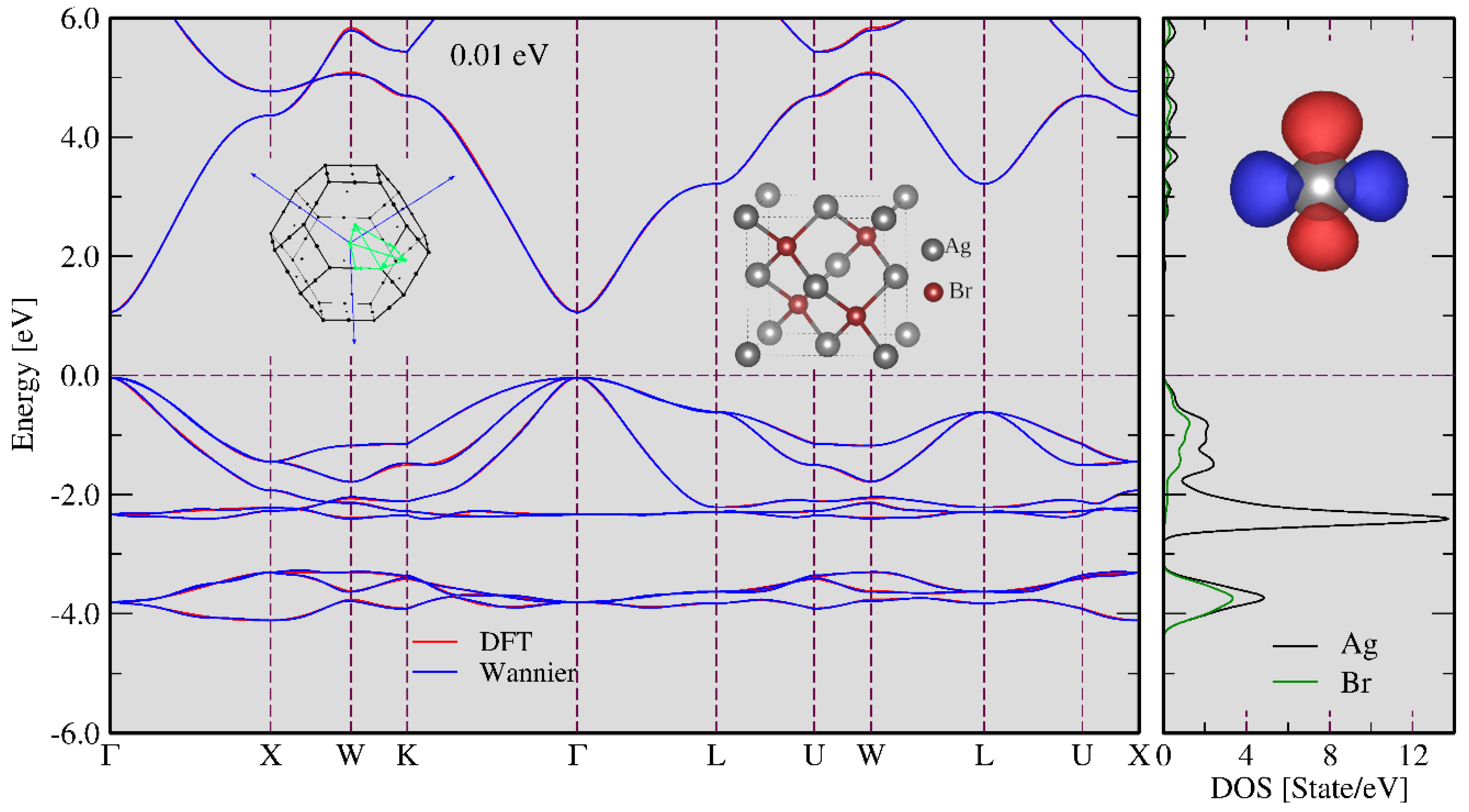
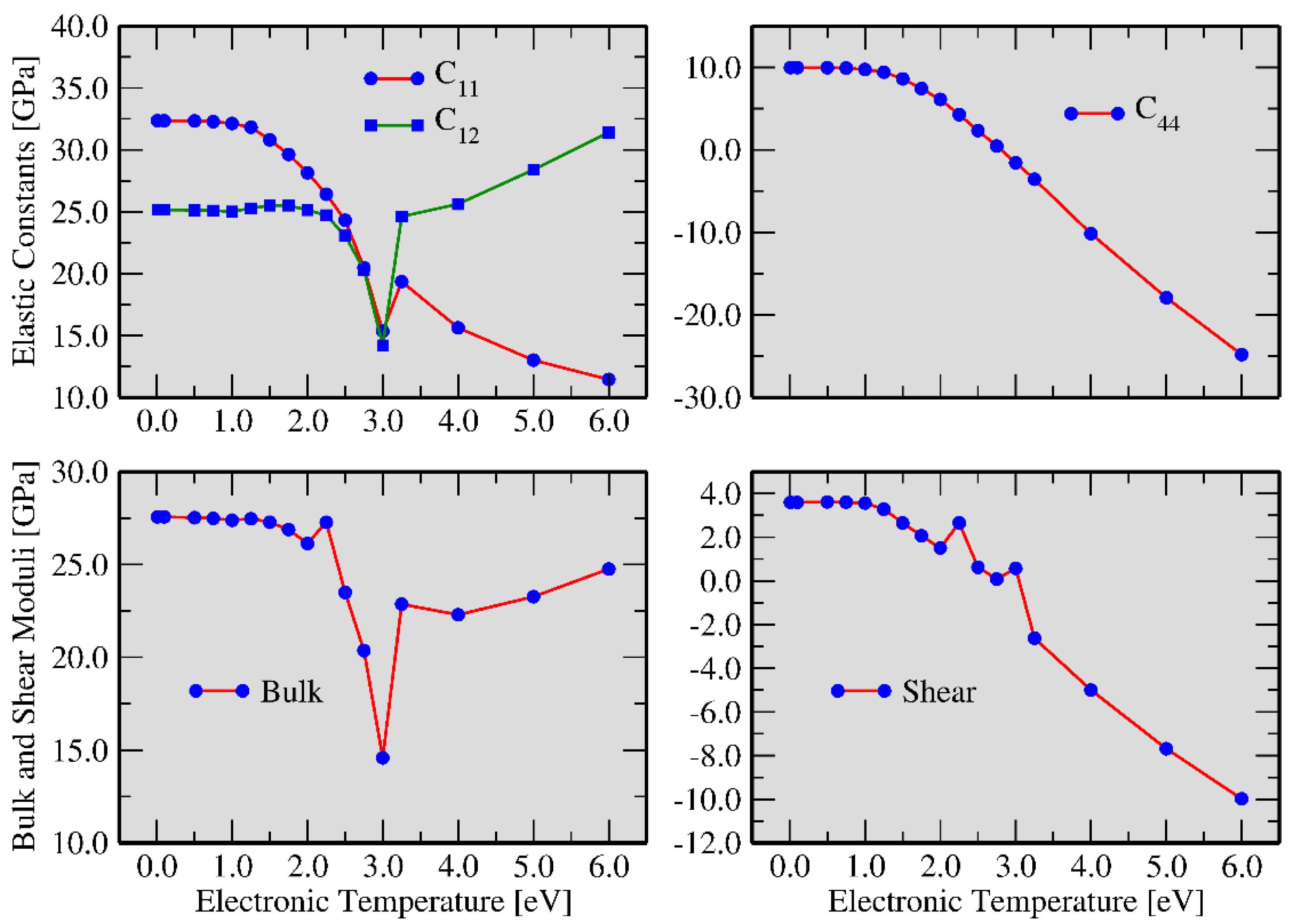
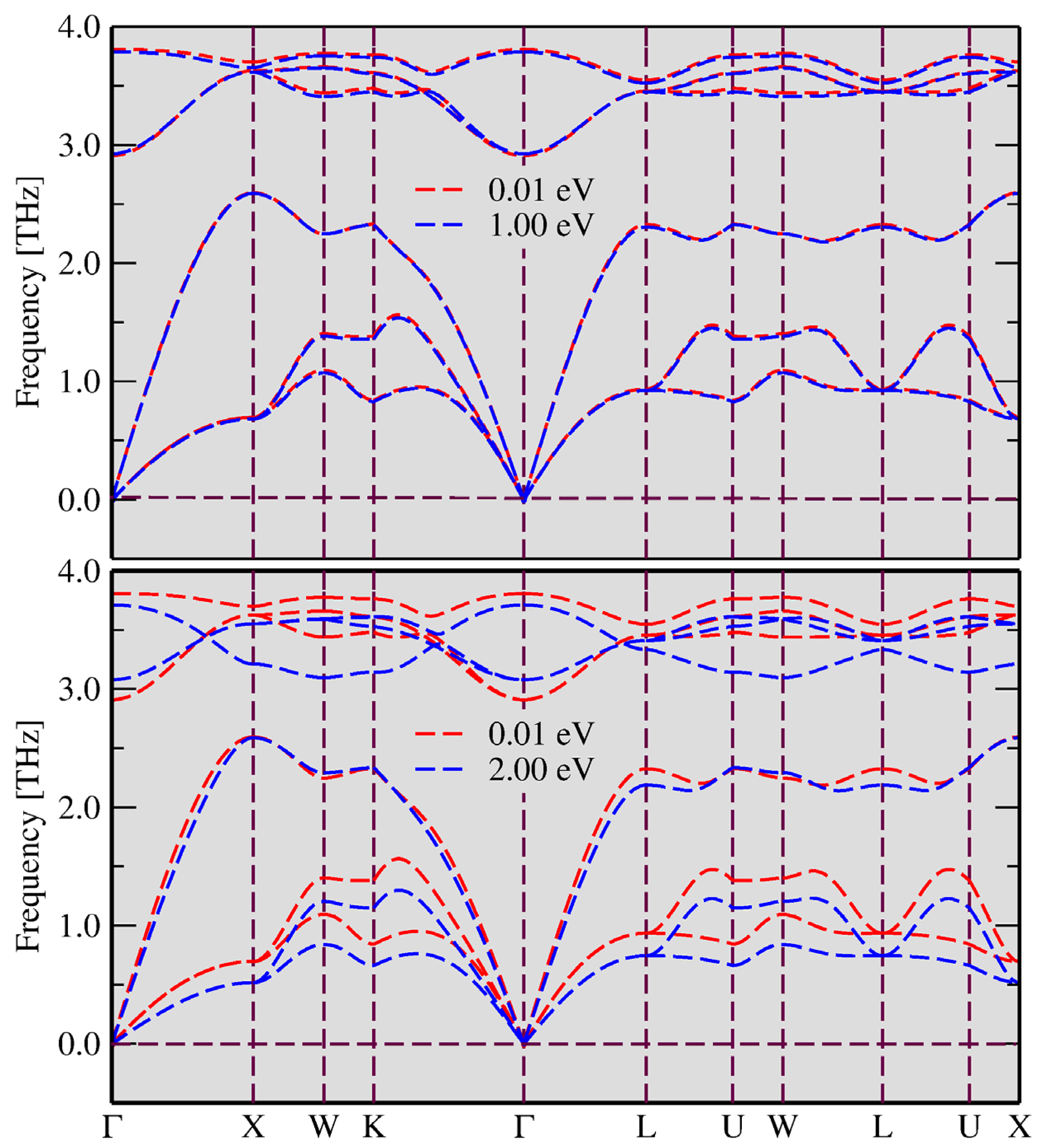
3.1. FT-DFT Investigation: Limits for Electronic Excitation
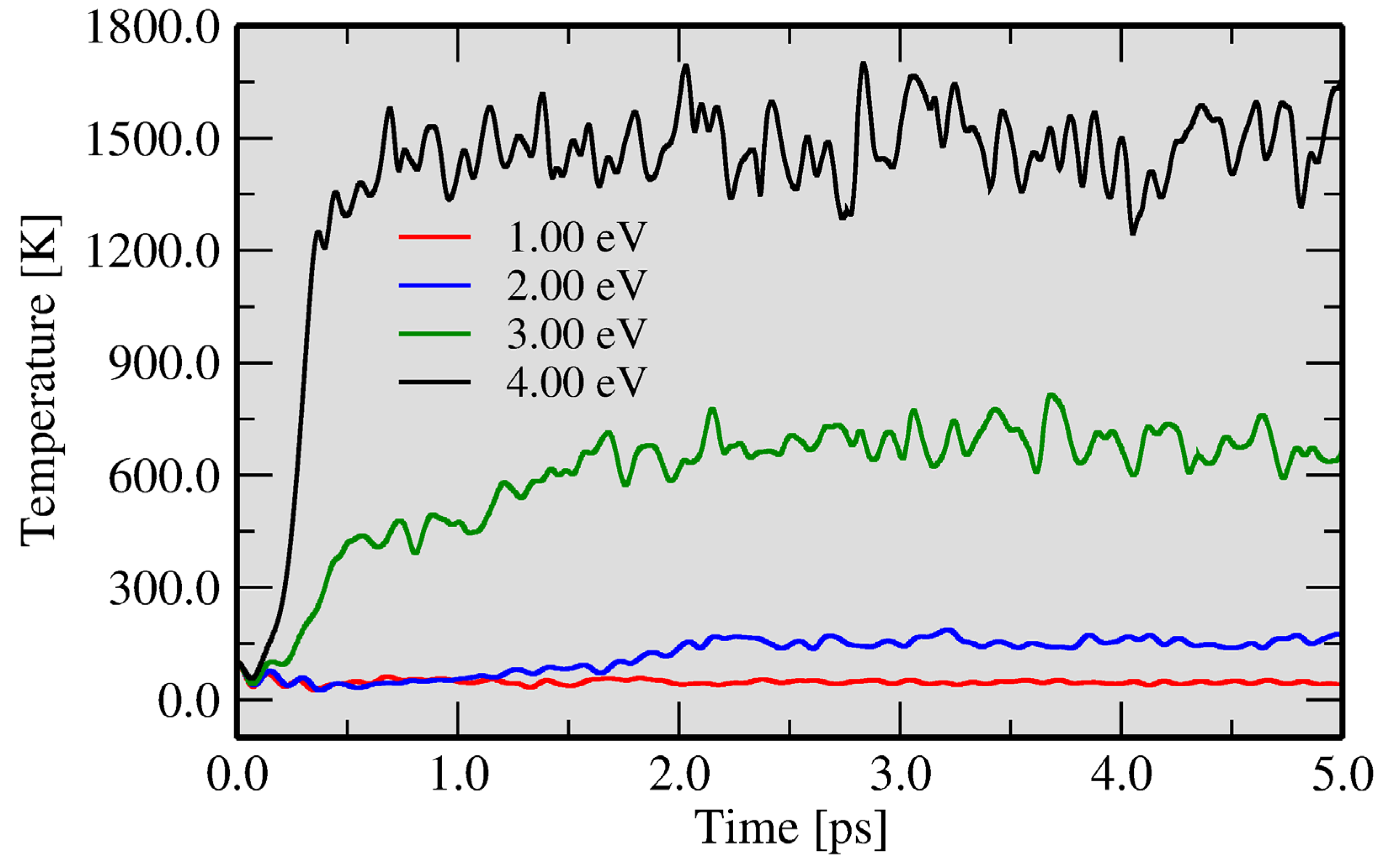
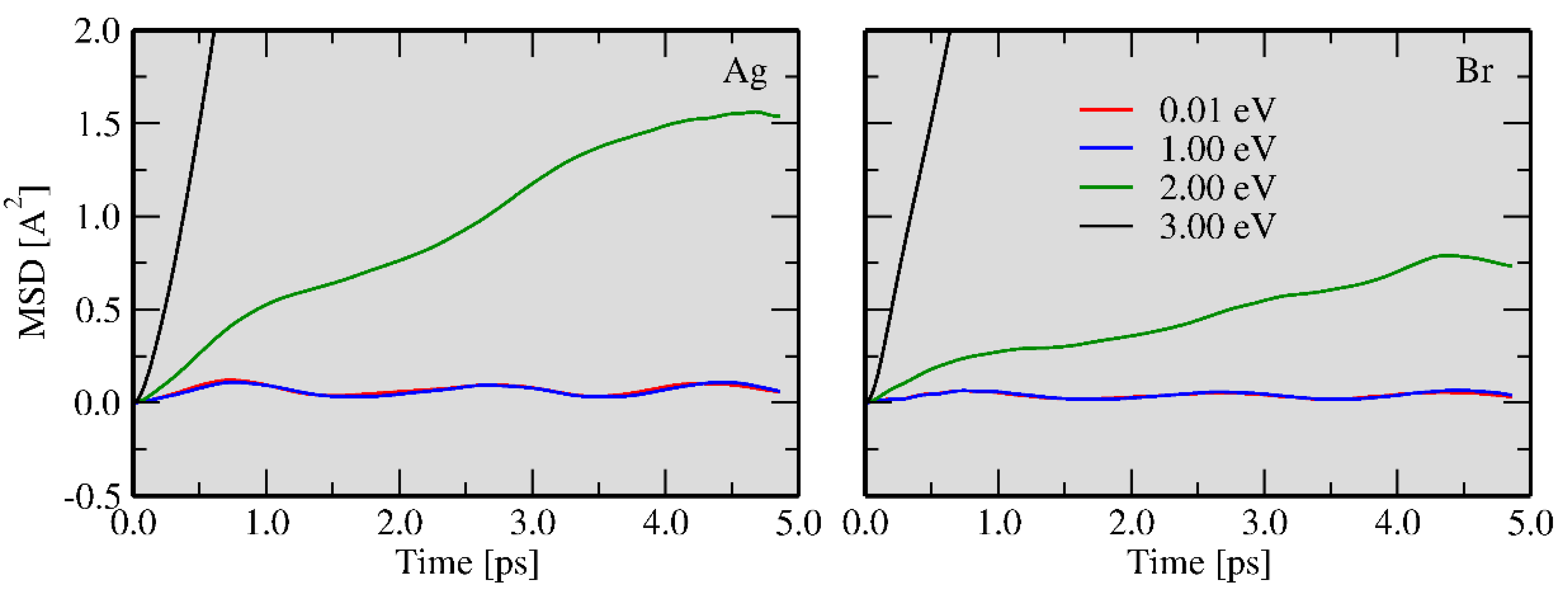
3.2. Two Temperature Model: Ab Initio Molecular Dynamics with Excited Atoms
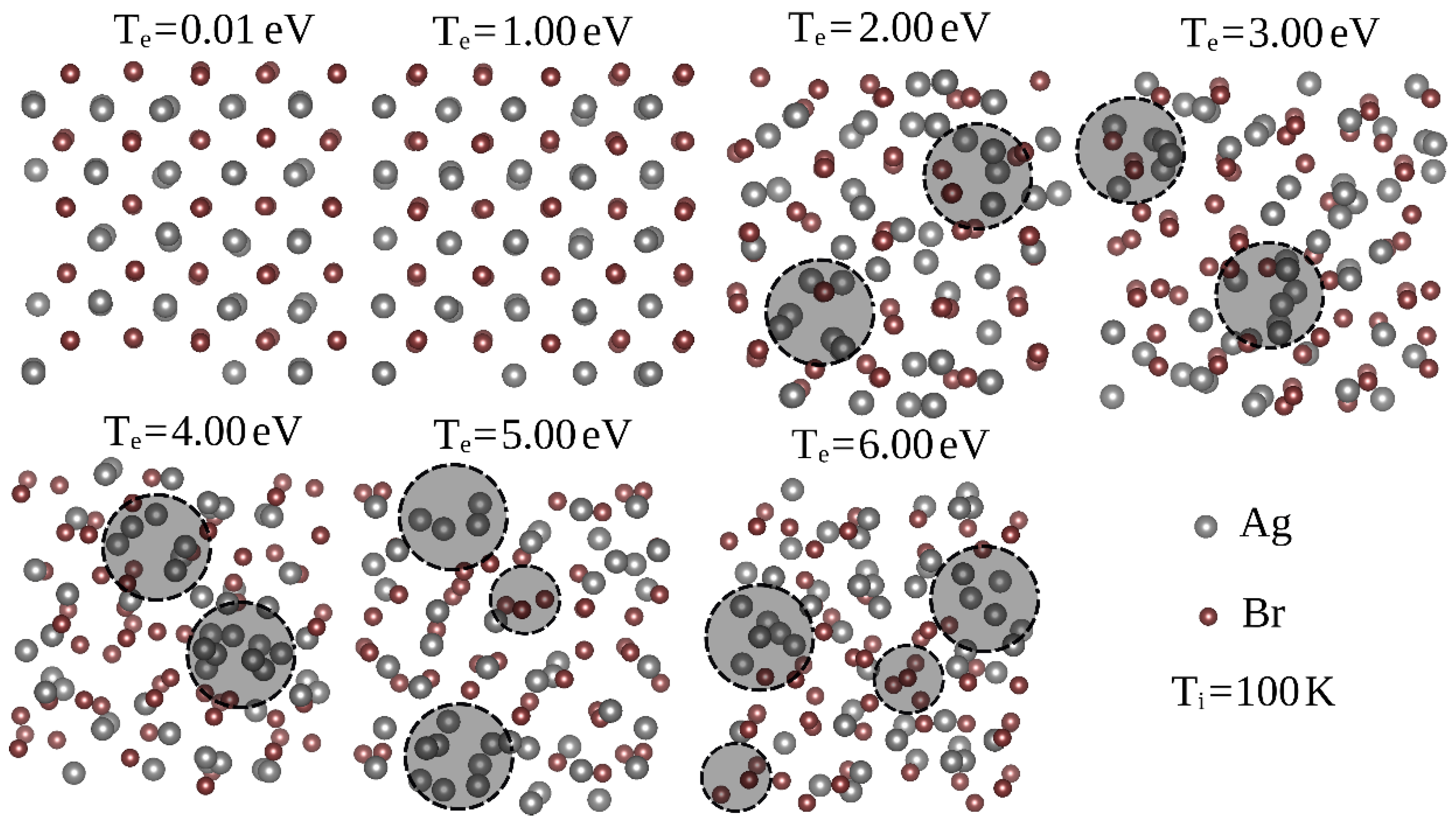
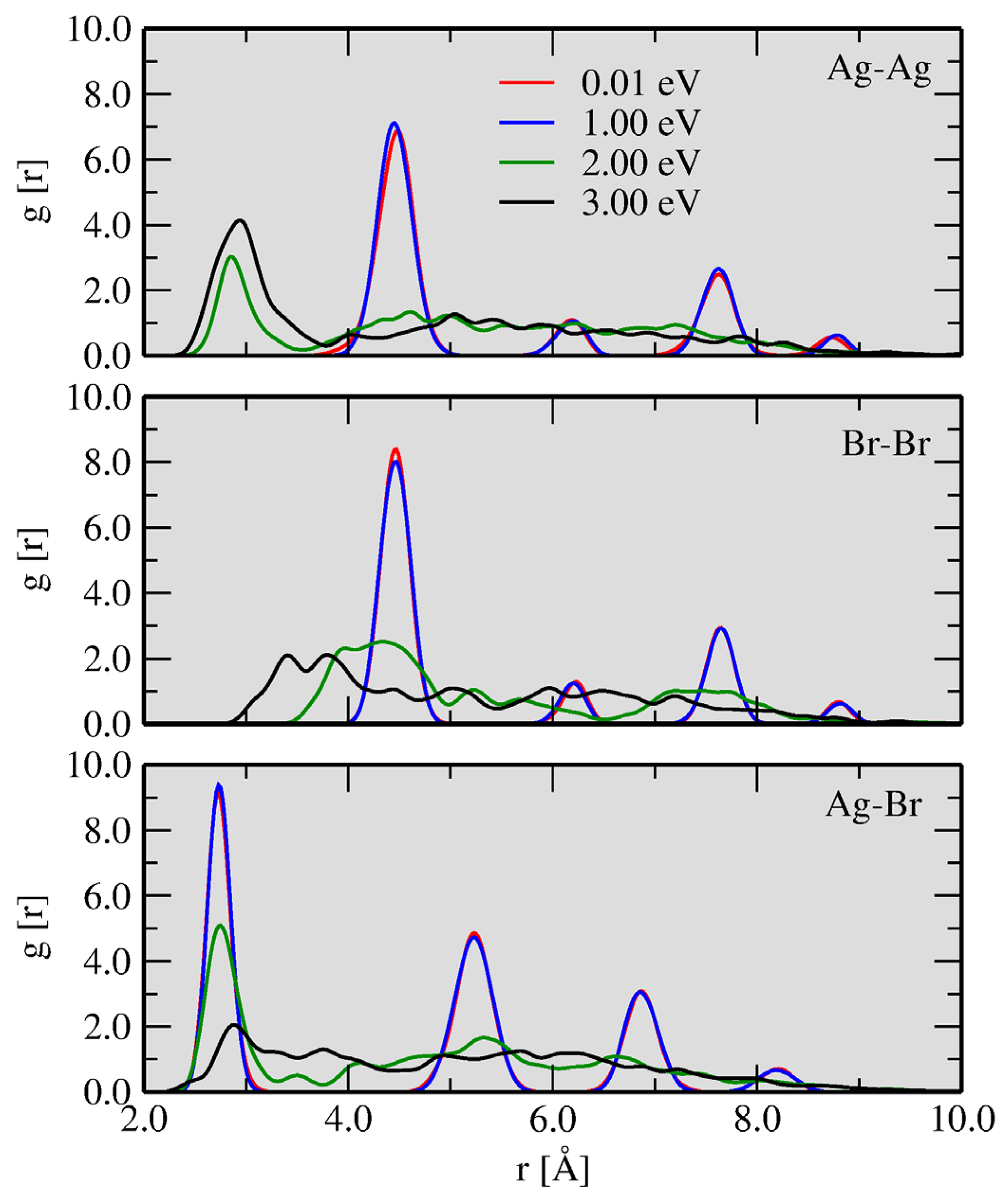
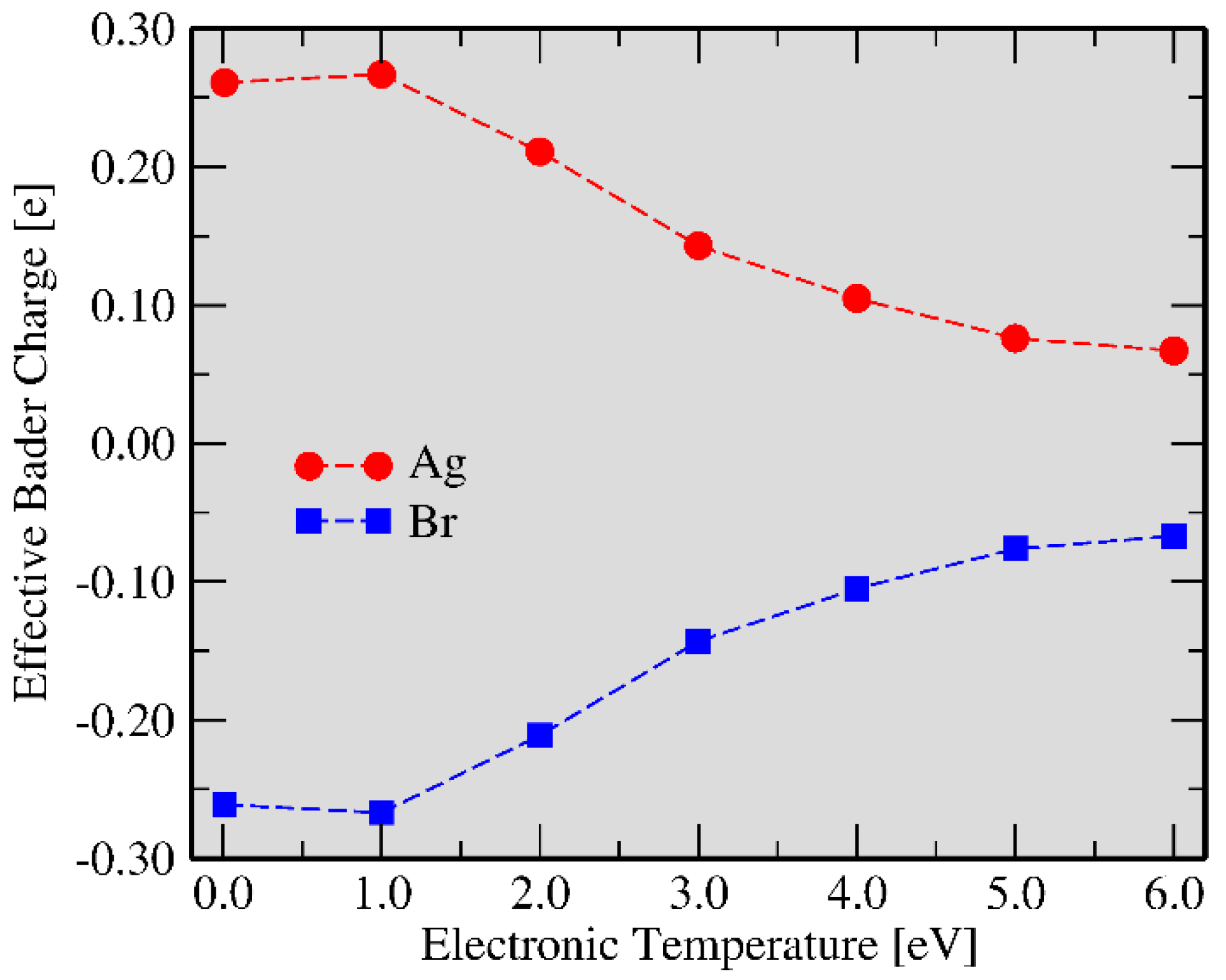
3.3. Ag and Br Clustering Processes: Charge Flow
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biasin, E.; Fox, Z.W.; Andersen, A.; Ledbetter, K.; Kjær, K.S.; Alonso-Mori, R.; Carlstad, J.M.; Chollet, M.; Gaynor, J.D.; Glownia, J.M.; et al. Direct observation of coherent femtosecond solvent reorganization coupled to intramolecular electron transfer. Nat. Chem. 2021, 13, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Jalil, S.A.; Lai, B.; ElKabbash, M.; Zhang, J.; Garcell, E.; Singh, S.; Guo, C. Spectral absorption control of femtosecond laser-treated metals and application in solar-thermal devices. Light Sci. Appl. 2020, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, M.; Aguirregabiria, G.; Ritzkowsky, F.; Rybka, T.; Marinica, D.C.; Aizpurua, J.; Borisov, A.G.; Leitenstorfer, A.; Brida, D. Sub-femtosecond electron transport in a nanoscale gap. Nat. Phys. 2019, 16, 341–345. [Google Scholar] [CrossRef]
- Skopintsev, P.; Ehrenberg, D.; Weinert, T.; James, D.; Kar, R.K.; Johnson, P.J.M.; Ozerov, D.; Furrer, A.; Martiel, I.; Dworkowski, F.; et al. Femtosecond-to-millisecond structural changes in a light-driven sodium pump. Nature 2020, 583, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Jiang, R.; Hu, X.; He, Z.; Wang, Y. Structure and properties of nano SiC coatings in-situ fabricated by laser irradiation. Ceram. Int. 2020, 46, 14747–14755. [Google Scholar] [CrossRef]
- Momoki, K.; Manabe, T.; Li, L.; Yan, J. Silicon nanoparticle generation and deposition on glass from waste silicon powder by nanosecond pulsed laser irradiation. Mater. Sci. Semicond. Process. 2020, 111, 104998. [Google Scholar] [CrossRef]
- Yang, H.; Cheng, J.; Liu, Z.; Liu, Q.; Zhao, L.; Wang, J.; Chen, M. Dynamic behavior modeling of laser-induced damage initiated by surface defects on KDP crystals under nanosecond laser irradiation. Sci. Rep. 2020, 10, 500. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, S.K.; Mazur, E. Inducing and probing non-thermal transitions in semiconductors using femtosecond laser pulses. Nat. Mater. 2020, 1, 217. [Google Scholar] [CrossRef]
- Izawa, Y.; Izawa, Y.; Setsuhara, Y.; Hashida, M.; Fujita, M.; Sasaki, R.; Nagai, H.; Yoshida, M. Ultrathin amorphous Si layer formation by femtosecond laser pulse irradiation. Appl. Phys. Lett. 2007, 90, 044107. [Google Scholar] [CrossRef]
- Shuleiko, D.V.; Potemkin, F.V.; Romanov, I.A.; Parhomenko, I.N.; Pavlikov, A.V.; Presnov, D.E.; Kashkarov, P.K. Femtosecond laser pulse modification of amorphous silicon films: Control of surface anisotropy. Laser Phys. Lett. 2018, 15, 056001. [Google Scholar] [CrossRef]
- Öktem, B.; Pavlov, I.; Ilday, S.; Kalaycıoğlu, H.; Rybak, A.; Yavaş, S.; Ilday, F.Ö. Nonlinear laser lithography for indefinitely large-area nanostructuring with femtosecond pulses. Nat. Photonics 2013, 7, 897–901. [Google Scholar] [CrossRef]
- Pan, C.-L.; Chen, K.-W.; Wang, Y.-C.; Kao, S.-H.; Wu, P. Room-temperature crystallization of amorphous silicon by near-UV femtosecond pulses. AIP Adv. 2020, 10, 055321. [Google Scholar] [CrossRef]
- Xu, C.; Jiang, L.; Li, X.; Li, C.; Shao, C.; Zuo, P.; Liang, M.; Qu, L.; Cui, T. Miniaturized high-performance metallic 1T-Phase MoS2 micro-supercapacitors fabricated by temporally shaped femtosecond pulses. Nano Energy 2019, 67, 104260. [Google Scholar] [CrossRef]
- Brahms, C.; Belli, F.; Travers, J.C. Infrared attosecond field transients and UV to IR few-femtosecond pulses generated by high-energy soliton self-compression. Phys. Rev. Res. 2020, 2, 043037. [Google Scholar] [CrossRef]
- Pu, G.; Yi, L.; Zhang, L.; Luo, C.; Li, Z.; Hu, W. Intelligent control of mode-locked femtosecond pulses by time-stretch-assisted real-time spectral analysis. Light Sci. Appl. 2020, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Nourbakhsh, Z.; Tancogne-Dejean, N.; Merdji, H.; Rubio, A. High Harmonics and Isolated Attosecond Pulses from MgO. Phys. Rev. Appl. 2021, 15, 014013. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Weber, W.J. Ab Initio Study of Electronic Excitation Effects on SrTiO3. J. Phys. Chem. C 2017, 121, 26622–26628. [Google Scholar] [CrossRef]
- Alavi, A.; Kohanoff, J.; Parrinello, M.; Frenkel, D. Ab Initio molecular dynamics with excited electrons. Phys. Rev. Lett. 1994, 73, 2599–2602. [Google Scholar] [CrossRef] [Green Version]
- Silvestrelli, P.L.; Alavi, A.; Parrinello, M.; Frenkel, D. Ab initio molecular dynamics simulation of laser melting of silicon. Phys. Rev. Lett. 1996, 77, 3149–3152. [Google Scholar] [CrossRef] [Green Version]
- Silvestrelli, P.L.; Alavi, A. Structural, dynamical, electronic, and bonding properties of laser-heated silicon: An ab initio molecular-dynamics study. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 56, 3806–3812. [Google Scholar] [CrossRef]
- Cabral, L.; Andrés, J.; Machado, T.R.; Picinin, A.; Rino, J.P.; Lopez-Richard, V.; Longo, E.; Gouveia, A.F.; Marques, G.E.; da Silva, E.Z.; et al. Evidence for the formation of metallic in after laser irradiation of InP. J. Appl. Phys. 2019, 126, 025902. [Google Scholar] [CrossRef]
- An, M.; Song, Q.; Yu, X.; Meng, H.; Ma, D.; Baoling, H.; Jin, Z.; Huang, B.; Ruiyang, L. Generalized Two-Temperature Model for Coupled Phonons in Nanosized Graphene. Nano Lett. 2017, 17, 5805–5810. [Google Scholar] [CrossRef] [Green Version]
- de Assis, M.; Robeldo, T.; Foggi, C.C.; Kubo, A.M.; Mínguez-Vega, G.; Condoncillo, E.; Beltran-Mir, H.; Torres-Mendieta, R.; Andrés, J.; Oliva, M.; et al. Ag Nanoparticles/α-Ag2WO4 Composite Formed by Electron Beam and Femtosecond Irradiation as Potent Antifungal and Antitumor Agents. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Assis, M.; Macedo, N.G.; Machado, T.R.; Ferrer, M.M.; Gouveia, A.F.; Cordoncillo, E.; Torres-Mendieta, R.; Beltrán-Mir, H.; Mínguez-Vega, G.; Leite, E.R.; et al. Laser/Electron Irradiation on Indium Phosphide (InP) Semiconductor: Promising Pathways to In Situ Formation of Indium Nanoparticles. Part. Part. Syst. Charact. 2018, 35, 1–10. [Google Scholar] [CrossRef]
- Assis, M.; Cordoncillo, E.; Torres-Mendieta, R.; Beltrán-Mir, H.; Mínguez-Vega, G.; Gouveia, A.F.; Leite, E.; Andrés, J.; Longo, E. Laser-induced formation of bismuth nanoparticles. Phys. Chem. Chem. Phys. 2018, 20, 13693–13696. [Google Scholar] [CrossRef]
- Añez, R.; Cabral, L.; da Silva, E.Z.; Longo, E.; Andrés, J.; San-Miguel, M.A. Unveiling the Ag-Bi miscibility at the atomic level: A theoretical insight. Comput. Mater. Sci. 2021, 197, 110612. [Google Scholar] [CrossRef]
- Torres-Mendieta, R.O.; Teixeira, M.M.; Mínguez-Vega, G.; de Souza, D.; Gobato, Y.G.; Assis, M.; Longo, E. Toward Expanding the Optical Response of Ag2CrO4and Bi2O3by Their Laser-Mediated Heterojunction. J. Phys. Chem. C 2020, 124, 26404–26414. [Google Scholar] [CrossRef]
- Ågren, R.; Zeberg, H. Low-Resistance silver bromide electrodes for recording fast ion channel kinetics under voltage clamp conditions. J. Neurosci. Methods 2020, 348, 108984. [Google Scholar] [CrossRef]
- Thakur, P.; Raizada, P.; Singh, P.; Kumar, A.; Khan, A.A.P.; Asiri, A.M. Exploring recent advances in silver halides and graphitic carbon nitride-based photocatalyst for energy and environmental applications. Arab. J. Chem. 2020, 13, 8271–8300. [Google Scholar] [CrossRef]
- Sharma, S.; Dutta, V.; Raizada, P.; Hosseini-Bandegharaei, A.; Thakur, V.K.; Kalia, S.; Nguyen, V.-H.; Singh, P. Recent advances in silver bromide-based Z-scheme photocatalytic systems for environmental and energy applications: A review. J. Environ. Chem. Eng. 2021, 9, 105157. [Google Scholar] [CrossRef]
- Sambhy, V.; MacBride, M.M.; Peterson, B.R.; Sen, A. Silver bromide nanoparticle/polymer composites: Dual action tunable antimicrobial materials. J. Am. Chem. Soc. 2006, 128, 9798–9808. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater Phys. 1999, 59, 1758–1775. [Google Scholar]
- Hafner, J. Materials simulations using VASP-a quantum perspective to materials science. Comput. Phys. Commun. 2007, 177, 6–13. [Google Scholar] [CrossRef]
- Pizzi, G.; Vitale, V.; Arita, R.; Bluegel, S.; Freimuth, F.; Géranton, G.; Gibertini, M.; Gresch, D.; Johnson, C.; Koretsune, T.; et al. Wannier90 as a community code: New features and applications. J. Physics Condens. Matter 2019, 32, 165902. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.L.; Persson, K.A.; Ceder, G. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; Payne, M.C. Ensemble density-functional theory for ab initio molecular dynamics of metals and finite-temperature insulators. Phys. Rev. Lett. 1997, 79, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Mermin, N.D. Thermal properties of the inhomogeneous electron gas. Phys Rev. 1965, 137, 1–3. [Google Scholar] [CrossRef]
- Boukhtouta, M.; Lamraoui, S.; Touam, S.; Meradji, H.; Ghemid, S.; Hassan, F.E.H. Phase stability and electronic properties of silver halides. Phase Transit. 2015, 88, 357–367. [Google Scholar] [CrossRef]
- Li, T.; Luo, S.; Yang, L. Microwave-assisted solvothermal synthesis of flower-like Ag/AgBr/BiOBr microspheres and their high efficient photocatalytic degradation for p-nitrophenol. J. Solid State Chem. 2013, 206, 308–316. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Ivlev, S.I.; Gaul, K.; Chen, M.; Karttunen, A.J.; Berger, R.; Kraus, F. Synthesis and Characterization of [Br 3][MF 6] (M=Sb, Ir), as well as Quantum Chemical Study of [Br 3] + Structure, Chemical Bonding, and Relativistic Effects Compared with [XBr 2] + (X=Br, I, At, Ts) and [TsZ 2] + (Z=F, Cl, Br, I, At, Ts). Chem. A Eur. J. 2019, 25, 5793–5802. [Google Scholar] [CrossRef]
- Focsa, C.; Li, H.; Bernath, P. Characterization of the Ground State of Br2 by Laser-Induced Fluorescence Fourier Transform Spectroscopy of the B3Π0+u-X1Σ+g System. J. Mol. Spectrosc. 2000, 200, 104–119. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral, L.; Andrés, J.; Longo, E.; San-Miguel, M.A.; da Silva, E.Z. Formation of Metallic Ag on AgBr by Femtosecond Laser Irradiation. Physchem 2022, 2, 179-190. https://doi.org/10.3390/physchem2020013
Cabral L, Andrés J, Longo E, San-Miguel MA, da Silva EZ. Formation of Metallic Ag on AgBr by Femtosecond Laser Irradiation. Physchem. 2022; 2(2):179-190. https://doi.org/10.3390/physchem2020013
Chicago/Turabian StyleCabral, Luís, Juan Andrés, Elson Longo, Miguel A. San-Miguel, and Edison Z. da Silva. 2022. "Formation of Metallic Ag on AgBr by Femtosecond Laser Irradiation" Physchem 2, no. 2: 179-190. https://doi.org/10.3390/physchem2020013