
Mitigation of Cadmium Toxicity through Modulation of the Frontline Cellular Stress Response



Abstract
:1. Introduction
2. Measures of Human Cadmium Exposure
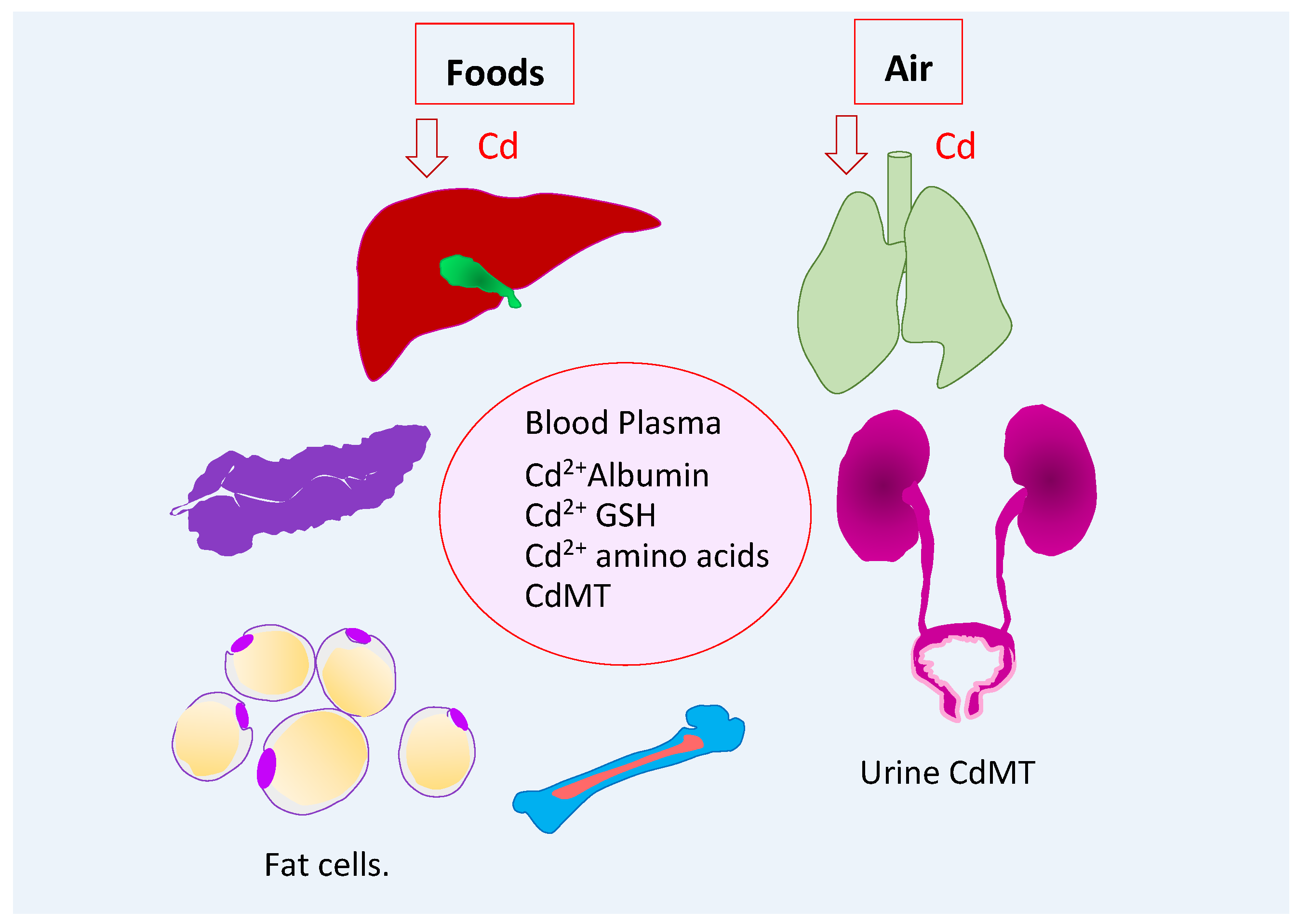
2.1. Entry, Distribuion, and Excretion of Cadmium
2.2. Endogenous Suppliers of Cadmium-Metallothionein Complexes
2.3. Blood Cadmium as an Indicator of Recent Exposure
2.4. Urinary Cadmium as an Indicator of Cumulative Lifetime Exposure
2.5. Roles for Zinc Transporters in the Biliary Excretion and Cytotoxicity of Cadmium
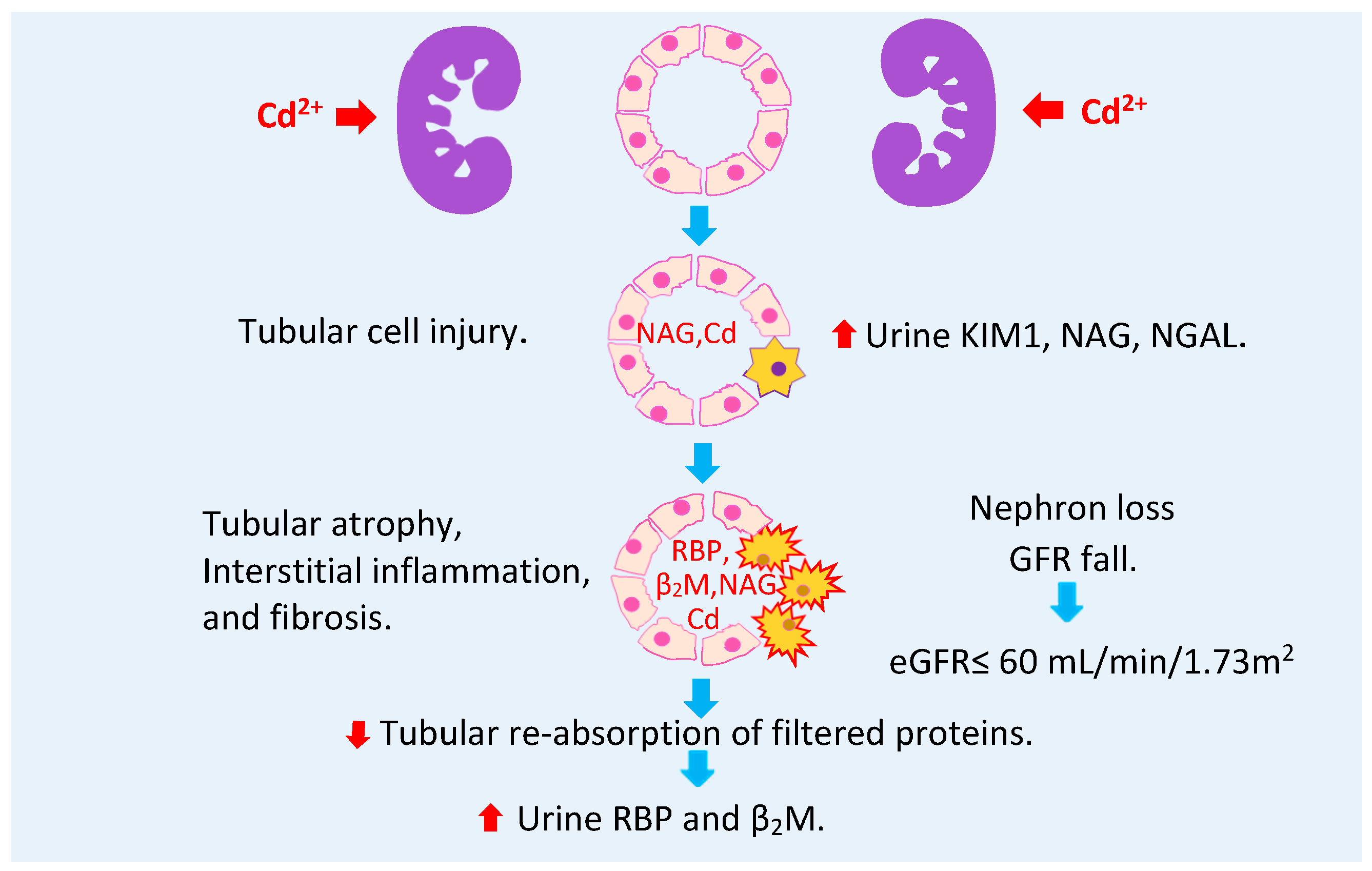
2.6. Urine Cadmium as a Warning Sign of Toxicity in Progress
3. Manifestation of Cadmium Toxicity
3.1. Cadmium and the Risk of Type 2 Diabetes
3.2. An Inverse Relationship between Cadmium Body Burden and Obesity
3.3. Cadmium-Induced Oxidative Stresss and Inflammation
4. Mitigation of the Cytotoxicity of Cadmium
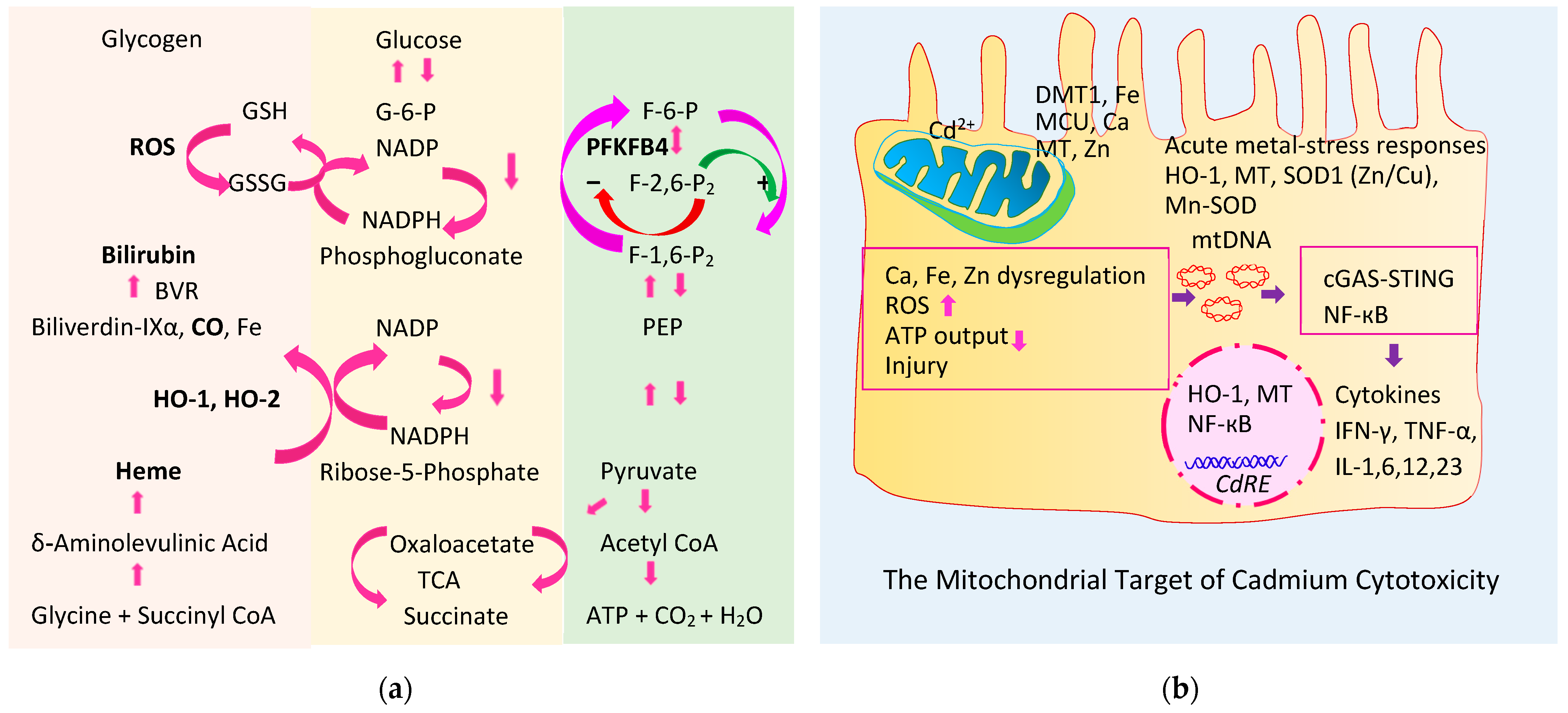
4.1. Heme Oxygenase-1 and Heme Oxygenase-2 (HO-1, HO-2)
4.2. Products of the Physiologic Heme Degrdation
4.2.1. Bilirubin
4.2.2. Carbon Monoxide
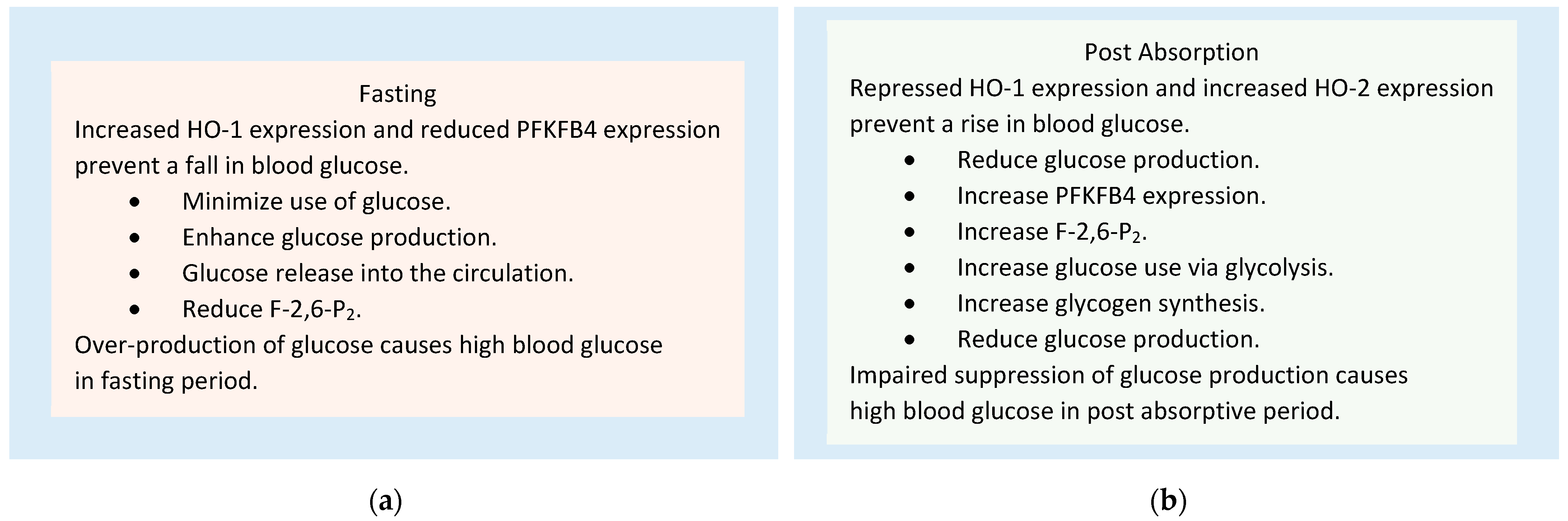
4.3. Role of HO-1, HO-2, and PFKFB4 in the Homeostasis of Blood Glucose
4.4. Exogenous HO-1 Inducers
5. Different HO-1 Gene Activation Mechanisms
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Järup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.G. Natural sources of metals to the environment. Hum. Ecologic. Risk Assess. 2010, 6, 945–963. [Google Scholar] [CrossRef]
- Verbeeck, M.; Salaets, P.; Smolders, E. Trace element concentrations in mineral phosphate fertilizers used in Europe: A balanced survey. Sci. Total Environ. 2020, 712, 136419. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Zhou, S.; Zhou, Y.; Jia, Z.; Guo, T.; Wang, J. Cadmium pollution of soil-rice ecosystems in rice cultivation dominated regions in China: A review. Environ. Pollut. 2021, 280, 116965. [Google Scholar] [CrossRef] [PubMed]
- McDowell, R.W.; Gray, C.W. Do soil cadmium concentrations decline after phosphate fertiliser application is stopped: A comparison of long-term pasture trials in New Zealand? Sci. Total Environ. 2022, 804, 150047. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Y.; Li, Y.; Zhao, H.; Wang, X.; Shen, Y.; Kuang, X. Correlation between environmental low-dose cadmium exposure and early kidney damage: A comparative study in an industrial zone vs. a living quarter in Shanghai, China. Environ. Toxicol. Pharmacol. 2020, 79, 103381. [Google Scholar] [CrossRef]
- Jung, M.S.; Kim, J.Y.; Lee, H.S.; Lee, C.G.; Song, H.S. Air pollution and urinary N-acetyl-β-glucosaminidase levels in residents living near a cement plant. Ann. Occup. Environ. Med. 2016, 28, 52. [Google Scholar] [CrossRef]
- Wu, S.; Deng, F.; Hao, Y.; Shima, M.; Wang, X.; Zheng, C.; Wei, H.; Lv, H.; Lu, X.; Huang, J.; et al. Chemical constituents of fine particulate air pollution and pulmonary function in healthy adults: The Healthy Volunteer Natural Relocation study. J. Hazard. Mater. 2013, 260, 183–191. [Google Scholar] [CrossRef]
- Świetlik, R.; Trojanowska, M. Chemical fractionation in environmental studies of potentially toxic particulate-bound elements in urban air: A critical review. Toxics 2022, 10, 124. [Google Scholar] [CrossRef]
- Repić, A.; Bulat, P.; Antonijević, B.; Antunović, M.; Džudović, J.; Buha, A.; Bulat, Z. The influence of smoking habits on cadmium and lead blood levels in the Serbian adult people. Environ. Sci. Pollut. Res. Int. 2020, 27, 751–760. [Google Scholar] [CrossRef]
- Pappas, R.S.; Fresquez, M.R.; Watson, C.H. Cigarette smoke cadmium breakthrough from traditional filters: Implications for exposure. J. Anal. Toxicol. 2015, 39, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sunderman, F.W., Jr. Nasal toxicity, carcinogenicity, and olfactory uptake of metals. Ann. Clin. Lab. Sci. 2001, 31, 3–24. [Google Scholar] [PubMed]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Mello, T.; Becatti, M.; Pazzagli, L.; Colzi, I.; Gonnelli, C.; Carrino, D.; Paternostro, F.; et al. Effects of cadmium on ZO-1 tight junction integrity of the blood brain barrier. Int. J. Mol. Sci. 2019, 20, 6010. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-induced oxidative stress: Focus on the central nervous system. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Gobe, G.C. Current health risk assessment practice for dietary cadmium: Data from different countries. Food Chem. Toxicol. 2017, 106, 430–445. [Google Scholar] [CrossRef]
- Satarug, S.; Gobe, G.C.; Vesey, D.A.; Phelps, K.R. Cadmium and lead exposure, nephrotoxicity, and mortality. Toxics 2020, 8, 86. [Google Scholar] [CrossRef]
- Satarug, S.; Phelps, K.R. Cadmium Exposure and Toxicity. In Metal Toxicology Handbook; Bagchi, D., Bagchi, M., Eds.; CRC Press: Boca Raton, FL, USA, 2021; pp. 219–274. [Google Scholar]
- Arnich, N.; Sirot, V.; Rivière, G.; Jean, J.; Noël, L.; Guérin, T.; Leblanc, J.-C. Dietary exposure to trace elements and health risk assessment in the 2nd French total diet study. Food Chem. Toxicol. 2012, 50, 2432–2449. [Google Scholar] [CrossRef]
- Sand, S.; Becker, W. Assessment of dietary cadmium exposure in Sweden and population health concern including scenario analysis. Food Chem. Toxicol. 2012, 50, 536–544. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Sabolić, I.; Breljak, D.; Skarica, M.; Herak-Kramberger, C.M. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 2010, 23, 897–926. [Google Scholar] [CrossRef]
- McKenna, I.M.; Gordon, T.; Chen, L.C.; Anver, M.R.; Waalkes, M.P. Expression of metallothionein protein in the lungs of Wistar rats and C57 and DBA mice exposed to cadmium oxide fumes. Toxicol. Appl. Pharmacol. 1998, 153, 169–178. [Google Scholar] [CrossRef]
- Echeverría, R.; Vrhovnik, P.; Salcedo-Bellido, I.; Iribarne-Durán, L.M.; Fiket, Ž.; Dolenec, M.; Martin-Olmedo, P.; Olea, N.; Arrebola, J.P. Levels and determinants of adipose tissue cadmium concentrations in an adult cohort from Southern Spain. Sci. Total Environ. 2019, 670, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Bellido, I.; Gómez-Peña, C.; Pérez-Carrascosa, F.M.; Vrhovnik, P.; Mustieles, V.; Echeverría, R.; Fiket, Ž.; Pérez-Díaz, C.; Barrios-Rodríguez, R.; Jiménez-Moleón, J.J.; et al. Adipose tissue cadmium concentrations as a potential risk factor for insulin resistance and future type 2 diabetes mellitus in GraMo adult cohort. Sci. Total Environ. 2021, 780, 146359. [Google Scholar] [CrossRef] [PubMed]
- El Muayed, M.; Raja, M.R.; Zhang, X.; MacRenaris, K.W.; Bhatt, S.; Chen, X.; Urbanek, M.; O’Halloran, T.V.; Lowe, W.L., Jr. Accumulation of cadmium in insulin-producing β cells. Islets 2012, 4, 405–416. [Google Scholar] [CrossRef]
- Satarug, S.; Baker, J.R.; Reilly, P.E.; Moore, M.R.; Williams, D.J. Cadmium levels in the lung, liver, kidney cortex, and urine samples from Australians without occupational exposure to metals. Arch. Environ. Health 2002, 57, 69–77. [Google Scholar] [CrossRef]
- Christensen, E.I.; Birn, H.; Storm, T.; Weyer, K.; Nielsen, R. Endocytic receptors in the renal proximal tubule. Physiology 2012, 27, 223–236. [Google Scholar] [CrossRef]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef]
- Krężel, A.; Maret, W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef]
- Krężel, A.; Maret, W. The bioinorganic chemistry of mammalian metallothioneins. Chem. Rev. 2021, 121, 14594–14648. [Google Scholar] [CrossRef] [PubMed]
- Boonprasert, K.; Ruengweerayut, R.; Aunpad, R.; Satarug, S.; Na-Bangchang, K. Expression of metallothionein isoforms in peripheral blood leukocytes from Thai population residing in cadmium-contaminated areas. Environ. Toxicol. Pharmacol. 2012, 34, 935–940. [Google Scholar] [CrossRef]
- Boonprasert, K.; Satarug, S.; Morais, C.; Gobe, G.C.; Johnson, D.W.; Na-Bangchang, K.; Vesey, D.A. The stress response of human proximal tubule cells to cadmium involves up-regulation of haemoxygenase 1 and metallothionein but not cytochrome P450 enzymes. Toxicol. Lett. 2016, 249, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Hennigar, S.R.; Kelley, A.M.; McClung, J.P. Metallothionein and zinc transporter expression in circulating human blood cells as biomarkers of zinc status: A systematic review. Adv. Nutr. 2016, 7, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.H.; Sens, M.A.; Todd, J.H.; Somji, S.; Sens, D.A. Expression of MT-3 protein in the human kidney. Toxicol. Lett. 1999, 105, 207–214. [Google Scholar] [CrossRef]
- Vašák, M.; Meloni, G. Mammalian metallothionein-3: New functional and structural insights. Int. J. Mol. Sci. 2017, 18, 1117. [Google Scholar] [CrossRef]
- Sabolić, I.; Škarica, M.; Ljubojević, M.; Breljak, D.; Herak-Kramberger, C.M.; Crljen, V.; Ljubešić, N. Expression and immunolocalization of metallothioneins MT1, MT2 and MT3 in rat nephron. J. Trace Elem. Med. Biol. 2018, 46, 62–75. [Google Scholar] [CrossRef]
- Misra, R.R.; Hochadel, J.F.; Smith, G.T.; Cook, J.C.; Waalkes, M.P.; Wink, D.A. Evidence that nitric oxide enhances cadmium toxicity by displacing the metal from metallothionein. Chem. Res. Toxicol. 1996, 9, 326–332. [Google Scholar] [CrossRef]
- Satarug, S.; Baker, J.R.; Reilly, P.E.; Esumi, H.; Moore, M.R. Evidence for a synergistic interaction between cadmium and endotoxin toxicity and for nitric oxide and cadmium displacement of metals in the kidney. Nitric Oxide 2000, 4, 431–440. [Google Scholar] [CrossRef]
- Zhu, J.; Meeusen, J.; Krezoski, S.; Petering, D.H. Reactivity of Zn-, Cd-, and apo-metallothionein with nitric oxide compounds: In vitro and cellular comparison. Chem. Res. Toxicol. 2010, 23, 422–431. [Google Scholar] [CrossRef]
- Lou, M.; Garay, R.; Alda, J.O. Cadmium uptake through the anion exchanger in human red blood cells. J. Physiol. 1991, 443, 123–136. [Google Scholar] [CrossRef]
- Horn, N.M.; Thomas, A.L. Interactions between the histidine stimulation of cadmium and zinc influx into human erythrocytes. J. Physiol. 1996, 496, 711–718. [Google Scholar] [CrossRef]
- Sagmeister, P.; Gibson, M.A.; McDade, K.H.; Gailer, J. Physiologically relevant plasma d,l-homocysteine concentrations mobilize Cd from human serum albumin. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1027, 181–186. [Google Scholar] [CrossRef]
- Scott, B.J.; Bradwell, A.R. Identification of the serum binding proteins for iron, zinc, cadmium, nickel, and calcium. Clin. Chem. 1983, 29, 629–633. [Google Scholar] [CrossRef]
- Järup, L.; Rogenfelt, A.; Elinder, C.G.; Nogawa, K.; Kjellström, T. Biological half-time of cadmium in the blood of workers after cessation of exposure. Scand. J. Work Environ. Health 1983, 9, 327–331. [Google Scholar] [CrossRef]
- Elinder, C.G.; Lind, B.; Kjellström, T.; Linnman, L.; Friberg, L. Cadmium in kidney cortex, liver, and pancreas from Swedish autopsies. Estimation of biological half time in kidney cortex, considering calorie intake and smoking habits. Arch. Environ. Health 1976, 31, 292–302. [Google Scholar] [CrossRef]
- Elinder, C.G.; Kjellstöm, T.; Lind, B.; Molander, M.L.; Silander, T. Cadmium concentrations in human liver, blood, and bile: Comparison with a metabolic model. Environ. Res. 1978, 17, 236–241. [Google Scholar] [CrossRef]
- Barregard, L.; Fabricius-Lagging, E.; Lundh, T.; Mölne, J.; Wallin, M.; Olausson, M.; Modigh, C.; Sallsten, G. Cadmium, mercury, and lead in kidney cortex of living kidney donors: Impact of different exposure sources. Environ. Res. 2010, 110, 47–54. [Google Scholar] [CrossRef]
- Akerstrom, M.; Barregard, L.; Lundh, T.; Sallsten, G. The relationship between cadmium in kidney and cadmium in urine and blood in an environmentally exposed population. Toxicol. Appl. Pharmacol. 2013, 268, 286–293. [Google Scholar] [CrossRef]
- Sun, H.; Wang, D.; Zhou, Z.; Ding, Z.; Chen, X.; Xu, Y.; Huang, L.; Tang, D. Association of cadmium in urine and blood with age in a general population with low environmental exposure. Chemosphere 2016, 156, 392–397. [Google Scholar] [CrossRef]
- Graf, P.; Sies, H. Hepatic uptake of cadmium and its biliary release as affected by dithioerythritol and glutathione. Biochem. Pharmacol. 1984, 33, 639–643. [Google Scholar] [CrossRef]
- Fujishiro, H.; Himeno, S. New insights into the roles of ZIP8, a cadmium and manganese transporter, and its relation to human diseases. Biol. Pharm. Bull. 2019, 42, 1076–1082. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.L.; Tan, H.W.; Wu, J.Y.; Chen, X.L.; Wang, X.Y.; Xu, Y.M.; Lau, A.T.Y. The impact of ZIP8 disease-associated variants G38R, C113S, G204C, and S335T on selenium and cadmium accumulations: The first characterization. Int. J. Mol. Sci. 2021, 22, 11399. [Google Scholar] [CrossRef]
- Aiba, I.; Hossain, A.; Kuo, M.T. Elevated GSH level increases cadmium resistance through down-regulation of Sp1-dependent expression of the cadmium transporter ZIP8. Mol. Pharmacol. 2008, 74, 823–833. [Google Scholar] [CrossRef]
- Ren, L.; Qi, K.; Zhang, L.; Bai, Z.; Ren, C.; Xu, X.; Zhang, Z.; Li, X. Glutathione might attenuate cadmium-induced liver oxidative stress and hepatic stellate cell activation. Biol. Trace Elem. Res. 2019, 191, 443–452. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C. The evolving role for zinc and zinc transporters in cadmium tolerance and urothelial cancer. Stresses 2021, 1, 105–118. [Google Scholar] [CrossRef]
- Satarug, S.; Garrett, S.H.; Somji, S.; Sens, M.A.; Sens, D.A. Zinc, zinc transporters, and cadmium cytotoxicity in a cell culture model of human urothelium. Toxics 2021, 9, 94. [Google Scholar] [CrossRef]
- Satarug, S.; Garrett, S.H.; Somji, S.; Sens, M.A.; Sens, D.A. Aberrant expression of ZIP and ZnT zinc transporters in UROtsa cells transformed to malignant cells by cadmium. Stresses 2021, 1, 78–89. [Google Scholar] [CrossRef]
- Takiguchi, M.; Cherrington, N.J.; Hartley, D.P.; Klaassen, C.D.; Waalkes, M.P. Cyproterone acetate induces a cellular tolerance to cadmium in rat liver epithelial cells involving reduced cadmium accumulation. Toxicology 2001, 165, 13–25. [Google Scholar] [CrossRef]
- Ohana, E.; Sekler, I.; Kaisman, T.; Kahn, N.; Cove, J.; Silverman, W.F.; Amsterdam, A.; Hershfinkel, M. Silencing of ZnT-1 expression enhances heavy metal influx and toxicity. J. Mol. Med. 2006, 84, 753–763. [Google Scholar] [CrossRef]
- Fujishiro, H.; Doi, M.; Enomoto, S.; Himeno, S. High sensitivity of RBL-2H3 cells to cadmium and manganese: An implication of the role of ZIP8. Metallomics 2011, 3, 710–718. [Google Scholar] [CrossRef]
- Fujishiro, H.; Ohashi, T.; Takuma, M.; Himeno, S. Suppression of ZIP8 expression is a common feature of cadmium-resistant and manganese-resistant RBL-2H3 cells. Metallomics 2013, 5, 437–444. [Google Scholar] [CrossRef]
- Nordberg, M.; Nordberg, G.F. Metallothionein and cadmium toxicology-historical review and commentary. Biomolecules 2022, 12, 360. [Google Scholar] [CrossRef]
- Wolf, C.; Strenziok, R.; Kyriakopoulos, A. Elevated metallothionein-bound cadmium concentrations in urine from bladder carcinoma patients, investigated by size exclusion chromatography-inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2009, 631, 218–222. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Ruangyuttikarn, W.; Nishijo, M.; Gobe, G.C.; Phelps, K.R. The source and pathophysiologic significance of excreted cadmium. Toxics 2019, 7, 55. [Google Scholar] [CrossRef]
- Akesson, A.; Lundh, T.; Vahter, M.; Bjellerup, P.; Lidfeldt, J.; Nerbrand, C.; Samsioe, G.; Strömberg, U.; Skerfving, S. Tubular and glomerular kidney effects in Swedish women with low environmental cadmium exposure. Environ. Health Perspect. 2005, 113, 1627–1631. [Google Scholar] [CrossRef]
- Barregard, L.; Sallsten, G.; Lundh, T.; Mölne, J. Low-level exposure to lead, cadmium and mercury, and histopathological findings in kidney biopsies. Environ. Res. 2022, 211, 113119. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kido, T.; Nakagawa, H.; Nishijo, M.; Honda, R.; Kobayashi, E.; Dochi, M.; Nogawa, K. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 2009, 14, 77–81. [Google Scholar] [CrossRef]
- Ishizaki, M.; Suwazono, Y.; Kido, T.; Nishijo, M.; Honda, R.; Kobayashi, E.; Nogawa, K.; Nakagawa, H. Estimation of biological half-life of urinary cadmium in inhabitants after cessation of environmental cadmium pollution using a mixed linear model. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 1273–1276. [Google Scholar] [CrossRef]
- Fransson, M.N.; Barregard, L.; Sallsten, G.; Akerstrom, M.; Johanson, G. Physiologically-based toxicokinetic model for cadmium using Markov-chain Monte Carlo analysis of concentrations in blood, urine, and kidney cortex from living kidney donors. Toxicol. Sci. 2014, 141, 365–376. [Google Scholar] [CrossRef]
- Hyder, O.; Chung, M.; Cosgrove, D.; Herman, J.M.; Li, Z.; Firoozmand, A.; Gurakar, A.; Koteish, A.; Pawlik, T.M. Cadmium exposure and liver disease among US adults. J. Gastrointest. Surg. 2013, 17, 1265–1273. [Google Scholar] [CrossRef]
- Hong, D.; Min, J.Y.; Min, K.B. Association between cadmium exposure and liver function in adults in the United States: A Cross-sectional study. J. Prev. Med. Public Health 2021, 54, 471–480. [Google Scholar] [CrossRef]
- Xu, Z.; Weng, Z.; Liang, J.; Liu, Q.; Zhang, X.; Xu, J.; Xu, C.; Gu, A. Association between urinary cadmium concentrations and liver function in adolescents. Environ. Sci. Pollut. Res. Int. 2022, 29, 39768–39776. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Il’yasova, D.; Ivanova, A. Urinary cadmium, impaired fasting glucose, and diabetes in the NHANES III. Diabetes Care 2003, 26, 468–470. [Google Scholar] [CrossRef]
- Wallia, A.; Allen, N.B.; Badon, S.; El Muayed, M. Association between urinary cadmium levels and prediabetes in the NHANES 2005-2010 population. Int. J. Hyg. Environ. Health 2014, 217, 854–860. [Google Scholar] [CrossRef]
- Shi, P.; Yan, H.; Fan, X.; Xi, S. A benchmark dose analysis for urinary cadmium and type 2 diabetes mellitus. Environ. Pollut. 2021, 273, 116519. [Google Scholar] [CrossRef]
- Navas-Acien, A.; Tellez-Plaza, M.; Guallar, E.; Muntner, P.; Silbergeld, E.; Jaar, B.; Weaver, V. Blood cadmium and lead and chronic kidney disease in US adults: A joint analysis. Am. J. Epidemiol. 2009, 170, 1156–1164. [Google Scholar] [CrossRef]
- Ferraro, P.M.; Costanzi, S.; Naticchia, A.; Sturniolo, A.; Gambaro, G. Low level exposure to cadmium increases the risk of chronic kidney disease: Analysis of the NHANES 1999–2006. BMC Public Health 2010, 10, 304. [Google Scholar] [CrossRef]
- Madrigal, J.M.; Ricardo, A.C.; Persky, V.; Turyk, M. Associations between blood cadmium concentration and kidney function in the U.S. population: Impact of sex, diabetes and hypertension. Environ. Res. 2018, 169, 180–188. [Google Scholar] [CrossRef]
- Zhu, X.J.; Wang, J.J.; Mao, J.H.; Shu, Q.; Du, L.Z. Relationships of cadmium, lead, and mercury levels with albuminuria in US Adults: Results from the National Health and Nutrition Examination Survey Database, 2009–2012. Am. J. Epidemiol. 2019, 188, 1281–1287. [Google Scholar] [CrossRef]
- Lin, Y.S.; Ho, W.C.; Caffrey, J.L.; Sonawane, B. Low serum zinc is associated with elevated risk of cadmium nephrotoxicity. Environ. Res. 2014, 134, 33–38. [Google Scholar] [CrossRef]
- Stumvoll, M.; Meyer, C.; Mitrakou, A.; Nadkarni, V.; Gerich, J.E. Renal glucose production and utilization: New aspects in humans. Diabetologia 1997, 40, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Gerich, J.E. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: Therapeutic implications. Diabet. Med. 2010, 27, 136–142. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Davidson, J.A.; Prato, S.D. The role of the kidneys in glucose homeostasis: A new path towards normalizing glycaemia. Diabetes Obes. Metab. 2012, 14, 5–14. [Google Scholar] [CrossRef]
- Xiao, L.; Li, W.; Zhu, C.; Yang, S.; Zhou, M.; Wang, B.; Wang, X.; Wang, D.; Ma, J.; Zhou, Y.; et al. Cadmium exposure, fasting blood glucose changes, and type 2 diabetes mellitus: A longitudinal prospective study in China. Environ. Res. 2021, 192, 110259. [Google Scholar] [CrossRef]
- Guo, F.F.; Hu, Z.Y.; Li, B.Y.; Qin, L.Q.; Fu, C.; Yu, H.; Zhang, Z.L. Evaluation of the association between urinary cadmium levels below threshold limits and the risk of diabetes mellitus: A dose-response meta-analysis. Environ. Sci. Pollut. Res. Int. 2019, 26, 19272–19281. [Google Scholar] [CrossRef]
- Filippini, T.; Wise, L.A.; Vinceti, M. Cadmium exposure and risk of diabetes and prediabetes: A systematic review and dose-response meta-analysis. Environ. Int. 2022, 158, 106920. [Google Scholar] [CrossRef]
- Padilla, M.A.; Elobeid, M.; Ruden, D.M.; Allison, D.B. An examination of the association of selected toxic metals with total and central obesity indices: NHANES 99-02. Int. J. Environ. Res. Public Health 2010, 7, 3332–3347. [Google Scholar] [CrossRef]
- Jain, R.B. Effect of pregnancy on the levels of blood cadmium, lead, and mercury for females aged 17-39 years old: Data from National Health and Nutrition Examination Survey 2003–2010. J. Toxicol. Environ. Health A 2013, 76, 58–69. [Google Scholar] [CrossRef]
- Noor, N.; Zong, G.; Seely, E.W.; Weisskopf, M.; James-Todd, T. Urinary cadmium concentrations and metabolic syndrome in U.S. adults: The National Health and Nutrition Examination Survey 2001–2014. Environ Int. 2018, 21, 349–356. [Google Scholar] [CrossRef]
- Wang, X.; Mukherjee, B.; Karvonen-Gutierrez, C.A.; Herman, W.H.; Batterman, S.; Harlow, S.D.; Park, S.K. Urinary metal mixtures and longitudinal changes in glucose homeostasis: The Study of Women’s Health Across the Nation (SWAN). Environ. Int. 2020, 145, 106109. [Google Scholar] [CrossRef]
- Shao, W.; Liu, Q.; He, X.; Liu, H.; Gu, A.; Jiang, Z. Association between level of urinary trace heavy metals and obesity among children aged 6-19 years: NHANES 1999–2011. Environ. Sci. Pollut. Res. Int. 2017, 24, 11573–11581. [Google Scholar] [CrossRef]
- Dhooge, W.; Den Hond, E.; Koppen, G.; Bruckers, L.; Nelen, V.; Van De Mieroop, E.; Bilau, M.; Croes, K.; Baeyens, W.; Schoeters, G.; et al. Internal exposure to pollutants and body size in Flemish adolescents and adults: Associations and dose-response relationships. Environ. Int. 2010, 36, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Garner, R.; Levallois, P. Cadmium levels and sources of exposure among Canadian adults. Health Rep. 2016, 27, 10–18. [Google Scholar] [PubMed]
- Akbar, L.; Zuk, A.M.; Martin, I.D.; Liberda, E.N.; Tsuji, L.J.S. Potential obesogenic effect of a complex contaminant mixture on Cree First Nations adults of Northern Québec, Canada. Environ. Res. 2021, 192, 110478. [Google Scholar] [CrossRef]
- Son, H.S.; Kim, S.G.; Suh, B.S.; Park, D.U.; Kim, D.S.; Yu, S.D.; Hong, Y.S.; Park, J.D.; Lee, B.K.; Moon, J.D.; et al. Association of cadmium with diabetes in middle-aged residents of abandoned metal mines: The first health effect surveillance for residents in abandoned metal mines. Ann. Occup. Environ. Med. 2015, 27, 20. [Google Scholar] [CrossRef]
- Nie, X.; Wang, N.; Chen, Y.; Chen, C.; Han, B.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Lu, M.; et al. Blood cadmium in Chinese adults and its relationships with diabetes and obesity. Environ. Sci Pollut. Res. Int. 2016, 23, 18714–18723. [Google Scholar] [CrossRef]
- Feng, X.; Zhou, R.; Jiang, Q.; Wang, Y.; Yu, C. Analysis of cadmium accumulation in community adults and its correlation with low-grade albuminuria. Sci. Total Environ. 2022, 834, 155210. [Google Scholar] [CrossRef]
- Gasser, M.; Lenglet, S.; Bararpour, N.; Sajic, T.; Wiskott, K.; Augsburger, M.; Fracasso, T.; Gilardi, F.; Thomas, A. Cadmium acute exposure induces metabolic and transcriptomic perturbations in human mature adipocytes. Toxicology 2022, 470, 153153. [Google Scholar] [CrossRef]
- Kawakami, T.; Sugimoto, H.; Furuichi, R.; Kadota, Y.; Inoue, M.; Setsu, K.; Suzuki, S.; Sato, M. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology 2010, 267, 20–26. [Google Scholar] [CrossRef]
- Kawakami, T.; Nishiyama, K.; Kadota, Y.; Sato, M.; Inoue, M.; Suzuki, S. Cadmium modulates adipocyte functions in metallothionein-null mice. Toxicol. Appl. Pharmacol. 2013, 272, 625–636. [Google Scholar] [CrossRef]
- Lee, D.H.; Lim, J.S.; Song, K.; Boo, Y.; Jacobs, D.R., Jr. Graded associations of blood lead and urinary cadmium concentrations with oxidative-stress-related markers in the U.S. population: Results from the third National Health and Nutrition Examination Survey. Environ. Health Perspect. 2006, 114, 350–354. [Google Scholar] [CrossRef]
- Lin, Y.S.; Rathod, D.; Ho, W.C.; Caffrey, J.L. Cadmium exposure is associated with elevated blood C-reactive protein and fibrinogen in the U.S. population: The third national health and nutrition examination survey (NHANES III, 1988–1994). Ann. Epidemiol. 2009, 19, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Pollack, A.Z.; Mumford, S.L.; Mendola, P.; Perkins, N.J.; Rotman, Y.; Wactawski-Wende, J.; Schisterman, E.F. Kidney biomarkers associated with blood lead, mercury, and cadmium in premenopausal women: A prospective cohort study. J. Toxicol. Environ. Health A 2015, 78, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Colacino, J.A.; Arthur, A.E.; Ferguson, K.K.; Rozek, L.S. Dietary antioxidant and anti-inflammatory intake modifies the effect of cadmium exposure on markers of systemic inflammation and oxidative stress. Environ. Res. 2014, 131, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Zota, A.R.; Needham, B.L.; Blackburn, E.H.; Lin, J.; Park, S.K.; Rehkopf, D.H.; Epe, E.S. Associations of cadmium and lead exposure with leukocyte telomere length: Findings from National Health and Nutrition Examination Survey, 1999–2002. Am. J. Epidemiol. 2014, 181, 127–136. [Google Scholar] [CrossRef]
- Patel, C.J.; Manrai, A.K.; Corona, E.; Kohane, I.S. Systematic correlation of environmental exposure and physiological and self-reported behaviour factors with leukocyte telomere length. Int. J. Epidemiol. 2017, 46, 44–56. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, J.; Xu, C.; Da, M.; Xu, Y.; Chen, Y.; Mo, X. Increased serum levels of cadmium are associated with an elevated risk of cardiovascular disease in adults. Environ. Sci. Pollut. Res. Int. 2022, 29, 1836–1844. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, D.; Shi, F.; Wang, F.; Liu, X.; Wen, H.; Mubarik, S.; Yu, C. Association of serum 25(OH)D, cadmium, CRP with all-cause, cause-specific mortality: A prospective cohort study. Front. Nutr. 2022, 9, 803985. [Google Scholar] [CrossRef]
- Shibahara, S. The heme oxygenase dilemma in cellular homeostasis: New insights for the feedback regulation of heme catabolism. Tohoku J. Exp. Med. 2003, 200, 167–186. [Google Scholar] [CrossRef]
- Shibahara, S.; Han, F.; Li, B.; Takeda, K. Hypoxia and heme oxygenases: Oxygen sensing and regulation of expression. Antiox. Redox Signal. 2007, 9, 2209–2225. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on heme oxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Quigley, J.G. Control of intracellular heme levels: Heme transporters and heme oxygenases. Biochim. Biophys. Acta 2011, 1813, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ishizawa, S.; Sato, M.; Yoshida, T.; Shibahara, S. Identification of a cis-acting element that is responsible for cadmium-mediated induction of the human heme oxygenase gene. J. Biol. Chem. 1994, 269, 22858–22867. [Google Scholar] [CrossRef]
- Zhang, F.; Guan, W.; Fu, Z.; Zhou, L.; Guo, W.; Ma, Y.; Gong, Y.; Jiang, W.; Liang, H.; Zhou, H. Relationship between serum indirect bilirubin level and insulin sensitivity: Results from two independent cohorts of obese patients with impaired glucose regulation and type 2 diabetes mellitus in China. Int. J. Endocrinol. 2020, 2020, 5681296. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.P.; Vitek, L.; Schwertner, H.A. Serum bilirubin and genes controlling bilirubin concentrations as biomarkers for cardiovascular disease. Clin. Chem. 2010, 56, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting heme oxygenase-1 in the arterial response to injury and disease. Antioxidants 2020, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Yu, Z.; Bai, L.; Hou, W.; Tang, S.; Zhang, W.; Chen, X.; Hu, Z.; Duan, Z.; Zheng, S. Association of serum bilirubin with metabolic syndrome and non-alcoholic fatty liver disease: A systematic review and meta-analysis. Front. Endocrinol. (Lausanne) 2022, 13, 869579. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.A.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Carbon monoxide: A critical quantitative analysis and review of the extent and limitations of its second messenger function. Clin. Pharmacol. 2015, 7, 37–56. [Google Scholar] [CrossRef]
- Stuckim, D.; Steinhausen, J.; Westhoff, P.; Krahl, H.; Brilhaus, D.; Massenberg, A.; Weber, A.P.M.; Reichert, A.S.; Brenneisen, P.; Stahl, W. Endogenous carbon monoxide signaling modulates mitochondrial function and intracellular glucose utilization: Impact of the heme oxygenase substrate hemin. Antioxidants 2020, 9, 652. [Google Scholar] [CrossRef]
- Stucki, D.; Stahl, W. Carbon monoxide—Beyond toxicity? Toxicol. Lett. 2020, 333, 251–260. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox. Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef]
- Shibahara, S.; Yoshizawa, M.; Suzuki, H.; Takeda, K.; Meguro, K.; Endo, K. Functional analysis of cDNAs for two types of human heme oxygenase and evidence for their separate regulation. J. Biochem. (Tokyo) 1993, 113, 214–218. [Google Scholar] [CrossRef]
- Bao, W.; Song, F.; Li, X.; Rong, S.; Yang, W.; Wang, D.; Xu, J.; Fu, J.; Zhao, Y.; Liu, L. Association between heme oxygenase-1 gene promoter polymorphisms and type 2 diabetes mellitus: A HuGE review and meta-analysis. Am. J. Epidemiol. 2010, 172, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.L.; Sun, L.; Wang, Y.X.; Sun, B.H.; Li, Y.F.; Jin, Y.L. Association between HO-1 gene promoter polymorphisms and diseases (Review). Mol. Med. Rep. 2022, 25, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, B.; Emmett, M.J.; Damle, M.; Sun, Z.; Feng, D.; Armour, S.M.; Remsberg, J.R.; Jager, J.; Soccio, R.E.; et al. Discrete functions of nuclear receptor Rev-erbα couple metabolism to the clock. Science 2015, 348, 1488–1492. [Google Scholar] [CrossRef]
- Everett, L.J.; Lazar, M.A. Nuclear receptor Rev-erbα: Up, down, and all around. Trends Endocrinol. Metab. 2014, 25, 586–592. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Medina, M.V.; Sapochnik, D.; Garcia Solá, M.; Coso, O. Regulation of the expression of heme oxygenase-1: Signal transduction, gene promoter activation, and beyond. Antioxid. Redox Signal. 2020, 32, 1033–1044. [Google Scholar] [CrossRef]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yin, L.; Hanniman, E.A.; Joshi, S.; Lazar, M.A. Negative feedback maintenance of heme homeostasis by its receptor, Rev-erb alpha. Genes Dev. 2009, 23, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Watanabe-Matsui, M. Wearing red for signaling: The Heme-Bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J. Exp. Med. 2014, 232, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Hanna, D.A.; Moore, C.M.; Liu, L.; Yuan, X.; Dominic, I.M.; Fleischhacker, A.S.; Hamza, I.; Ragsdale, S.W.; Reddi, A.R. Heme oxygenase-2 (HO-2) binds and buffers labile ferric heme in human embryonic kidney cells. J. Biol. Chem. 2022, 298, 101549. [Google Scholar] [CrossRef]
- Fleischhacker, A.S.; Carter, E.L.; Ragsdale, S.W. Redox regulation of heme oxygenase-2 and the transcription factor, Rev-Erb, through heme regulatory motifs. Antioxid. Redox Signal. 2018, 29, 1841–1857. [Google Scholar] [CrossRef]
- Fleischhacker, A.S.; Gunawan, A.L.; Kochert, B.A.; Liu, L.; Wales, T.E.; Borowy, M.C.; Engen, J.R.; Ragsdale, S.W. The heme-regulatory motifs of heme oxygenase-2 contribute to the transfer of heme to the catalytic site for degradation. J. Biol. Chem. 2020, 295, 5177–5191. [Google Scholar] [CrossRef]
- Sodhi, K.; Inoue, K.; Gotlinger, K.H.; Canestraro, M.; Vanella, L.; Kim, D.H.; Manthati, V.L.; Koduru, S.R.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J. Pharmacol. Exp. Ther. 2009, 331, 906–916. [Google Scholar] [CrossRef]
- Yao, H.; Peterson, A.L.; Li, J.; Xu, H.; Dennery, P.A. Heme oxygenase 1 and 2 differentially regulate glucose metabolism and adipose tissue mitochondrial respiration: Implications for metabolic dysregulation. Int. J. Mol. Sci. 2020, 21, 7123. [Google Scholar] [CrossRef]
- Burgess, A.P.; Vanella, L.; Bellner, L.; Gotlinger, K.; Falck, J.R.; Abraham, N.G.; Schwartzman, M.L.; Kappas, A. Heme oxygenase (HO-1) rescue of adipocyte dysfunction in HO-2 deficient mice via recruitment of epoxyeicosatrienoic acids (EETs) and adiponectin. Cell Physiol. Biochem. 2012, 29, 99–110. [Google Scholar] [CrossRef]
- Li, B.; Takeda, K.; Ishikawa, K.; Yoshizawa, M.; Sato, M.; Shibahara, S.; Furuyama, K. Coordinated expression of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 4 and heme oxygenase 2: Evidence for a regulatory link between glycolysis and heme catabolism. Tohoku J. Exp. Med. 2012, 228, 27–41. [Google Scholar] [CrossRef]
- Okar, D.A.; Manzano, A.; Navarro-Sabatè, A.; Riera, L.; Bartrons, R.; Lange, A.J. PFK-2/FBPase-2: Maker and breaker of the essential biofactor fructose-2, 6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Han, F.; Takeda, K.; Ishikawa, K.; Ono, M.; Date, F.; Yokoyama, S.; Furuyama, K.; Shinozawa, Y.; Urade, Y.; Shibahara, S. Induction of lipocalin-type prostaglandin D synthase in mouse heart under hypoxemia. Biochem. Biophys. Res. Commun. 2009, 385, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K.; Garrick, M.D. Iron and cadmium entry into renal mitochondria: Physiological and toxicological implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- Hahn, D.; Shin, S.H.; Bae, J.S. Natural antioxidant and anti-inflammatory compounds in foodstuff or medicinal herbs inducing heme oxygenase-1 expression. Antioxidants 2020, 9, 1191. [Google Scholar] [CrossRef]
- Stec, D.E.; Hinds, T.D., Jr. Natural product heme oxygenase inducers as treatment for nonalcoholic fatty Liver disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef]
- Keller, A.; Wallace, T.C. Tea intake and cardiovascular disease: An umbrella review. Ann. Med. 2021, 53, 929–944. [Google Scholar] [CrossRef]
- Han, K.C.; Wong, W.C.; Benzie, I.F. Genoprotective effects of green tea (Camellia sinensis) in human subjects: Results of a controlled supplementation trial. Br. J. Nutr. 2011, 105, 171–179. [Google Scholar] [CrossRef]
- Choi, S.W.; Yeung, V.T.F.; Collins, A.R.; Benzie, I.F.F. Redox-linked effects of green tea on DNA damage and repair, and influence of microsatellite polymorphism in HMOX-1: Results of a human intervention trial. Mutagenesis 2015, 30, 129–137. [Google Scholar] [CrossRef]
- Ho, C.K.; Choi, S.W.; Siu, P.M.; Benzie, I.F. Effects of single dose and regular intake of green tea (Camellia sinensis) on DNA damage, DNA repair, and heme oxygenase-1 expression in a randomized controlled human supplementation study. Mol. Nutr. Food Res. 2014, 58, 1379–1383. [Google Scholar] [CrossRef]
- Satarug, S.; Wisedpanichkij, R.; Takeda, K.; Li, B.; Na-Bangchang, K.; Moore, M.R.; Shibahara, S. Prostaglandin D2 induces heme oxygenase-1 mRNA expression through the DP2 receptor. Biochem. Biophys. Res. Commun. 2008, 377, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Spik, I.; Brénuchon, C.; Angéli, V.; Staumont, D.; Fleury, S.; Capron, M.; Trottein, F.; Dombrowicz, D. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J. Immunol. 2005, 174, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tashiro, S.; Sun, J.; Doi, H.; Satomi, S.; Igarashi, K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J. Biol. Chem. 2003, 278, 49246–49253. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organs | NHANES Datasets | Adverse Effects and Risk Estimates | References |
|---|---|---|---|
| Liver | 1988–1994 n 12,732, ≥20 yrs | In women, liver inflammation was associated with urinary Cd levels ≥ 0.83 μg/g creatinine (OR 1.26). In men, liver inflammation, NAFLD and NASH were associated with urinary Cd ≥ 0.65 μg/g creatinine with respective OR values of 2.21, 1.30, and 1.95. | Hyder et al., 2013 [70] |
| Liver | 1999–2015 n 11, 838, ≥20 yrs | Elevated plasma ALT and AST was associated with a 10-fold increment of urinary Cd with respective OR values of 1.36 and 1.31. | Hong et al., 2021 [71] |
| Liver | 1999–2016 n 4411 adolescents | Elevated plasma ALT and AST were associated with urinary Cd quartile 4 with respective OR values of 1.40 and 1.64. The effect was larger in boys than girls. | Xu et al., 2022 [72] |
| Pancreas | 1988–1994 n 8722, ≥40 yrs | Risks of prediabetes and diabetes were associated with urinary Cd levels 1–2 μg/g creatinine with respective OR values of 1.48 and 1.24. | Schwartz et al., 2003 [73] |
| Pancreas | 2005–2010 n 2398, ≥40 yrs | An increased risk of prediabetes was associated with urinary Cd levels ≥ 0.7 µg/g creatinine after adjustment for covariates. | Wallia et al., 2014 [74] |
| Pancreas | 1999–2006 n 4530 adults | BMDL5 and BMDL10 of urinary Cd levels derived from diabetes endpoint were of 0.198 and 0.365 μg/g creatinine, respectively. | Shi et al., 2021 [75] |
| Kidneys | 1999–2006 n 14,778, aged ≥ 20 yrs | Reduced GFR a (OR 1.32), albuminuria b (OR 1.92), and reduced GFR plus albuminuria (OR 2.91) were associated with blood Cd levels ≥ 0.6 μg/L with respective OR values of 1.32, 1.92, and 2.91. | Navas-Acien et al., 2009 [76] |
| Kidneys | 1999–2006 n 5426, aged ≥ 20 yrs | Albuminuria (OR 1.63) was associated with urinary Cd levels > 1 µg/g creatinine plus blood Cd levels > 1 µg/L (OR 1.63). Reduced eGFR (OR 1.48) and albuminuria (OR 1.41) were associated with blood Cd levels > 1 µg/L with respective OR values of 1.48 and 1.41. | Ferraro et al., 2010 [77] |
| Kidneys | 2007–2012 n 12,577, aged ≥ 20 yrs | Reduced eGFR (OR 1.80) and albuminuria (OR 1.60) were associated with blood Cd levels > 0.61 μg/L with respective OR values of 1.80 and 1.60. | Madrigal et al., 2019 [78] |
| Kidneys | 2009–2012 n 2926, aged ≥ 20 yrs | An elevated albumin excretion was associated with urinary Cd levels > 0.220 μg/L and blood Cd levels > 0.243 μg/L. | Zhu et al., 2019 [79] |
| Kidneys | 2011–2012 n 1545, aged ≥ 20 yrs | Reduced eGFR (OR 2.21) and albuminuria (OR 2.04) were associated with blood Cd levels > 0.53 μg/L with respective OR values of 2.21 and 2.04. | Lin et al., 2014 [80] |
| Biomarkers | Datasets | Findings | References |
|---|---|---|---|
| Serum GGT. | NHANES III, n 10,098, aged ≥ 20 yrs. | Serum GGT was positively associated with urinary Cd levels between 0.002 and 23.4 μg/g creatinine. Serum vitamins C and E and carotenoids were inversely associated with GGT. | Lee et al., 2006 [101] |
| Serum CRP and fibrinogen | NHANES III, n 6497, aged 40–79 yrs. | Elevations of serum CRP and fibrinogen were associated with urinary Cd levels ≥ 0.93 µg/g creatinine with respective OR values of 1.24 and 2.12. | Lin et al., 2009 [102] |
| Serum bilirubin | Healthy women, Buffalo, New York n 259, aged 18–44 yrs. | A reduction in serum bilirubin by 4.9% was associated with a 2-fold increase in blood Cd. Median Cd level (interquartile range) was 0.3 (0.19–0.43) μg/L. | Pollack et al., 2015 [103] |
| CRP, GGT, ALP, bilirubin and white cell count. | NHANES 2003–2010, n 3056 women, n 3288 men. | Serum CRP, GGT, and ALP levels were increased, respectively, by 47.5%, 8.8% and 3.7%, in urinary Cd quartiles 4 vs. 1. Consumption of anti-oxidative and anti-inflammatory nutrients were associated with an increase in serum bilirubin by 3% and reductions, respectively, in CRP, GGT, ALP, and white blood cell count by 7.4%, 3.3%, 5.2%, and 2.5%. | Colacino et al., 2014 [104] |
| Telomere length | NHANES 1999–2002, n 2093 with urinary Cd data, n 6796 with blood Cd plus Pb data. | Telomere shortening was associated with urinary and blood Cd levels but not blood Pb. | Zota et al., 2014 [105] |
| Telomere length | NHANES 1999–2002, n 7120 non-smokers, n 2296 smokers | A shorter telomere was associated with higher Cd exposure, CRP, trunk fat, and inactivity. A longer telomere was associated with retinyl stearate. | Patel et al., 2016 [106] |
| CRP and cardiovascular disease | NHANES 1999–2016 n 38,223 | CRP, triglycerides, total cholesterol, and white cell count were associated with elevated blood Cd levels. An increased risk of cardiovascular disease was associated blood Cd (OR 1.45). | Ma et al., 2022 [107] |
| Mortality | NHANES 2001–2010 Prospective, n 20,221, mean follow-up 9.1 years, n 2945 with diabetes | Risk of dying from all caused was increased by 49%, comparing blood Cd levels > 0.6 vs. < 0.24 µg/L. Cd, CRP, and 25(OH)D were associated with all-cause mortality among those with type 2 diabetes. | Liu et al., 2022 [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satarug, S.; Vesey, D.A.; Gobe, G.C. Mitigation of Cadmium Toxicity through Modulation of the Frontline Cellular Stress Response. Stresses 2022, 2, 355-372. https://doi.org/10.3390/stresses2030025
Satarug S, Vesey DA, Gobe GC. Mitigation of Cadmium Toxicity through Modulation of the Frontline Cellular Stress Response. Stresses. 2022; 2(3):355-372. https://doi.org/10.3390/stresses2030025
Chicago/Turabian StyleSatarug, Soisungwan, David A. Vesey, and Glenda C. Gobe. 2022. "Mitigation of Cadmium Toxicity through Modulation of the Frontline Cellular Stress Response" Stresses 2, no. 3: 355-372. https://doi.org/10.3390/stresses2030025