Composition, Thermal Expansion and Phase Transitions in Framework Silicates: Revisitation and Review of Natural and Synthetic Analogues of Nepheline-, Feldspar- and Leucite-Mineral Groups

Abstract
:1. Introduction
2. Research Topic 1: Natural Nepheline Chemistry and Structural Formulae
2.1. Background to a New Approach on Reporting Nepheline Compositional Relationships
| 24 Ne (nepheline, NaAlSiO4) | Na2 | Na6 | Al8 | Si8 | O32 |
| 24 Ks (kalsilite, KAlSiO4) | K2 | K6 | Al8 | Si8 | O32 |
| 24 An′ (20 An anorthite, CaAl2Si2O8 + 4 □ ) | □4 | Ca4 | Al8 | Si8 | O32 |
| 24 Q’ (16 Q tridymite, SiO2 + 8 □) | □2 | □6 | Si8 | Si8 | O32 |
2.2. Calculation of Nepheline Endmember Molecules
2.2.1. Ne—Qz Solid Solution Series
2.2.2. Ne—An’ (CaNe) Solid Solution Series
2.2.3. Ne—An’ (CaNe)—Q Solid Solutions
2.2.4. Natural Nephelines
2.3. Assessment of Igneous Rock Nepheline Compositions
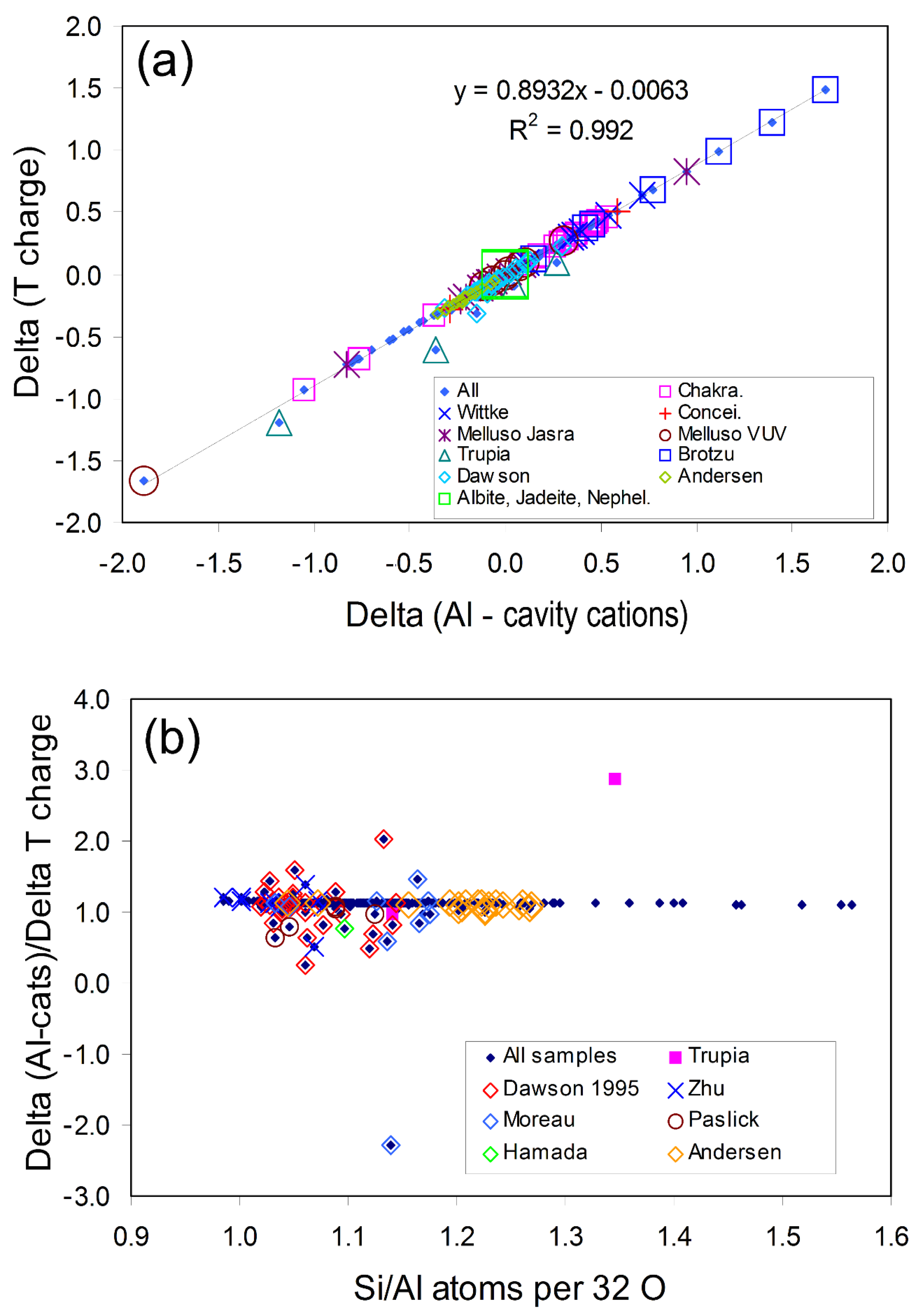
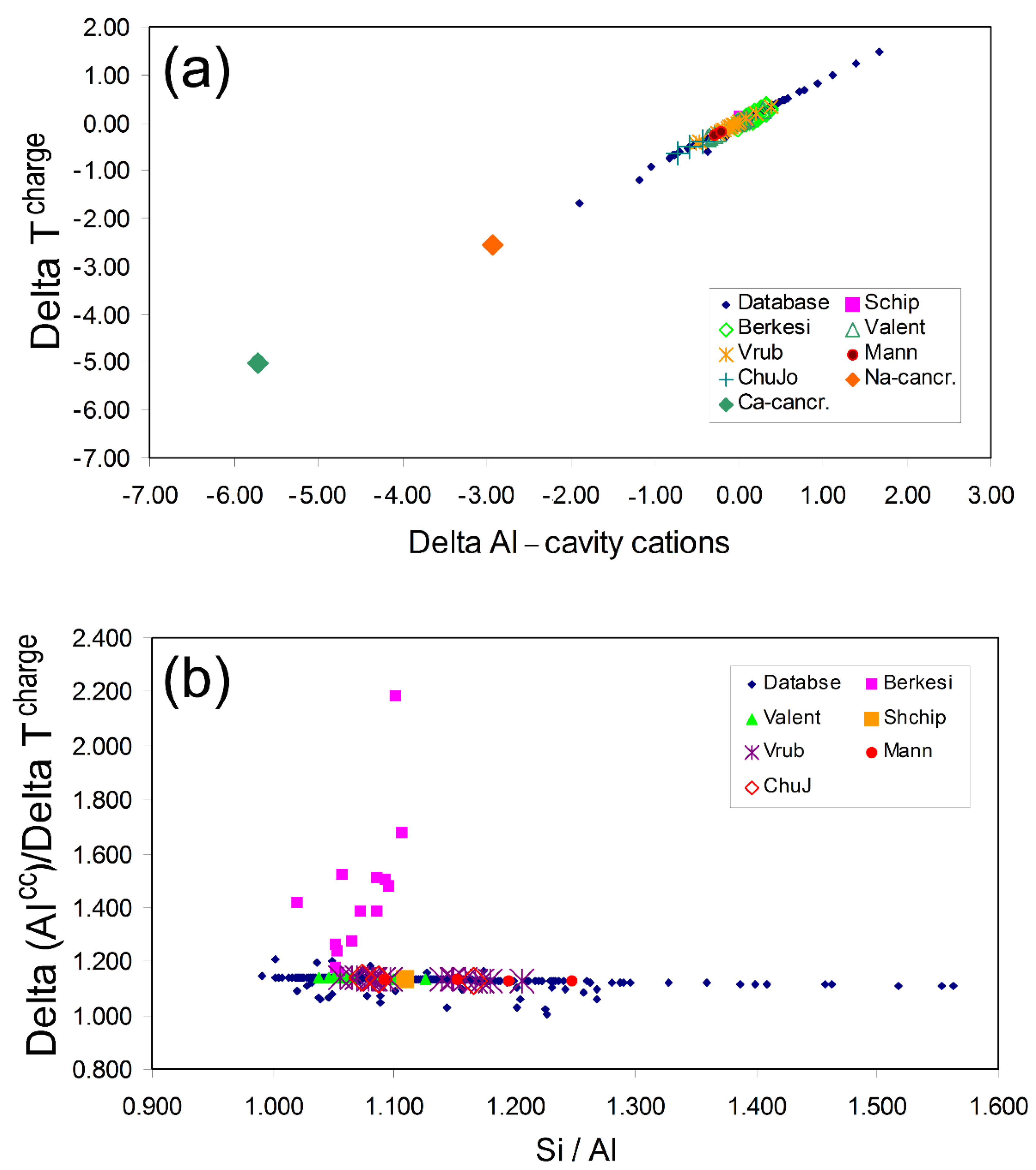
2.3.1. Understanding the Dependence of ∆Alcc/∆Tcharge on Composition
2.3.2. Compositions of Naturally Occurring Nephelines and Some Ideal “Nepheline” Analogues
2.3.3. Possible Significance of a Cancrinite Compon ent Existing in Nepheline
3. Research Topic 2: Nepheline and Structural Analogues Including Sr-Ba Aluminates
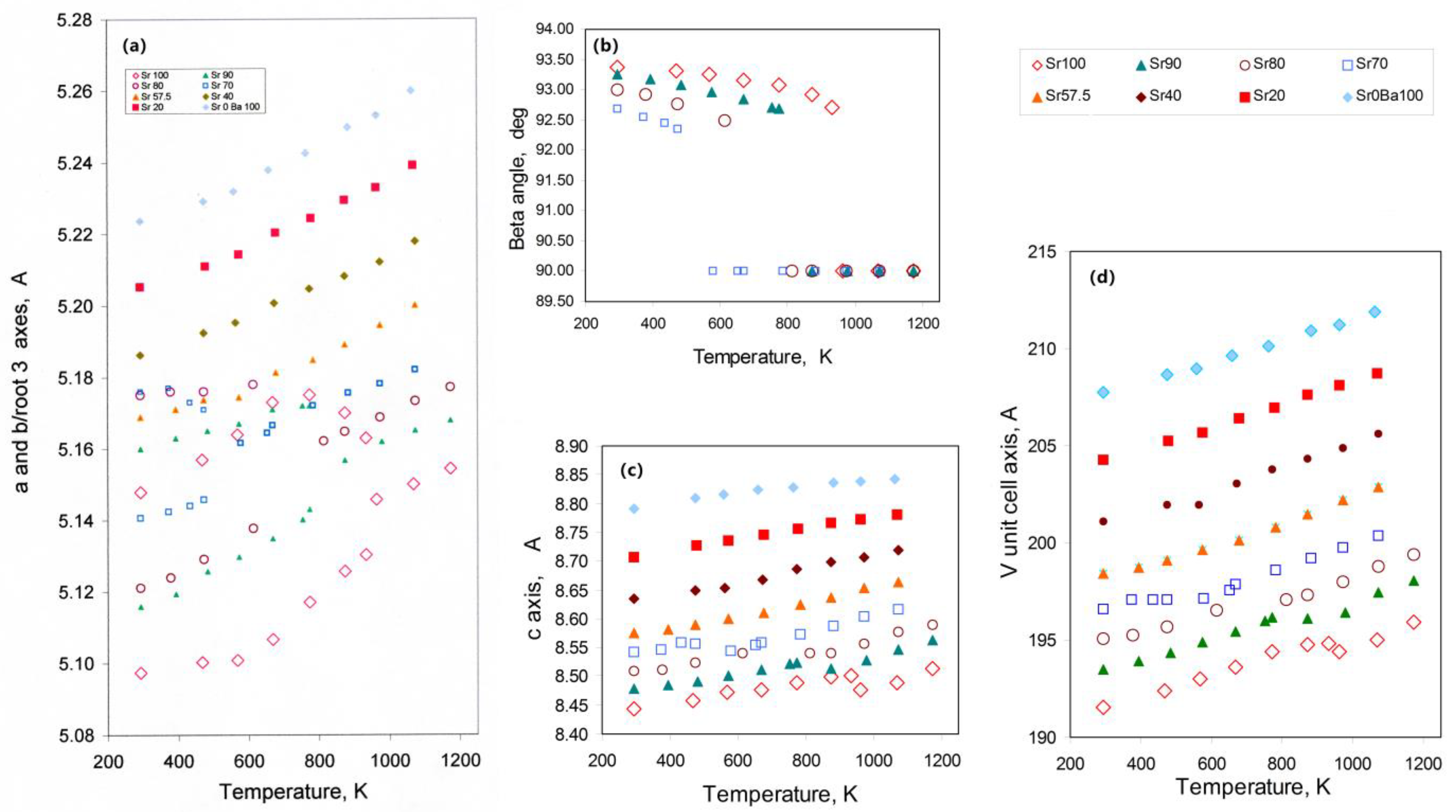
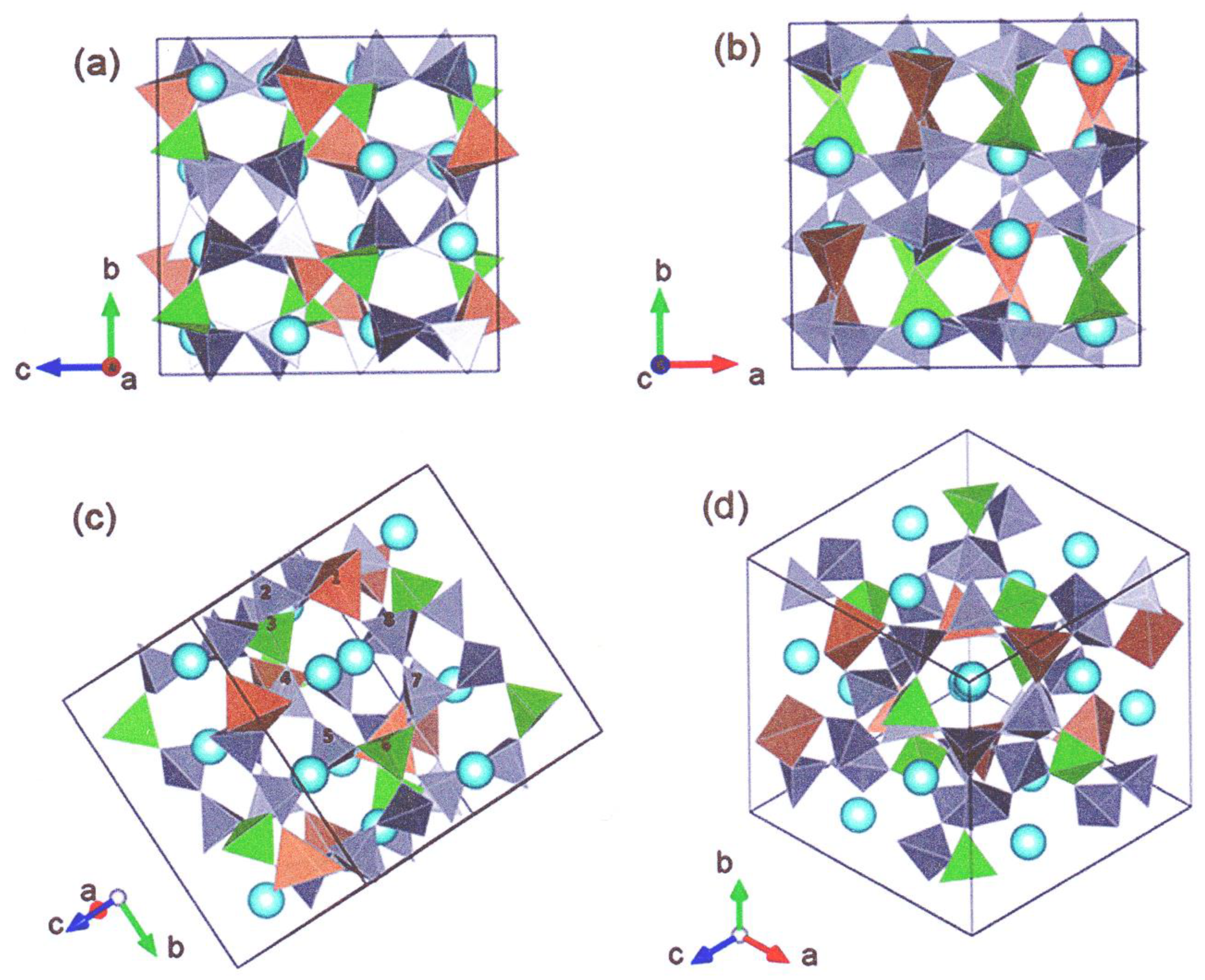
3.1. The Nepheline/Kalsilite-Analogue SrAl2O4—BaAl2O4 System
3.1.1. Thermal Expansion and Phase Transition Data for the SrAl2O4—BaAl2O4 System
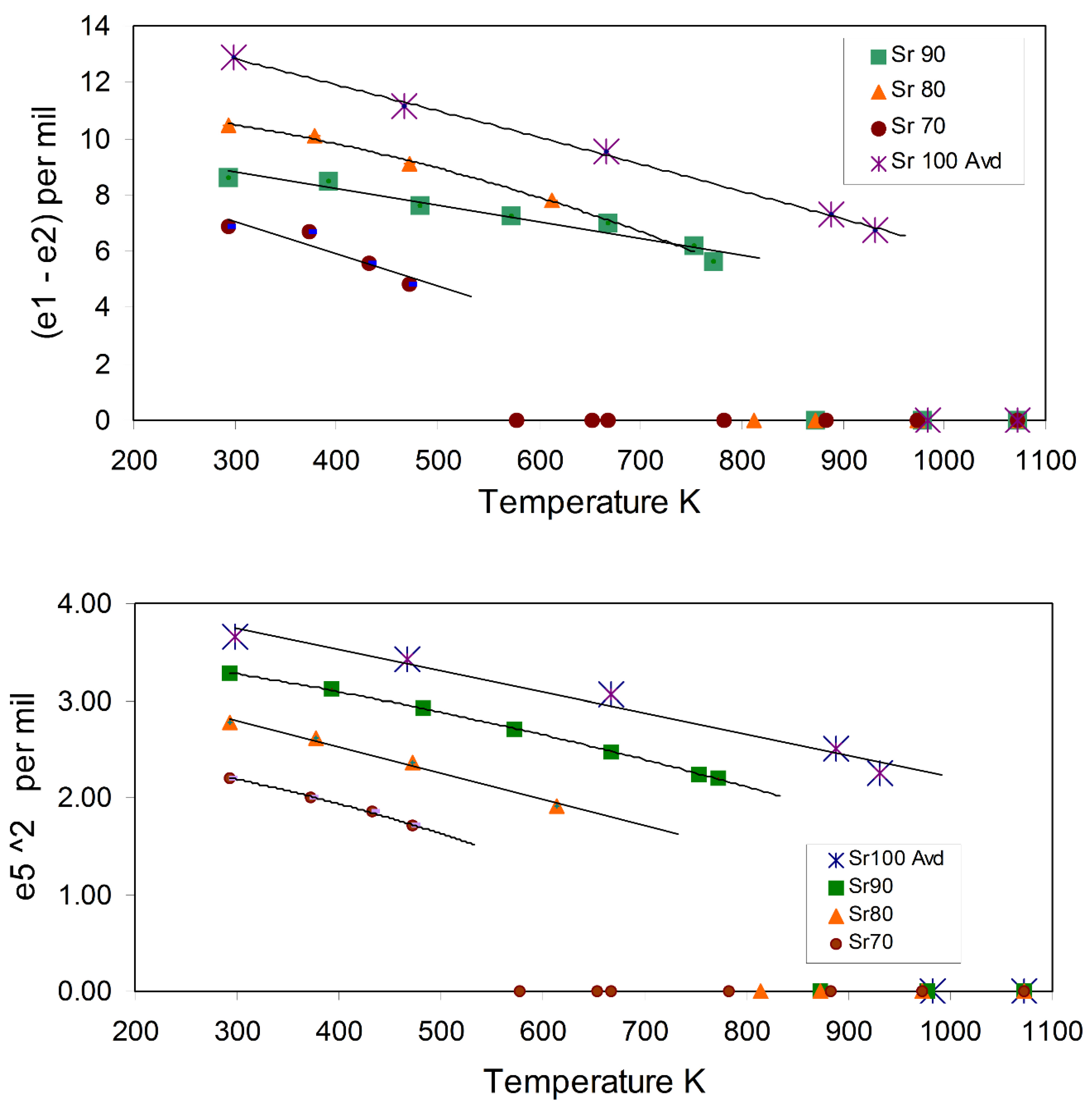
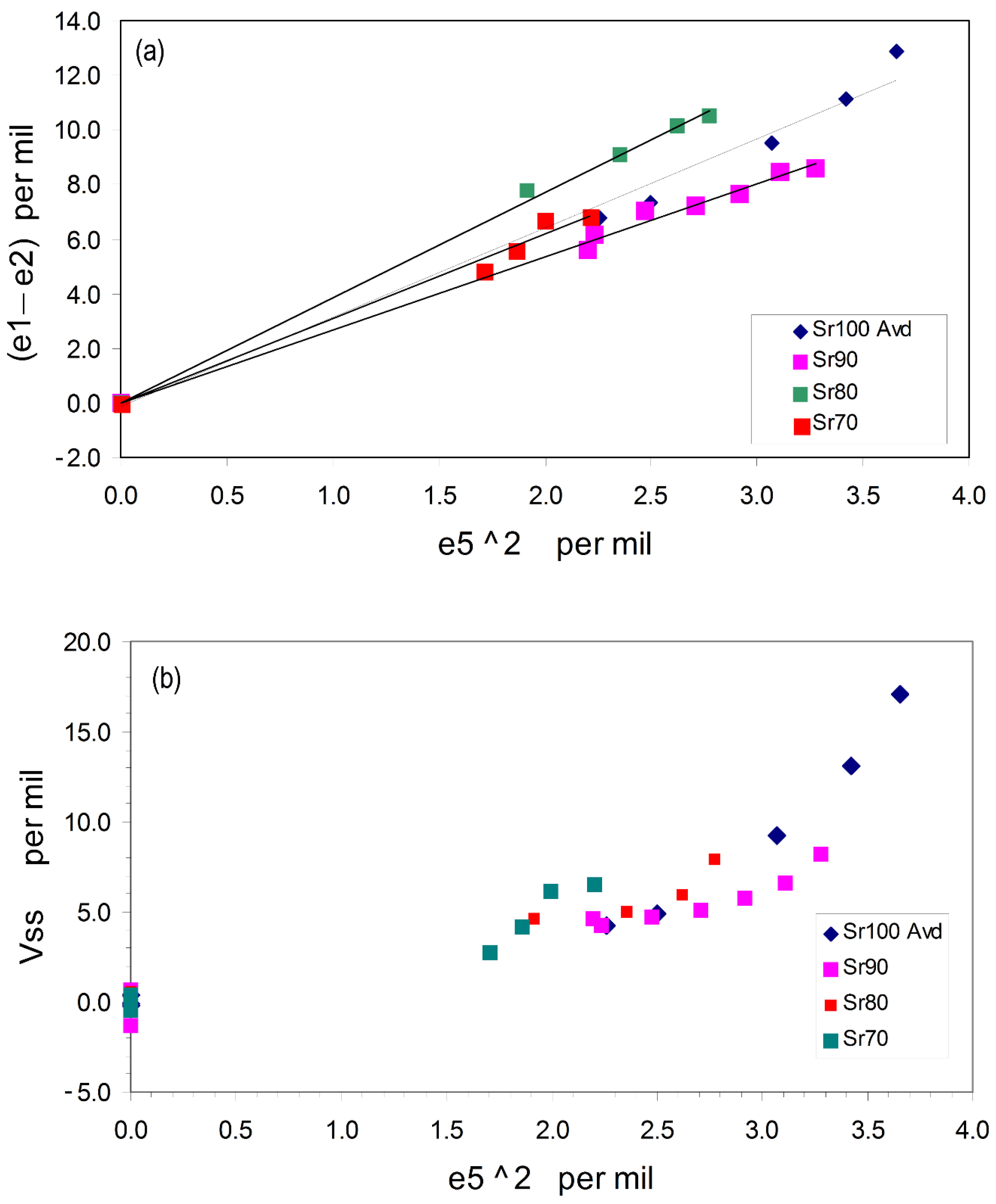
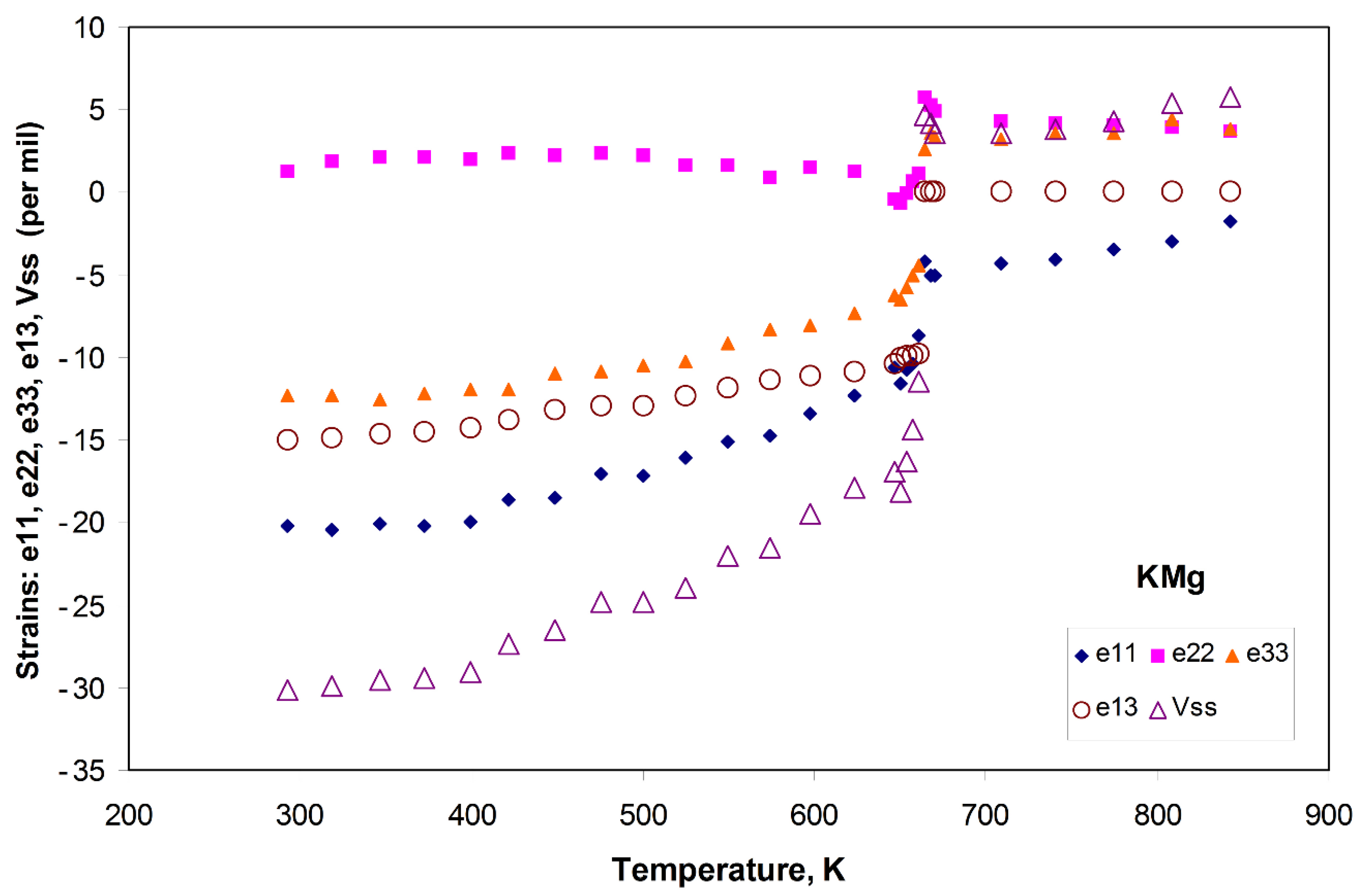
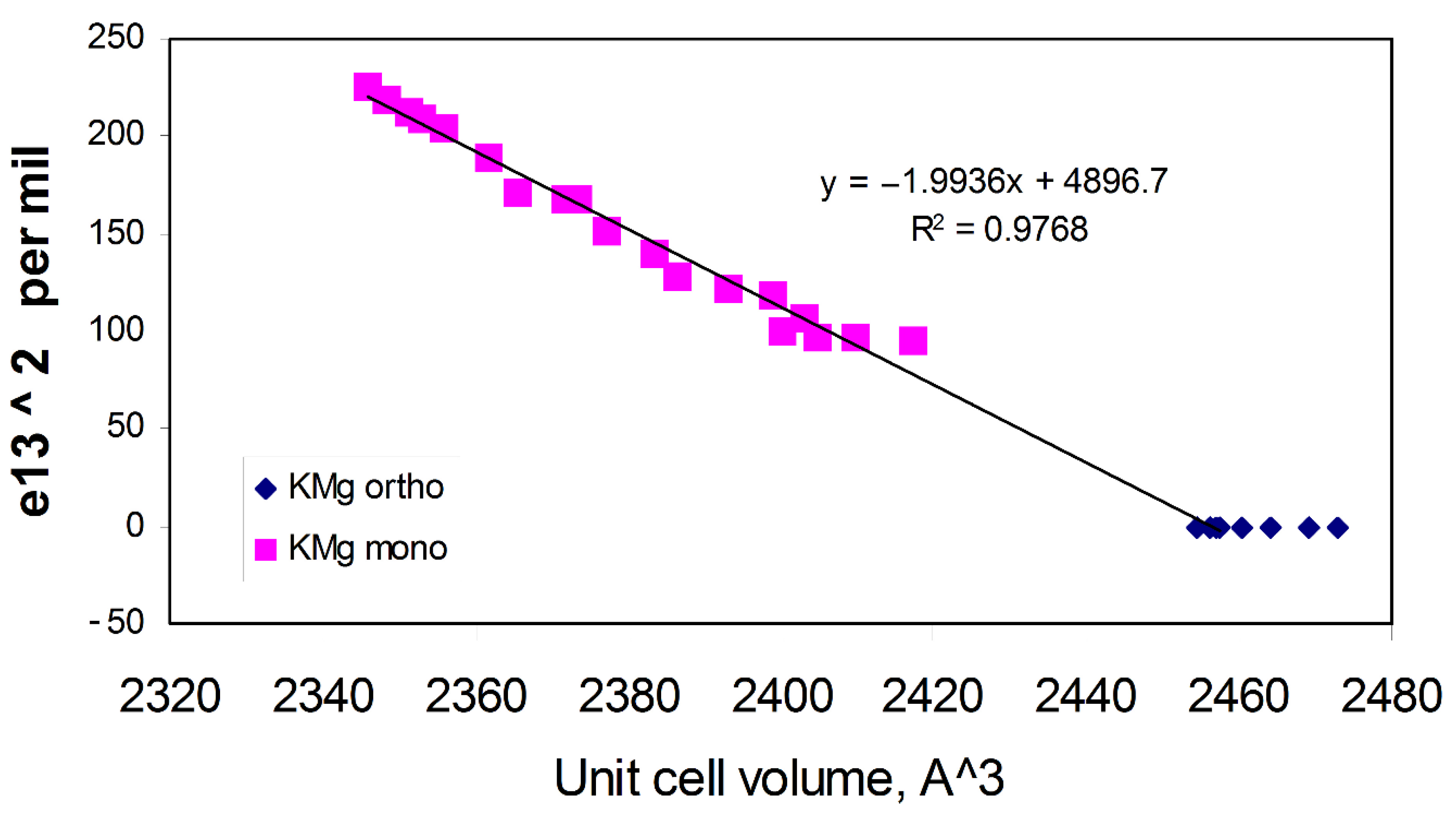
3.1.2. Spontaneous Strain Analyses for the System BaAl2O4—SrAl2O4.
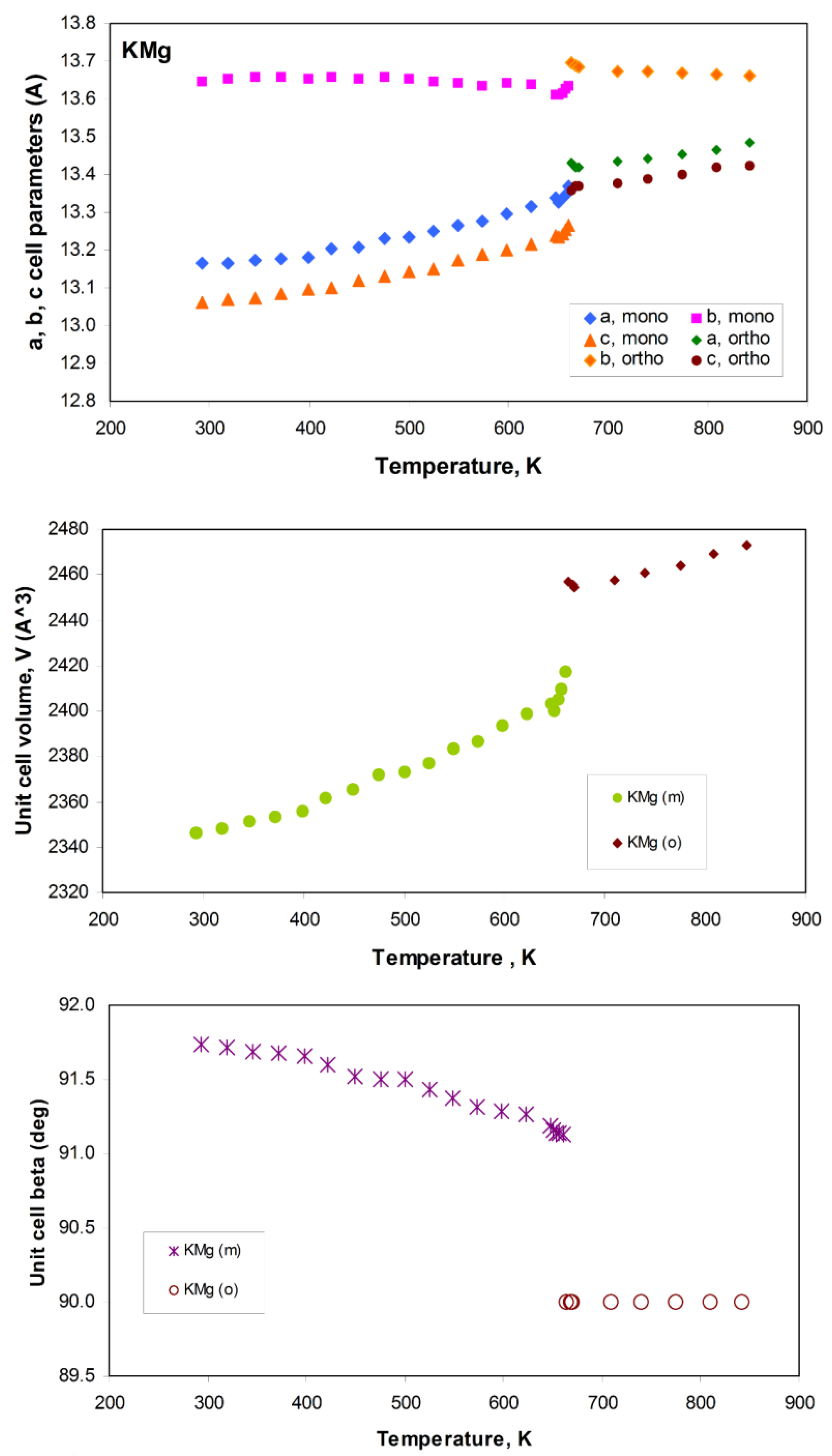
4. Research Topic 3: Thermal Expansion and Phase Transitions in the Feldspar Group
Synthetic Feldspar Analogues
5. Research Topic 4: The Leucite/Pollucite Group of Materials and Their Variable Stoichiometries and Structures
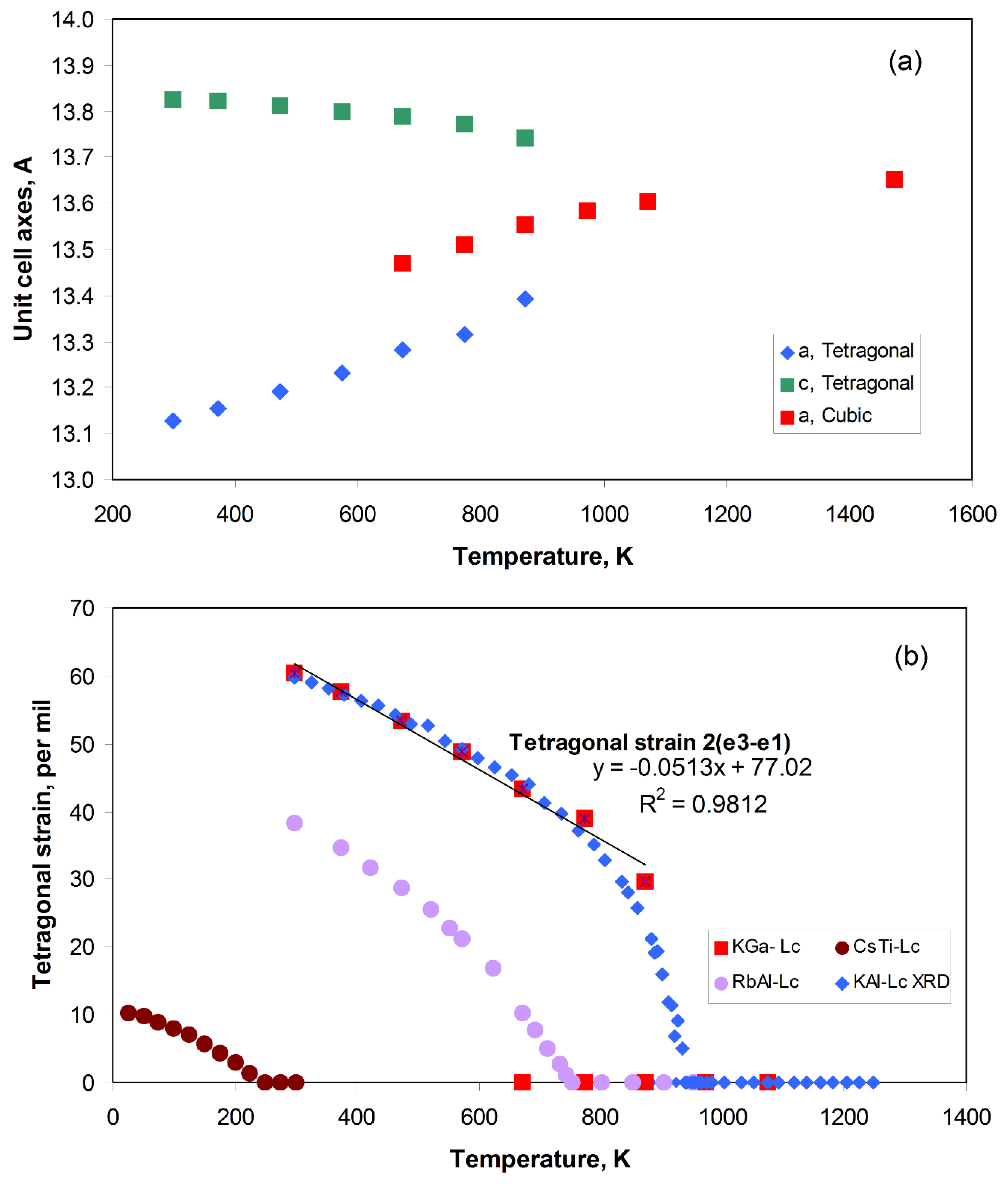
Leucite Structure Phase Transitions
6. Final Comment
Funding
Acknowledgments
Conflicts of Interest
References
- Zharadyik, J.; Jirásek, J.; Starý, J.; Sivek, M. Production, reserves, and processing of feldspar and feldspathoid rocks in the Czech Republic from 2005 to 2019–An overview. Minerals 2020, 10, 722. [Google Scholar] [CrossRef]
- Hogan, M.A.; Risbud, S.H. Gel-derived amorphous cesium-aluminosilicate powders for formation of pollucite glass-ceramics. J. Mater. Sci. 1991, 6, 217–219. [Google Scholar] [CrossRef]
- Deshkar, A.; Marcial, J.; Southern, S.A.; Kobera, L.; Bryce, D.L.; McCloy, J.S.; Goel, A. Understanding the structural origin of crystalline phase transformations in nepheline (NaAlSiO4)-based glass-ceramics. J. Amer. Ceram. Soc. 2017, 100, 2859–2878. [Google Scholar] [CrossRef]
- Maeda, K.; Hirose, M.; Kobayashi, T. High performance transparent glass-ceramics for optical components. Ceram. Soc. Jpn. 2015, 123, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Koenig, C.J. Use of nepheline syenite in sanitary porcelain. J. Am. Ceram. Soc. 2006, 22, 38–46. [Google Scholar] [CrossRef]
- Diella, V.; Adamo, H.; Pagliari, L.; Francescon, F. Effect of particle size and starting composition in Na-feldspar/kaolinite system at high temperature. J. Eur. Ceram. Soc. 2015, 35, 1327–1335. [Google Scholar] [CrossRef]
- Bernasconi, A.; Marinoni, N.; Pavese, A.; Francescon, F. Feldspar and firing cycle effects on the evolution of sanitary-ware body. Ceram. Int. 2014, 40, 6389–6398. [Google Scholar] [CrossRef]
- Roth, G.; Bȍhm, H. Ionic conductivity of sodium-nepheline single crystals. Solid State Ion. 1986, 18–19, 553–556. [Google Scholar] [CrossRef]
- Palmer, D.C.; Salje, E.K.H. Phase transitions in leucite: Dielectric properties and transition mechanism. Phys. Chem. Miner. 1990, 17, 444–452. [Google Scholar] [CrossRef]
- Jones, R.J.; Thrall, M.; Henderson, C.M.B. Complex impedance spectroscopy and transport properties of a natural leucite as a function of temperature and pressure. Miner. Mag. 2010, 74, 507–519. [Google Scholar] [CrossRef]
- Kumar, R.; Rakiewicz, E.F.; Rajagopalan, K. Preparation and characterization of of fluid cracking catalysts containing pollucite. J. Catal. 1993, 143, 304–307. [Google Scholar] [CrossRef]
- Nishioka, N.; Yanigisawa, K.; Yamasaki, N. Solidification of sludge ash by hydrothermal hot-pressing. Res. J. Water Pollut. Fed. 1990, 62, 926–932. [Google Scholar]
- Ng, E.-P.; Aleid Ghadah, M.S.; Rigolet, S.; Daou, T.J.; Mintova, S.; Chuan Ling, T. Micro- and macroscopic observations of the nucleation process and crystal growth of nanosized Cs-pollucite in an organotemplate-free hydrosol. New J. Chem. 2019, 43, 17433–17440. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Nishioka, M.; Yamasaki, N. Immobilization of cesium into pollucite structure by hydrothermal hot-pressing. J. Nucl. Sci. Tech. 1987, 24, 51–60. [Google Scholar] [CrossRef]
- Gatta, G.S.; Rinaldi, R.; Mclntyre, G.J.; Nenert, G.; Bellatreccia, F.; Guastoni, A.; Della Ventura, G. On the crystal structure and crystal chemistry of pollucite, (Cs,Na)16Al16Si32O96.H2O. Am. Miner. 2009, 94, 1560–1568. [Google Scholar] [CrossRef]
- Orlova, A.I.; Ojovan, M.I. Ceramic mineral waste-forms for nuclear waste immobilization. Materials 2019, 12, 2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesar, P.F.; Yoshimura, H.N.; Miranda, C.Y.; Okada, C.Y. Correlation between fracture toughness and leucite content in dental porcelains. J. Dent. 2005, 33, 721–729. [Google Scholar] [CrossRef]
- Nakashima, J.; Taira, T.; Sawasa, T. In vitro wear of four ceramic materials and human enamel on enamel antagonist. Eur. J. Oral Sci. 2016, 124, 219–300. [Google Scholar] [CrossRef]
- Cattell, M.J.; Patzig, C.; Bissasu, S.; Tsoutsos, A. Nucleation efficacy and flexural strength of novel leucite glass ceramics. Dent. Mater. 2020, 36, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Linvill, M.L.; Vandersande, J.W.; Pohl, R.O. Thermal conductivity of feldspars. Bull. Min. 1984, 107, 521–527. [Google Scholar] [CrossRef]
- Wu, X.; Zeng, Y.-F. Compensation effect for electrical conductivity and its application to estimate oxygen diffusivity in minerals. J. Geophys. Res. Solid Earth 2003, 108, 12. [Google Scholar] [CrossRef]
- Hu, H.; Dai, L.; Li, H.; Jiang, J. Electrical conductivity of K-feldspar at high-temperature and pressure. Min. Pet. 2014, 108, 609–618. [Google Scholar] [CrossRef]
- Buerger, M.J. The stuffed derivatives of the silica structures. Am. Min. 1954, 39, 600–614. [Google Scholar]
- Schairer, J.F.; Bowen, N.L. Preliminary report on equilibrium relations between feldspathoids, alkali feldspars and silica. Am. Geophys. Union Trans. 1935, 16, 325–328. [Google Scholar] [CrossRef]
- Bannister, F.A.; Hey, M.H. A chemical, optical and X-ray study of nepheline and kaliophilite. Min. Mag. 1931, 22, 569–608. [Google Scholar] [CrossRef]
- Bowen, N.L. The composition of nephelite. Am. J. Sci. 1912, 33, 49–54. [Google Scholar] [CrossRef]
- Greig, J.W.; Barth, T.F.W. The system Na2O.Al2O3.2SiO2 (nephelite, carnegieite)–Na2O.Al2O3.6SiO2 (albite). Am. J. Sci. 1938, 35A, 93–112. [Google Scholar]
- Bowen, N.L. The binary system: Na2Al¬2Si2O8 (nephelite, carnegieite)–CaAl2Si2O8 (anorthite). Am. J. Sci. 1912, 33, 551–573. [Google Scholar] [CrossRef]
- Goldsmith, J.R. Some aspects of the system NaAlSiO4-CaO.Al2O3. Am. Miner. 1949, 34, 471–493. [Google Scholar]
- Hahn, T.; Buerger, M.J. The detailed structure of nepheline, KNa3Al4Si4O16. Z. Krist. 1955, 106, 308–338. [Google Scholar] [CrossRef]
- Donnay, G.; Schairer, J.F.; Donnay, J.D.H. Nepheline solid solutions. Miner. Mag. 1959, 32, 93–109. [Google Scholar] [CrossRef]
- Hamilton, D.L.; MacKenzie, W.S. Nepheline solid solution in the system NaAlSiO4–KAlSiO4–SiO2. J. Pet. 1960, 1, 56–72. [Google Scholar] [CrossRef]
- Blancher, S.; D’Arco, P.; Fonteilles, M.; Pascal, M.L. Evolution of nepheline from mafic to highly differentiated members of the alkaline series: The Messum complex, Namibia. Min. Mag. 2010, 74, 413–432. [Google Scholar] [CrossRef]
- Barth, T.F.W. The composition of nepheline. Schweiz. Miner. Pet. Mitt. 1963, 43, 153–164. [Google Scholar]
- Dollase, W.A.; Thomas, W.M. The crystal chemistry of silica-rich, alkali-deficient nepheline. Contrib. Min. Pet. 1978, 66, 311–318. [Google Scholar] [CrossRef]
- Roedder, E.W. The system K2O-MgO-SiO2. Am. J. Sci. 1951, 249, 81–130, 224–248. [Google Scholar] [CrossRef]
- Torres-Martinez, L.M.; West, A.R. Pollucite- and leucite-related phases: A2BX5O12 and ACX2O6 (A = K,Rb,Cs; B = Be,Mg,Fe,Co,Ni,Zn,Cd; C = B, Al, Ga,Fe,Cr; X = Si, Ge). Zeitsschrift. Anorg. Allg. Chem. 1989, 573, 223–230. [Google Scholar] [CrossRef]
- Torres-Martinex, L.M.; Gard, J.A.; Howie, R.A.; West, A.R. Synthesis of Cs2BeSi5O12 with a pollucite structure. J. Solid State Chem. 1984, 51, 100–103. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Bell, A.M.T.; Kohn, S.C.; Page, C.S. Leucite-pollucite structure-type variability and the structure of a synthetic end-member wairakite (CaAl2Si4O12H2O). Miner. Mag. 1998, 62, 165178. [Google Scholar] [CrossRef]
- Roedder, E. A reconnaissance of the liquidus relationships in the system K2O.2SiO2–FeO–SiO2. Am. J. Sci. Bowen Vol. 1952, 250, 435–456. [Google Scholar]
- Liu, B.; Barbier, J. Structures of the stuffed-tridymite derivatives, BaMSiO4 (M = Co, Zn, Mg). J. Solid State Chem. 1993, 102, 115–125. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Taylor, D. The structural behaviour of the nepheline family: 1. Sr and Ba aluminates (MAl2O4), Miner. Mag. 1982, 45, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Roux, J. Etude des solutions solides des néphélines (Na,K)AlSiO4 et (Na,Rb)AlSiO4). Geochim. Cosmochim. Acta 1974, 38, 1213–1224. [Google Scholar] [CrossRef]
- Peterson, T.D. Peralkaline nephelinites. 1. Comparative petrology of Shombole and Oldoinyo Lengai, East Africa. Contrib. Miner. Pet. 1989, 101, 458–478. [Google Scholar] [CrossRef]
- Rossi, G.; Oberti, R.; Smith, D.C. The crystal structure of a K-poor, Ca-rich silicate with the nepheline framework, and crystal-chemical relationships in the compositional space (K,Na,Ca,□)8(Al,Si)16O32. Eur. J. Miner. 1989, 1, 59–70. [Google Scholar] [CrossRef]
- Hamada, M.; Akasaka, M.; Ohfuji, H. Crystal chemistry of K-rich nepheline in nephelinite from Hamada Shimane Province. Miner. Mag. 2019, 83, 239–247. [Google Scholar] [CrossRef]
- Morgan, G.B., VI; London, D. Effect of current density on the electron microprobe analysis of alkali aluminosilicate glasses. Am. Miner. 2005, 90, 1131–1138. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Pierozynski, W.J. An experimental study of Sr, Ba and Rb partitioning between alkali feldspar and silicate liquid in the system nepheline- kalsilite-quartz at 0.1 GPa P(H2O). Miner. Mag. 2012, 76, 157–190. [Google Scholar] [CrossRef]
- Okumiya, M.; Yamaguchi, G. The crystal structure of κ’-Al2O3, the new intermediate phase. Bull. Chem. Soc. Jpn. 1971, 44, 1567–1570. [Google Scholar] [CrossRef] [Green Version]
- Levin, I.; Brandon, D. Metastable alumina polymorphs: Crystal structures and transition sequences. J. Am. Ceram. Soc. 1998, 81, 1995–2012. [Google Scholar] [CrossRef]
- Deer, W.A.; Howie, R.A.; Zussman, J. Rock-Forming Minerals, Framework Silicates: Silica Minerals, Feldspathoids and the Zeolites; The Geological Society: London, UK, 2004; Volume 4B, 982p. [Google Scholar]
- Deer, W.A.; Howie, R.A.; Zussman, J. An Introduction to the Rock-Forming Minerals, 2nd ed.; Longman Scientific & Technical: Harlow, UK, 1992; 696p. [Google Scholar]
- Edgar, A.D. Studies on cancrinite: II–Stability fields and cell dimensions of calcium and potassium-rich cancrinites. Can. Miner. 1964, 8, 53–67. [Google Scholar]
- Sirbescu, M.; Jenkins, D.M. Experiments on the stability of cancrinite in the system Na2O-CaO-Al2O3-SiO2-CO2-H2O. Am. Miner. 1999, 84, 1850–1860. [Google Scholar] [CrossRef]
- Pant, A.K. A reconsideration of the crystal structure of β-Na2Si2O5. Acta Cryst. 1968, B24, 1077–1083. [Google Scholar] [CrossRef]
- Kahlenberg, V. Structural chemistry of anhydrous sodium silicate—A review. Chimia 2010, 64, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.M.B. Nepheline solid solution compositions: Stoichiometry revisited, reviewed, clarified and rationalised. Mineral. Mag. 2020. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Gibb, F.G.F. Felsic mineral crystallization trends in differentiating alkaline basic magmas. Contrib. Miner. Pet. 1983, 84, 355–364. [Google Scholar] [CrossRef]
- Wilkinson, J.F.G.; Hensel, H.D. Nephelines and analcimes in some alkaline igneous rocks. Contrib. Miner. Pet. 1994, 118, 79–91. [Google Scholar] [CrossRef]
- Mann, U.; Marks, M.; Markl, G. Influence of oxygen fugacity on mineral compositions in peralkaline melts. The Katzenbuckel volcano, Southwest Germany. Lithos 2006, 91, 262–285. [Google Scholar] [CrossRef]
- Vulic, P.; Balić-Žunić, T.; Belmonte, L.J.; Kahlenberg, V. Crystal chemistry of nephelines from ijolites and nepheline-rich pegmatites: Influence of composition and genesis on the crystal structure investigated by X-ray diffraction. Miner. Pet. 2011, 101, 185–194. [Google Scholar] [CrossRef]
- Vrublevskii, V.V.; Nikiforov, A.V.; Sugorakova, A.M.; Kozulina, T.V. Petrogenesis and tectonic setting of the Cambrian Kharly alkaline-carbonatite complex (Sangilen Plateau, Southern Siberia): Implications for the Early Paleozoic evolution of magmatism in the western Central Asian Orogenic Belt. J. Asian Earth Sci. 2020, 188, 104163. [Google Scholar] [CrossRef]
- Valentin, E.; Botelho, N.F.; Dantas, E.L. Monte Santo suite, an example of Ediacaran-Cambrian deformed alakline rocks in the Aragaia Belt Central Brazil. Implications for Western Gondwana evolution. Lithos 2020, 365–367, 105552. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Dawson, J.B. The 24th September ash eruption of the carbonatite volcano Oldoinyo Lengai, Tanzania: Mineralogy of the ash and implications for formation of a new magma type. Miner. Mag. 2007, 71, 483–492. [Google Scholar] [CrossRef]
- Berkesi, M.; Bali, E.; Bodnar, R.J.; Szabó, A.; Guzmics, T. Carbonatite and highly peralkaline nephelinite melts from Oldoinyo Lengai volcano, Tanzania. The role of natrite-normative fluid degassing. Gondwana Res. 2020, 85, 76–83. [Google Scholar] [CrossRef]
- Shchipalkina, N.V.; Pekov, I.V.; Koshiykova, N.N.; Britvin, S.N.; Zubkova, N.V.; Varlamov, D.A.; Sidorov, E.G. Unusual mineralization in fumarolic sublimates of the Tolbachik volcano, Kamchatka, Russia–Part 2: Tectosilicates. Eur. J. Miner. 2020, 32, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Kerraouch, I.; Ebert, S.; Patzek, M.; Bischoff, A.; Zolensky, M.E.; Pack, A.; Schmitt-Kopplin, P.; Belhai, D.; Bendaoud, A.; Le, L. A light, chondritic xenolith in the Murchison (CM) chondrite–Formation by fluid-assisted percolation during metasomatism. Geochemistry 2019, 79, 125518. [Google Scholar] [CrossRef]
- Grundy, H.D.; Hassan, I. The crystal structure of a carbonate-rich cancrinite. Can. Miner. 1982, 20, 239–251. [Google Scholar]
- Hassan, I.; Grundy, H.D. The character of the cancrinite-vishnevite solid-solution series. Can. Miner. 1984, 22, 333–340. [Google Scholar]
- Hassan, I.; Grundy, H.D. The crystal structure of basic cancrinite, ideally Na8[Al6Si6O24](OH)2.3H2O. Can. Miner. 1991, 29, 377–383. [Google Scholar]
- McConnell, J.D.C. Electron diffraction study of subsidiary maxima of scattered intensity in nepheline. Miner. Mag. 1962, 33, 114–124. [Google Scholar] [CrossRef] [Green Version]
- McConnell, J.D.C. Time-temperature study of the intensity of satellite reflections in nepheline. Am. Miner. 1981, 66, 990–996. [Google Scholar]
- Antao, S.M.; Hassan, I. Nepheline: Structure of three samples from the Bancroft area, Ontario, obtained using synchrotron high-resolution powder X-ray diffraction. Can. Miner. 2010, 48, 69–80. [Google Scholar] [CrossRef]
- Friese, K.; Grzechnik, A.; Petřīč, V.; Schönleber, A.; van Smaalen, S.; Morgenroth, W. Modulated structure of nepheline. Acta Cryst. 2011, B67, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, C.M.B.; Roux, J. Inversions in sub-potassic nephelines. Contrib. Miner. Petrol. 1977, 61, 279–298. [Google Scholar] [CrossRef]
- Schneider, O.W.; Stoeck, H. The NaAlSiO4 nepheline-carnegieite solid-state transformation. Zeits. Krist. 1994, 209, 113–117. [Google Scholar] [CrossRef]
- Tanaka, E.; Ishii, Y.; Tsukasaki, H.; Tanigucha, H.; Mori, S. Structural changes and microstructures in stuffed-tridymite compounds Ba1-xSrxAl2O4. Jpn. J. Appl. Phys. 2014, 53, 09PB01. [Google Scholar] [CrossRef]
- Shivaruma, N.J.; Coetsee, E.; Roos, W.D.; Nagabhushana, K.R.; Swart, H.C. Charge carrier trapping processes in un-doped and BaAl2O4:Eu3+ nanophosphor for thermoluminescent dosimeter applications. J. Phys. D Appl. Phys. 2020, 53, 475305. [Google Scholar] [CrossRef]
- Rodehorst, U.; Carpenter, M.A.; Marion, S.; Henderson, C.M.B. Structural phase transitions and mixing behaviour of the Ba-aluminate (BaAl2O4)–Sr-aluminate (SrAl2O4) solid solution. Miner. Mag. 2003, 67, 989–1013. [Google Scholar] [CrossRef]
- Ishii, Y.; Tsukasaki, H.; Kawaguchi, S.; Ouchi, Y.; Mori, S. Structural investigation of the SrAl2O4–BaAl2O4 solid solution system with unstable domain walls. J. Solid State Chem. 2017, 249, 149–153. [Google Scholar] [CrossRef]
- Wolten, G.M. Diffusionless phase transformations in zirconia and hafnia. J. Amer. Ceram. Soc. 1963, 46, 418–422. [Google Scholar] [CrossRef]
- Garvie, R.C. The occurrence of metastable tetragonal zirconia as a crystallite size effect. J. Phys. Chem. 1965, 69, 1238–1243. [Google Scholar] [CrossRef]
- Megaw, H.D. Temperature changes in the crystal structure of barium titanium oxide. Proc. R. Soc. A 1947, 189, 261–283. [Google Scholar]
- Parker, T.J.; Burfoot, J.C. Cubic-tetragonal phase transitions in high resistivity BaTiO3 single crystals. Br. J. Appl. Phys. (J. Phys. D) 1969, 2, 1168–1170. [Google Scholar] [CrossRef]
- Leadbetter, A.J.; Wright, A.F. The α-β transition in cristobalite phases of SiO2 and AlPO4 I. X-ray studies. Phil. Mag. 1976, 33105–33112. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Henderson, C.M.B. Tetragonal-cubic phase transition in KGaSi2O6 synthetic leucite analogue and its probable mechansim. J. Solid State Chem. 2020, 284, 121142. [Google Scholar] [CrossRef]
- Avdeev, M.; Yakovlev, S.; Yaramchenko, A.; Kharton, V. Transitions between P21, P63(√3A), and P6322 modifications of SrAl2O4 by in situ high-temperature X-ray and neutron diffraction. J. Solid State Chem. 2007, 180, 3535–3544. [Google Scholar] [CrossRef]
- Carpenter, M.A.; Salje, E.K.H.; Graeme-Barber, A. Spontaneous strain as a determinant of thermodynamic properties for phase transitions in minerals. Eur. J. Miner. 1998, 10, 621–691. [Google Scholar] [CrossRef]
- Redfern, S.A.T.; Henderson, C.M.B. Monoclinic-orthorhombic phase transition in the K2MgSi5O12 leucite analog. Am. Miner. 1996, 81, 369–374. [Google Scholar] [CrossRef]
- Ardit, M.; Martucci, A.; Cruciani, G. Monoclinic-orthorhombic phase transition in ZSM-5 zeolite: Spontaneous strain variation and thermodynamic properties. J. Phys. Chem. C 2015, 119, 7351–7359. [Google Scholar] [CrossRef]
- Megaw, H.D. The architecture of feldspars. In The Feldsparsi; MacKenzie, W.S., Zussman, J., Eds.; NATO Advanced Study Institute, 1972; Manchester University Press: Manchester, UK, 1974; pp. 2–24. [Google Scholar]
- Henderson, C.M.B. An elevated temperature X-ray study of synthetic disordered Na-K alkali feldspars. Contrib. Miner. Petrol. 1979, 79, 71–79. [Google Scholar] [CrossRef]
- Henderson, C.M.B. Thermal expansion of alkali feldspars II. Rb-sanidine and maximum microcline. In Progress in Experimental Petrology; NERC: Swindon, UK, 1978; pp. 51–54. [Google Scholar]
- Henderson, C.M.B. Thermal expansion of feldspars III. RbGaSi3O8-sanidine, Sr-feldspar and an ordered microcline-albite solid solutions (Or62.4, mole %). In Progress in Experimental Petrology; NERC Publication Series D, Sixth Progress Report; NERC Publication: Swindon, Wiltshire, UK, 1984; pp. 78–83. [Google Scholar]
- Hovis, G.L.; Morabito, J.R.; Spooner, A.; Mott, A.; Person, E.L.; Henderson, C.M.B.; Roux, J.; Harlov, D. A simple predictive model for the thermal expansion of Al,Si3 feldspars. Am. Miner. 2008, 93, 1568–1573. [Google Scholar] [CrossRef]
- Bruno, E.; Pentinghaus, H. Substitutions of cations in natural and synthetic feldspars. In The Feldspars; MacKenzie, W., Zussman, J., Eds.; NATO Advance Study Institute, 1972; Manchester University Press: Manchester, UK, 1974; pp. 574–614. [Google Scholar]
- Kyono, A.; Kimata, M. Refinement of the crystal structure of a synthetic non-stoichiometric Rb-feldspar. Miner. Mag. 2001, 65, 523–531. [Google Scholar] [CrossRef]
- Henderson, C.M.B.; Bell, A.M.T.; Knight, K.S. Variable stoichiometry in tectosilicates having the leucite/pollucite-type structure with particular emphasis on modeling the interframework cavity cation environment. J. Solid State Chem. 2017, 251, 90–104. [Google Scholar] [CrossRef]
- Ohashi, Y. A program to calculate the strain tensor from two sets of unit cell parameters. In Comparative Crystal Chemistry; Hazen, R.M., Finger, L.W., Eds.; Wiley: Chichester, UK; New York, NY, USA, 1982; pp. 92–102. [Google Scholar]
- Benna, P.; Bruno, E. Single crystal in situ high-temperature investigation on strontium feldspar. Am. Miner. 2001, 86, 690–696. [Google Scholar] [CrossRef]
- Waldbaum, D.R.; Robie, R.A. Calorimetric investigations of Na-K mixing and polymorphism in the alkali feldspars. Z. Krist. 1971, 134, 381–420. [Google Scholar]
- Winter, J.K.; Okamura, F.P.; Ghose, S. A high-temperature structural study of high-albite, monalbite and the analbite-monalbite phase transition. Am. Miner. 1979, 64, 409–423. [Google Scholar]
- Kroll, H.; Bambauer, H.-U.; Schirmer, U. The high-albite-monalbite andanalbite-monalbite transitions. Am. Miner. 1980, 65, 1192–1211. [Google Scholar]
- Openshaw, R.E.; Henderson, C.M.B.; Brown, W.L. A room-temperature phase transition in maximum microcline. Phys. Chem. Miner. 1979, 5, 95–104. [Google Scholar] [CrossRef]
- Grundy, H.D.; Brown, W.L. A high-temperature X-ray study of low and high plagiclase feldspars. In The Feldsparsi; MacKenzie, W.S., Zussman, J., Eds.; NATO Advance Study Institute, 1972; Manchester University Press: Manchester, UK, 1974; pp. 162–173. [Google Scholar]
- Tribaudino, M.; Angel, R.J.; Camara, F.; Nestola, F.; Pasqual, D.; Margiolaki, I. Thermal expansion of plagioclase feldspars. Contrib. Miner. Petrol. 2010, 160, 899–908. [Google Scholar] [CrossRef]
- Hovis, G.L.; Medford, A.; Conlon, M.; Tether, A.; Romanoski, A. Principles of thermal expansion in the feldspars. Am. Miner. 2010, 95, 1060–1068. [Google Scholar] [CrossRef]
- Winter, J.K.; Ghose, S.; Okamura, F.P. A high-temperature study of the thermal expansion and the anisotropy of the sodium atom in low albite. Am. Miner. 1977, 62, 921–931. [Google Scholar]
- Ohashi, Y.; Finger, L.W. An effect of temperature on the feldspar structure: Crystal structure of sanidine at 800 °C. Carnegie Inst. Wash. Yearb. 1975, 74, 569–572. [Google Scholar]
- Benna, P.; Tribaudino, M.; Bruno, E. High-temperature in situ structural investigation on lead feldspar. Am. Miner. 1999, 84, 120–129. [Google Scholar] [CrossRef]
- Brown, W.L.; Openshaw, R.E.; McMillan, P.F.; Henderson, C.M.B. A review of the expansion behaviour of alkali feldspars: Coupled variations in cell parameters and possible phase transitions. Am. Miner. 1984, 69, 1058–1071. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of intratomic distances in halides and chalcogenides. Acta Cryst. 1976, A32, 733–767. [Google Scholar]
- Fleet, M.E. Tetrahedral-site occupancies in sodium aluminum-gallium feldspar solid solutions [Na(Al1-xGax)Si3O8]. J. Solid State Chem. 1991, 92, 295–300. [Google Scholar] [CrossRef]
- Tribaudino, M.; Bruno, M.; Nestola, F.; Pasqual, D.; Angel, R.J. Thermoelastic and thermodynamic properties of plagioclase feldspars from thermal expansion measurements. Am. Miner. 2011, 96, 992–1002. [Google Scholar] [CrossRef]
- Brown, J.M.; Angel, R.J.; Ross, N.L. Elasticity of plagioclase feldspars. J. Geophys. Res. 2016, 121, 663–675. [Google Scholar] [CrossRef]
- Baerlocher, C.; Meier, W.M.; Olson, D.H. Atlas of zeolite framework types. In Structure Commission of the International Zeolite Association, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2001; 308p. [Google Scholar]
- Mazzi, F.; Galli, E.; Gottardi, G. The crystal structure of tetragonal leucite. Am. Miner. 1976, 61, 108–115. [Google Scholar]
- Beger, R.M. The crystal structure and chemical composition of pollucite. Zeits. Krist. 1969, 129, 280–302. [Google Scholar] [CrossRef]
- Teerstra, D.K.; Cȇrny, P. First natural occurrence of end-member pollucite: A product of low-temperature re-equilibration. Eur. J. Miner. 1995, 7, 1137–1148. [Google Scholar] [CrossRef] [Green Version]
- Hori, H.; Nagashima, K.; Yamada, M.; Miyawaki, R.; Marabashi, T. Ammonioleucite, a product of low temperature equilibrium. Am. Miner. 1986, 71, 1022–1027. [Google Scholar]
- Agakhanov, A.A.; Pautov, L.A.; Karpenko, V.Y.; Sokolova, E.; Hawthorne, F.C. Kirchhoffite, CsBSi2O6, a new mineral species from the Darai-Pioz alkaline massif, Tajikistan: Description and crystal structure. Can. Miner. 2012, 50, 523–529. [Google Scholar] [CrossRef]
- Ferraris, G.; Jones, D.W.; Yerken, J. A neutron diffraction study of the crystal structure of analcime, NaAlSi2O6.H2O. Zeits Krist. 1972, 135, 240–252. [Google Scholar] [CrossRef]
- Takéuchi, Y.; Mazzi, F.; Haga, N.; Galli, E. The crystal structure of wairakite. Am. Miner. 1979, 64, 993–1001. [Google Scholar]
- Holakovsky, J.; Kratochvilova, I.; Kocirik, M. Assessment of percolation thresholds for channel systems in MFI zeolite based on the MFI and diamond lattice. Microporous Mesoporous Mater. 2006, 91, 170–171. [Google Scholar] [CrossRef]
- Coombs, D.S. Recommended nomenclature for zeolite minerals: Report of the Subcommittee on Zeolites of International Mineralogical Association. Committee on new minerals and mineral names. Can. Miner. 1977, 35, 1571–1600. [Google Scholar]
- Henderson, C.M.B.; Charnock, J.M.; Bell, A.M.T.; van der Laan, G.C.M.B. X-ray absorption study of 3d transition metals and Mg in glasses and analogue materials in AFe3+Si2O6 and A2X2+Si5O12, where A = K, Rb, or Cs and X = Mg, Mn, Fe, Cu, Ni, Cu, or Zn. J. Non-Cryst. Solids 2016, 451, 23–48. [Google Scholar] [CrossRef]
- Balmer, M.L.; Huang, Q.; Wong-Ng, W.; Roth, R.S.; Santoro, A. Neutron and X-ray diffraction study of the crystal structure of CsTiSi2O6.5. J. Solid State Chem. 1997, 130, 97–102. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sumino, S.; Tamai, S.; Yanase, I. Phase transition and lattice thermal expansion of Cs-deficient pollucite, Cs1-xAl1-xSi2+x (x >= 0.25), compounds. J. Am. Ceram. Soc. 2006, 89, 3157–3161. [Google Scholar] [CrossRef]
- Carmichael, I.S.E. The mineralogy and petrology of the volcanic rocks from the Leucite Hills, Wyoming. Contrib. Miner. Petrol. 1967, 15, 24–66. [Google Scholar] [CrossRef]
- MacKenzie, W.S.; Richardson, D.M.; Wood, B.J. Solid solution of SiO2 in leucite. Bull. Soc. Miner. Cristallogr. 1974, 97, 257–260. [Google Scholar]
- Krzhizhanovskaya, M.G.; Boubnova, R.S.; Filatov, S.K.; Meyer, D.; Pauffler, P. Transformations of the crystal structure in a series of rubidium borosilicate solid solutions from the X-ray powder diffraction data. Glass Phys. Chem. 2003, 29, 599–607. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. Isotropy Subgroups of the 230 Crystallographic Space Groups; World Scientific: Singapore, 1998. [Google Scholar]
- Bell, A.M.T.; Knight, K.S.; Henderson, C.M.B.; Fitch, A.N. Revision of the structure of CsCuSi5O12 leucite as orthorhombic Pbca. Acta Cryst. 2010, B66, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Henderson, C.M.B. The thermal expansion of the leucite group of minerals. Am. Miner. 1968, 53, 1476–1489. [Google Scholar]
- Palmer, D.C.; Dove, M.T.; Ibberson, R.M.; Powell, B.M. Structural behavior, crystal chemistry, and phase transitions in substituted leucite: High-resolution neutron powder diffraction studies. Am. Miner. 1997, 82, 16–29. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Henderson, C.M.B. Rietveld refinement of the structures of dry-synthesized MFe3+Si2O6 leucites (M = K, Rb, Cs) by synchrotron X-ray powder diffraction. Acta Cryst. 1994, C50, 1531–1536. [Google Scholar] [CrossRef]
- Lange, R.A.; Carmichael, I.S.E.; Stebbins, J.F. Phase transitions in leucite (KAlSi2O6), orthorhombic KAlSiO4, and their iron analogues (KFeSi2O6, KFeSiO4). Am. Miner. 1986, 71, 937–945. [Google Scholar]
- Palmer, D.C.; Salje, E.K.H.; Schmahl, W.W. Phase transitions in leucite: X-ray diffraction studies. Phys. Chem. Miner. 1989, 16, 714–719. [Google Scholar] [CrossRef]
- Xu, H.; Navrotsky, A.; Balmer, M.L.; Su, Y. Crystal chemistry and phase transitions in substituted pollucites along the CsAlSi2O6–CsTiSi2O6.5 join: A powder synchrotron X-ray diffractometry study. J. Am. Ceram. Soc. 2002, 85, 1235–1242. [Google Scholar] [CrossRef]
- Bell, A.M.T.; Redfern, S.A.T.; Henderson, C.M.B.; Kohn, S.C. Structures of synthetic K2MgSi5O12 leucites by integrated X-ray powder diffraction, electron diffraction and 29Si MAS NMR methods. Acta Cryst. 1994, B50, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Bell, A.M.T.; Redfern, S.A.T.; Henderson, C.M.B.; Kohn, S.C. Structural relations and tetrahedral ordering pattern of synthetic orthorhombic Cs2CdSi5O12 leucite: A combined synchrotron X-ray powder diffraction and multinuclear MAS NMR study. Acta Cryst. 1994, 50, 560–566. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Atoms and Vacancies Per 32 O | Composition | wt.% | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mol % Ne | mol % Q’ | mol % Ne | mol. % Q’ | Na2O | Al2O3 | SiO2 | Total | ||||||||
| Mol % | Cation vacancies □Si In cavity sites | Na in Ne | Al in Ne | Si in Ne | Excess Si’ In framework | =Na × 100/(Na + Sixs/2) or Na × 100/8 | =Si’/2 × 100/(Na + Sixs/2) or □Si × 100/8 | =3 Na × 100/24 | =1.5 × Si × 100/24 | ||||||
| Ne100 | 0 | 8 | 8 | 8 | 0 | 32 | 100 | 0 | 100 | 0 | 21.82 | 35.89 | 42.30 | 100.00 | |
| Ne90Qz10 | 0.8 | 7.2 | 7.2 | 7.2 | 1.6 | 32 | 90 | 10 | 90 | 10 | 19.94 | 32.80 | 47.25 | 100.00 | |
| Ne87.5Qz12.5 | 1 | 7 | 7 | 7 | 2 | 32 | 87.5 | 12.5 | 87.5 | 12.5 | 19.46 | 32.02 | 48.52 | 100.00 | |
| Ne80Qz20 | 1.6 | 6.4 | 6.4 | 6.4 | 3.2 | 32 | 80 | 20 | 80 | 20 | 18.01 | 29.62 | 52.37 | 100.00 | |
| Ne75Qz25 | 2 | 6 | 6 | 6 | 4 | 32 | 75 | 25 | 75 | 25 | 17.02 | 27.99 | 54.99 | 100.00 | |
| Ne70Qz30 | 2.4 | 5.6 | 5.6 | 5.6 | 4.8 | 32 | 70 | 30 | 70 | 30 | 16.01 | 26.34 | 57.65 | 100.00 | |
| Ne62.5Qz35.5 | 3 | 5 | 5 | 5 | 6 | 32 | 62.5 | 37.5 | 62.5 | 37.5 | 14.47 | 23.81 | 61.72 | 100.00 | |
| Ne60Qz40 | 3.2 | 4.8 | 4.8 | 4.8 | 6.4 | 32 | 60 | 40 | 60 | 40 | 13.95 | 22.95 | 63.10 | 100.00 | |
| Ne50Qz50 | 4 | 4 | 4 | 4 | 8 | 32 | 50 | 50 | 50 | 50 | 11.82 | 19.44 | 68.74 | 100.00 | |
| Ne40Qz60 | 4.8 | 3.2 | 3.2 | 3.2 | 9.6 | 32 | 40 | 60 | 40 | 60 | 9.61 | 15.82 | 74.57 | 100.00 | |
| Ne37.5Qz67.5 | 5 | 3 | 3 | 3 | 10 | 32 | 37.5 | 62.5 | 37.5 | 62.5 | 9.05 | 14.89 | 76.06 | 100.00 | |
| Ne30Qz70 | 5.6 | 2.4 | 2.4 | 2.4 | 11.2 | 32 | 30 | 70 | 30 | 70 | 7.34 | 12.07 | 80.60 | 100.00 | |
| Ne25Qz75 | 6 | 2 | 2 | 2 | 12 | 32 | 25 | 75 | 25 | 75 | 6.17 | 10.14 | 83.69 | 100.00 | |
| Ne20Qz80 | 6.4 | 1.6 | 1.6 | 1.6 | 12.8 | 32 | 20 | 80 | 20 | 80 | 4.98 | 8.19 | 86.84 | 100.00 | |
| Ne12.5Qz87.5 | 7 | 1 | 1 | 1 | 14 | 32 | 12.5 | 87.5 | 12.5 | 87.5 | 3.15 | 5.18 | 91.66 | 100.00 | |
| Ne10Qz90 | 7.2 | 0.8 | 0.8 | 0.8 | 14.4 | 32 | 10 | 90 | 10 | 90 | 2.53 | 4.17 | 93.30 | 100.00 | |
| Qz100 | 8 | 0 | 0 | 0 | 16 | 32 | 0 | 100 | 0 | 100 | 0.00 | 0.00 | 100.00 | 100.00 | |
| Atoms and Vacancies Per 32 O | Mol % Nepheline Endmember Molecules | wt.% Oxides | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| □Ca | Ca | Na | Al | Si | O | Ne | An’ | An’ Barth 1963 | CaNe This work | Ne Barth 1963 | CaO | Na2O | Al2O3 | SiO2 | Total | |
| Na × 100/(Na + 2Ca) or Na × 100/8 | (Ca + □Ca) × 100/24 | 5Ca × 100/24 | 6Ca × 100/24 | 3Na × 100/24 | ||||||||||||
| Ne100 | 0 | 0 | 8 | 8 | 8 | 32 | 100 | 0 | 0.00 | 0.00 | 100.00 | 0.00 | 21.82 | 35.89 | 42.30 | 100.00 |
| Ne90An10 | 0.4 | 0.4 | 7.2 | 8 | 8 | 32 | 90 | 10 | 8.33 | 10.00 | 90.00 | 1.98 | 19.67 | 35.96 | 42.38 | 100.00 |
| Ne80An20 | 0.8 | 0.8 | 6.4 | 8 | 8 | 32 | 80 | 20 | 16.67 | 20.00 | 80.00 | 3.96 | 17.52 | 36.04 | 42.47 | 100.00 |
| Ne75Ne25 | 1 | 1 | 6 | 8 | 8 | 32 | 75 | 25 | 20.83 | 25.00 | 75.00 | 4.96 | 16.45 | 36.08 | 42.52 | 100.00 |
| Ne70Ne30 | 1.2 | 1.2 | 5.6 | 8 | 8 | 32 | 70 | 30 | 25.00 | 30.00 | 70.00 | 5.96 | 15.37 | 36.11 | 42.56 | 100.00 |
| Ne60Ne40 | 1.6 | 1.6 | 4.8 | 8 | 8 | 32 | 60 | 40 | 33.33 | 40.00 | 60.00 | 7.96 | 13.20 | 36.19 | 42.65 | 100.00 |
| Ne50An50 | 2 | 2 | 4 | 8 | 8 | 32 | 50 | 50 | 41.67 | 50.00 | 50.00 | 9.97 | 11.02 | 36.26 | 42.74 | 100.00 |
| Ne40An60 | 2.4 | 2.4 | 3.2 | 8 | 8 | 32 | 40 | 60 | 50.00 | 60.00 | 40.00 | 11.99 | 8.84 | 36.34 | 42.83 | 100.00 |
| Ne30An70 | 2.8 | 2.8 | 2.4 | 8 | 8 | 32 | 30 | 70 | 58.33 | 70.00 | 30.00 | 14.02 | 6.64 | 36.42 | 42.92 | 100.00 |
| Ne25An75 | 3 | 3 | 2 | 8 | 8 | 32 | 25 | 75 | 62.50 | 75.00 | 25.00 | 15.04 | 5.54 | 36.46 | 42.97 | 100.00 |
| Ne20An80 | 3.2 | 3.2 | 1.6 | 8 | 8 | 32 | 20 | 80 | 66.67 | 80.00 | 20.00 | 16.06 | 4.44 | 36.49 | 43.01 | 100.00 |
| Ne10An90 | 3.6 | 3.6 | 0.8 | 8 | 8 | 32 | 10 | 90 | 75.00 | 90.00 | 10.00 | 18.10 | 2.22 | 36.57 | 43.10 | 100.00 |
| 100An’ | 4 | 4 | 0 | 8 | 8 | 32 | 0 | 100 | 83.33 | 100.00 | 0.00 | 20.16 | 0.00 | 36.65 | 43.19 | 100.00 |
| Numbers of Atoms and Vacancies Per 32 Oxygens | Mol. %, Per 8 Cavity Atoms + Vacancies Per 32 O | wt.% Oxides | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vacs □Ca | Vacs □Si | Na | Ca | Al in Ne, An | Si in Ne, An | Excess Si’ | O | Ne | An | Q’ | Na | Ca | Al | Si | |
| Ne | 0 | 0 | 8 | 0 | 8 | 8 | 0 | 32 | 100 | 0 | 0 | 21.82 | 0.00 | 35.89 | 42.30 |
| An | 4 | 0 | 4 | 8 | 8 | 0 | 32 | 100 | 0 | 100 | 0.00 | 20.16 | 36.65 | 43.19 | |
| Qz | 0 | 8 | 0 | 0 | 0 | 0 | 16 | 32 | 0 | 100 | 100 | 0.00 | 0.00 | 0.00 | 100.00 |
| Ne90An5Qz5 | 0.2 | 0.4 | 7.2 | 0.2 | 7.6 | 7.6 | 0.8 | 32 | 90 | 5 | 5 | 19.81 | 1.00 | 34.39 | 44.80 |
| Ne80An10Qz10 | 0.4 | 0.8 | 6.4 | 0.4 | 7.2 | 7.2 | 1.6 | 32 | 80 | 10 | 10 | 17.76 | 2.01 | 32.87 | 47.35 |
| Ne70An20Qz10 | 0.8 | 0.8 | 5.6 | 0.8 | 7.2 | 7.2 | 1.6 | 32 | 70 | 20 | 10 | 15.58 | 4.03 | 32.94 | 47.45 |
| Ne70An10Qz20 | 0.4 | 1.6 | 5.6 | 0.4 | 6.4 | 6.4 | 3.2 | 32 | 70 | 10 | 20 | 15.79 | 2.04 | 29.69 | 52.48 |
| Ne60An20Qz20 | 0.8 | 1.6 | 4.8 | 0.8 | 6.4 | 6.4 | 3.2 | 32 | 60 | 20 | 20 | 13.56 | 4.09 | 29.75 | 52.60 |
| Ne50An30Qz20 | 1.2 | 1.6 | 4 | 1.2 | 6.4 | 6.4 | 3.2 | 32 | 50 | 30 | 20 | 11.33 | 6.15 | 29.81 | 52.71 |
| Ne50An20Qz30 | 0.8 | 2.4 | 4 | 0.8 | 5.6 | 5.6 | 4.8 | 32 | 50 | 20 | 30 | 11.49 | 4.16 | 26.45 | 57.90 |
| Ne40An40Qz20 | 1.6 | 1.6 | 3.2 | 1.6 | 6.4 | 6.4 | 3.2 | 32 | 40 | 40 | 20 | 9.08 | 8.22 | 29.88 | 52.82 |
| Ne20An30Qz50 | 1.2 | 4.0 | 1.6 | 1.2 | 4.0 | 4.0 | 8.0 | 32 | 20 | 30 | 50 | 4.76 | 6.46 | 19.57 | 69.21 |
| Ne50An20Qz30 | 0.8 | 2.4 | 4 | 0.8 | 5.6 | 5.6 | 4.8 | 32 | 50 | 20 | 30 | 11.49 | 4.16 | 26.45 | 57.90 |
| Basis | Formulae | |
|---|---|---|
| Excess Si Si’ | Si’a = Sitotal − Na − K − 2Ca | 1 |
| Si’b = Sitotal − Al | 2 | |
| Total sites, 24 | Qxs % = (24 − 3Na − 3K − 6Ca) × 100/24 | 3 |
| Ne % = 3Na × 100/24 | 4 | |
| Ks % = 3K × 100/24 | 5 | |
| CaNe % = 6Ca × 100/24 | 6 | |
| KsM % = 6M2+ × 100/24 | 7 | |
| Framework sites only, 16 | QSi % = Si’a × 100/16 | 8 |
| Q(Si-Al) = Si’b × 100/16 | 9 | |
| Cavity sites only, 8 | Qcav1 % = (8 − Na − K − 2Ca) × 100/8 | 10 |
| Qcav2 % = (24 − total Si − Al − Fe3+ − Na − K − 2Ca) × 100/8 | 11 | |
| Ne % = Na × 100/8 | 12 | |
| Ks % = K × 100/8 | 13 | |
| CaNe % = 2Ca × 100/8 | 14 | |
| Stoichiometric Na in Ne | NaNe = 8 − K − 2Ca − (Si − Al)/2 | 15 |
| Table 1 This Work Ne60Qz40 | Table 2 This Work Ne80An20 | Table 3 This Work Ne50An30Qz20 | D & T Syn [35] | H & G Crinan [58] | W & H Theral [59] | D & T Ne Sy [35] | Mann et al. Phonol [60] | Vulic et al. Ijolite [61] | Vruble Carb. [62] | Bl et al. Theral [33] | Valentin Ne Sy [63] | Mitchell Dawson Carb. ash [64] | Berkesi Nephelin [65] | Shch. et Volcan. [66] | Kerra Meteor. Glass [67] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wt.% | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| SiO2 | 63.10 | 42.47 | 52.71 | 53.69 | 49.8 | 51.82 | 45.26 | 45.54 | 44.57 | 44.44 | 44.11 | 44.63 | 42.44 | 43.33 | 44.02 | 44.5 |
| Al2O3 | 22.95 | 36.04 | 29.81 | 29.14 | 30.9 | 29.81 | 32.46 | 30.67 | 33.76 | 33.01 | 33.52 | 33.53 | 32.28 | 32.19 | 33.51 | 34.5 |
| Fe2O3 | 0 | 0.3 | 0.40 | 0.63 | 0.80 | 0.08 | 0.09 | 0.22 | 0.13 | 1.99 | 2.17 | 0.25 | 1.04 | |||
| MgO | 0.05 | 0.02 | ||||||||||||||
| CaO | 3.96 | 6.15 | 0 | 0.6 | 0 | 0 | 0.04 | 0.33 | 0.24 | 2.49 | 0.36 | 0.12 | 0.07 | 1.45 | ||
| Na2O | 13.95 | 17.52 | 11.33 | 17.05 | 16.0 | 16.65 | 16.64 | 16.90 | 16.90 | 16.03 | 15.58 | 15.98 | 15.92 | 15.09 | 19.00 | 17.5 |
| K2O | 1.05 | 2.6 | 2.15 | 5.71 | 4.98 | 5.12 | 6.11 | 3.90 | 5.80 | 7.66 | 7.29 | 2.01 | 2.79 | |||
| Total | 100.00 | 100.00 | 100.00 | 100.93 | 100.2 | 100.83 | 100.70 | 100.75 | 100.75 | 99.91 | 99.76 | 100.43 | 100.46 | 100.00 | 98.79 | 102.9 |
| Mol %, original paper | ||||||||||||||||
| Ne | ~61.5 | 63.6 | 62.3 | ~71.3 | 72 | 66.2 | 71.5 | 73.7 | 74.1 | |||||||
| Ks | ~2.5 | 6.7 | 5.3 | ~16.1 | 14 | 25.2 | 11.5 | 17.6 | 23.5 | |||||||
| Q | ~36.0 | 28.4 | 32.4 | ~12.6 | 14 | 7.0 | 5.3 | 8.7 | 1.93 | |||||||
| An | 1.3 | 1.6 | 11.4 | 0.62 | ||||||||||||
| Atoms/32(O) | ||||||||||||||||
| Si | 11.199 | 8.000 | 9.601 | 9.757 | 9.245 | 9.514 | 8.601 | 8.681 | 8.443 | 8.514 | 8.404 | 8.488 | 8.246 | 8.380 | 8.422 | 8.209 |
| Al | 4.801 | 8.000 | 6.399 | 6.241 | 6.761 | 6.450 | 7.270 | 6.890 | 7.538 | 7.453 | 7.527 | 7.516 | 7.394 | 7.337 | 7.556 | 7.484 |
| Fe3+ | 0 | 0.042 | 0.055 | 0.049 | 0.370 | 0.011 | 0.013 | 0.032 | 0.019 | 0.291 | 0.316 | 0.036 | 0.305 | |||
| M-2+ | 0.019 | 0.156 | ||||||||||||||
| Ca | 0.800 | 1.200 | 0 | 0.119 | 0 | 0 | 0.008 | 0.067 | 0.049 | 0.508 | 0.073 | 0.025 | 0.015 | 0.286 | ||
| Na | 4.800 | 6.400 | 4.000 | 6.007 | 5.759 | 5.927 | 6.131 | 6.246 | 6.207 | 5.954 | 5.744 | 5.892 | 5.999 | 5.658 | 7.048 | 6.245 |
| K | 0.243 | 0.616 | 0.504 | 1.384 | 1.211 | 1.237 | 1.493 | 0.948 | 1.407 | 1.899 | 1.798 | 0.491 | 0.655 | |||
| Total cations | 20.800 | 23.202 | 22.000 | 22.248 | 22.541 | 22.449 | 23.477 | 23.415 | 23.504 | 23.477 | 23.163 | 23.395 | 23.858 | 23.522 | 23.552 | 23.341 |
| ∆Alcc | 0 | 0 | 0 | −0.010 | 0.189 | 0.075 | −0.155 | −0.211 | −0.030 | −0.080 | −0.150 | 0.088 | −0.263 | 0.167 | 0.053 | 0.317 |
| ∆T valency | 0 | 0 | 0 | 0.009 | 0.169 | 0.068 | −0.137 | −0.187 | −0.026 | −0.070 | −0.132 | 0.077 | −0.231 | 0.113 | 0.047 | −0.006 |
| Mole % | ||||||||||||||||
| Ne | 60.0 | 80.0 | 50.0 | 75.07 | 71.46 | 73.39 | 75.51 | 73.38 | 77.45 | 74.27 | 71.40 | 73.42 | 71.35 | 66.78 | 87.65 | 74.26 |
| Nf | 0.52 | 0.69 | 1.13 | 4,70 | 0.14 | 0.16 | 0.40 | 0.23 | 3.64 | 3.95 | 0.45 | 3.81 | ||||
| Ks | 3.04 | 7.70 | 6.29 | 17.30 | 15.14 | 15.97 | 18.67 | 11.25 | 17.59 | 23.74 | 22.48 | 6.13 | 4.28 | |||
| KsM | 0.48 | 3.91 | ||||||||||||||
| CaNe | 20.0 | 30.0 | − | 2.98 | − | 0 | 0.26 | 1.67 | 1.23 | 12.71 | 1.83 | 0.62 | 0.36 | 7.15 | ||
| Qxs | 40.0 | 20.0 | 21.87 | 17.33 | 19.62 | 6.06 | 6.53 | 5.27 | 5.68 | 3.64 | 6.92 | 0.64 | 6.43 | 5.77 | 6.60 | |
| Cn | −0.046 | 0.93 | 0.36 | −0.73 | −0.99 | −0.14 | −0.374 | −0.70 | 0.41 | −1.23 | 0.78 | 0.25 | 1.483 | |||
| QSi | 40.0 | 80.0 | 20.0 | 21.91 | 16.45 | 19.27 | 6.79 | 7.52 | 5.40 | 6.05 | 4.35 | 6.51 | 1.88 | 5.59 | 5.52 | 4.61 |
| Qcavity | 40.0 | 80.0 | 20.0 | 21.87 | 16.74 | 19.39 | 6.06 | 7.19 | 5.36 | 5.92 | 4.11 | 6.65 | 1.47 | 5.79 | 5.60 | 4.66 |
| Q(Si-Al) | 40.0 | 80.0 | 20.0 | 21.97 | 15.26 | 18.80 | 7.76 | 8.84 | 5.59 | 6.55 | 5.28 | 5.96 | 3.52 | 4.54 | 5.19 | 2.63 |
| Mole % | ||||||||||||||||
| Ne | 60.0 | 100.0 | 71.5 | 75.09 | 74.20 | 74.08 | 76.64 | 78.28 | 78.91 | 75.37 | 82.25 | 75.03 | 75.46 | 70.98 | 88.10 | 84.08 |
| Ks | 0 | 0 | 3.04 | 7.93 | 6.29 | 17.30 | 15.18 | 15.73 | 18.90 | 13.57 | 17.92 | 23.89 | 22.56 | 6.13 | 8.82 | |
| Qxs | 40.0 | 28.5 | 22.87 | 17.87 | 19.62 | 6.06 | 6.54 | 5.36 | 5.75 | 4.17 | 7.05 | 0.65 | 6.45 | 5.77 | 7.10 | |
| Mol %Corrected Na | ||||||||||||||||
| Ne | 74.89 | 76.13 | 74.83 | 75.10 | 76.19 | 78.62 | 74.56 | 80.56 | 75.92 | 72.83 | 73.10 | 88.62 | 88.36 | |||
| Ks | 3.04 | 7.91 | 6.29 | 17.30 | 15.21 | 15.74 | 18.92 | 13.61 | 17.90 | 23.98 | 22.63 | 6.13 | 8.82 | |||
| Q(Si-Al) | 21.96 | 15.96 | 18.88 | 7.54 | 8.57 | 5.65 | 6.53 | 5.83 | 6.19 | 3.19 | 4.26 | 5.26 | 2.82 | |||
| Mol. % | ||||||||||||||||
| Na-cancr. | 0.08 | −1.6 | −0.01 | 1.29 | 1.7 | 0.24 | 0.65 | 1.23 | −0.72 | 2.15 | −1.4 | −0.4 | −2.98 | |||
| Temp. K | a, Å | b, Å | c, Å | α o | β o | γ o | V, Ǻ3 |
|---|---|---|---|---|---|---|---|
| RbAlSi3O8 | |||||||
| 293 | 8.844 | 13.044 | 7.190 | 116.35 | 743.3 | ||
| 393 | 8.849 | 13.046 | 7.191 | 116.27 | 744.4 | ||
| 473 | 8.852 | 13.042 | 7.190 | 116.26 | 744.4 | ||
| 576 | 8.861 | 13.041 | 7.190 | 116.21 | 745.5 | ||
| 673 | 8.875 | 13.035 | 7.188 | 116.15 | 746.5 | ||
| 778 | 8.890 | 13.031 | 7.193 | 116.04 | 748.7 | ||
| 883 | 8.901 | 13.024 | 7.193 | 116.02 | 749.4 | ||
| 978 | 8.916 | 13.023 | 7.197 | 116.00 | 751.1 | ||
| 1073 | 8.926 | 13.022 | 7.198 | 115.94 | 752.3 | ||
| 1178 | 8.934 | 13.019 | 7.199 | 115.89 | 753.3 | ||
| 1273 | 8.946 | 13.012 | 7.195 | 115.80 | 754.0 | ||
| RbGaSi3O8 | |||||||
| 293 | 8.919 | 13.089 | 7.254 | 116.43 | 758.3 | ||
| 363 | 8.930 | 13.084 | 7.253 | 116.40 | 759.1 | ||
| 458 | 8.941 | 13.078 | 7.253 | 116.34 | 760.1 | ||
| 573 | 8.955 | 13.075 | 7.256 | 116.30 | 761.6 | ||
| 668 | 8.965 | 13.070 | 7.257 | 116.26 | 762.7 | ||
| 773 | 8.976 | 13.067 | 7.260 | 116.21 | 764.0 | ||
| 873 | 8.987 | 13.066 | 7.260 | 116.16 | 765.3 | ||
| 973 | 8.997 | 13.062 | 7.264 | 116.11 | 766.6 | ||
| 1078 | 9.006 | 13.061 | 7.264 | 116.05 | 767.7 | ||
| 1173 | 9.015 | 13.058 | 7.267 | 115.98 | 769.1 | ||
| 1273 | 9.025 | 13.055 | 7.265 | 115.95 | 769.7 | ||
| SrAl2Si2O8 | |||||||
| 293 | 8.386 | 12.970 | 14.255 | 115.41 | 1400.3 | ||
| 393 | 8.391 | 12.971 | 14.269 | 115.49 | 1402.0 | ||
| 478 | 8.393 | 12.969 | 14.274 | 115.42 | 1403.2 | ||
| 588 | 8.398 | 12.978 | 14.282 | 115.42 | 1405.9 | ||
| 678 | 8.406 | 12.975 | 14.285 | 115.42 | 1407.2 | ||
| 723 | 8.405 | 12.980 | 14.289 | 115.40 | 1408.4 | ||
| 773 | 8.410 | 12.977 | 14.291 | 115.42 | 1408.6 | ||
| 828 | 8.415 | 12.982 | 14.295 | 115.41 | 1410.6 | ||
| 873 | 8.417 | 12.981 | 14.296 | 115.40 | 1411.1 | ||
| 918 | 8.419 | 12.980 | 14.301 | 115.37 | 1412.0 | ||
| 968 | 8.421 | 12.998 | 14.303 | 115.38 | 1412.3 | ||
| 1073 | 8.430 | 12.983 | 14.310 | 115.40 | 1414.7 | ||
| 1173 | 8.432 | 12.986 | 14.316 | 115.36 | 1416.4 | ||
| 1273 | 8.440 | 12.993 | 14.321 | 115.35 | 1419.4 | ||
| Or62.4, Max. micro. 6434 | |||||||
| 293 | 8.428 | 12.947 | 7.202 | 91.21 | 115.84 | 87.66 | 706.7 |
| 373 | 8.459 | 12.958 | 7.208 | 91.12 | 115.90 | 87.63 | 710.0 |
| 473 | 8.480 | 12.958 | 7.207 | 91.08 | 115.84 | 87.66 | 712.2 |
| 573 | 8.503 | 12.954 | 7.213 | 90.97 | 115.86 | 87.73 | 714.4 |
| 673 | 8.519 | 12.954 | 7.210 | 91.00 | 115.84 | 87.73 | 715.6 |
| 773 | 8.531 | 12.950 | 7.210 | 90.95 | 115.81 | 87.73 | 716.5 |
| 873 | 8.548 | 12.956 | 7.213 | 90.93 | 115.80 | 87.75 | 718.7 |
| 973 | 8.575 | 12.956 | 7.214 | 90.97 | 115.78 | 87.74 | 721.1 |
| 1073 | 8.587 | 12.960 | 7.210 | 90.97 | 115.72 | 87.70 | 722.3 |
| 1173 | 8.605 | 12.956 | 7.213 | 91.04 | 115.68 | 87.66 | 724.1 |
| 1273 | 8.608 | 12.958 | 7.210 | 91.06 | 115.69 | 87.63 | 724.0 |
| Sample (Or as mol. %) | Temp. °C | αa | αb | αc | αV |
|---|---|---|---|---|---|
| Or19 Henderson 1979 | 20–560 | 14.4 | 6.3 | 4.1 | 31.4 |
| 560–995 | 24.1 | 2.3 | 2.2 | 32.8 | |
| Or38 | 20–1000 | 19.8 | 1.6 | 1.3 | 25.3 |
| Or100 | 20–1000 | 15.3 | −0.6 | −0.1 | 17.2 |
| Or86 OF | 25–800 | 21.0 | −0.8 | 0.2 | 23.3 |
| Microcline, Or62.4 this work | 20–100 | 21.8 | 0.9 | 1.1 | 25.0 |
| Microcline Or100 OHB | 20–1005 | 16.2 | −1.9 | 0.1 | 17.1 |
| Or0.25 high albite Winter 1979 | 24–1060 | 13.1 | 8.5 | 4.5 | 29.6 |
| Or0.25 low albite, Winter 1977 | 25–970 | 16.2 | 6.2 | 2.4 | 31.0 |
| RbAlSi3O8, this work | 20–905 | 11.5 | −2.2 | 1.4 | 15.2 |
| RbGaSi3O8, this work | 20–1000 | 12.1 | −2.7 | 1.5 | 15.3 |
| Ab98 | 296–936 | 15.4 | 5.6 | 1.9 | 29.0 |
| An27 | 296–936 | 10.8 | 4.8 | 2.9 | 22.7 |
| An35 | 296–936 | 9.8 | 4.5 | 2.9 | 20.8 |
| An46 | 296–936 | 8.9 | 4.4 | 3.2 | 19.7 |
| An60 | 296–936 | 7.8 | 4.0 | 3.1 | 17.8 |
| An78 | 296–936 | 7.5 | 2.9 | 1.4 | 15.1 |
| An89 | 296–936 | 7.2 | 3.1 | 2.7 | 15.9 |
| An96 | 296–936 | 8.1 | 2.8 | 2.6 | 16. |
| An100 | 296–936 | 8.5 | 2.6 | 2.5 | 16.5 |
| An48.5 Hovis et al., 2010 | 22–850 | 8.8 | 3.9 | 4.3 | 21.3 |
| An95.5 Hovis et al., 2010 | 6.4 | 3.4 | 2.2 | 14.3 | |
| An57 Grundy and Brown, 1974 | 20–950 | 5.7 | 2.4 | 1.5 | 10.1 |
| An93 | 20–950 | 5.7 | 2.8 | −0.5 | 9.8 |
| An100 | 20–850 | 5.7 | 2.1 | 0.7 | 9.2 |
| SrAl2Si2O8 this work | 20–1000 | 6.6 | 1.8 | 4.7 | 13,9 |
| SrAl2Si2O8 Benna and Bruno | 20–670 | 9.0 | 1.4 | 5.0 | 17.0 |
| PbAl2Si2O8 Benna et al. 1999 | 20–700 | 8.0 | 1.6 | 2.5 | 12.6 |
| Sample | Principal Exp. Coeff.× 106 °C−1 | Orientation of Principal Axes;Angle (in Degrees) to | Sample | Principal Exp. Coeff.× 106 °C−1 | Orientation of Principal Axes; Angle (in Degrees) | ||||
|---|---|---|---|---|---|---|---|---|---|
| +a | +b | +c | +a | +b | +c | ||||
| Or19 Henderson 79 | 20–560 | Or19 Henderson 79 | 560–995 | ||||||
| α 1 | 53.7 (0.8) | 80 (1) | 49 (1) | 57 (1) | α 1 | 28.3 (1.4) | 24 (3) | 90 | 92 (3) |
| α 2 | 12.7 (0.9) | 11 (1) | 99 (1) | 121(1) | α 2 | 2.3 (0.5) | 90 | 0 | 90 |
| α 3 | −35 (0.6) | 87 (1) | 43 (1) | 131(1) | α 3 | 2.2 (0.8) | 114 (3) | 90 | 2 (1) |
| α p | 31.4 (1.6) | α p | 32.8 (1.7) | ||||||
| Or38 Henderson 79 | 500–1000 | Or100 Henderson 79 | 500–1000 | ||||||
| α 1 | 23.6 (0.9) | 23 (1) | 80 | 93 (1) | α 1 | 19.3 (0.5) | 26 (1) | 90 | 90 (1) |
| α 2 | 1.8 (0.4) | 90 | 0 | 90 | α 2 | 0.5 (0.3) | 90 | 0 | 90 |
| α 3 | 1.0 (0.4) | 113 (1) | 90 | 3 (1) | α 3 | 0.4 (0.4) | 116 (1) | 90 | 0 (1) |
| α p | 26.4 (1) | α p | 20.1 (0.7 | ||||||
| Low albite Or0.25 Winter et al. 1977 | 293–970 | High albite Or0/25 Winter et al. 1979 | 20–1080 | ||||||
| α 1 | 2.5 (3) | 48 (1) | 64(1) | 77 (1) | α 1 | 4.39 (4) | 80 (1) | 50(1) | 57 (1) |
| α 2 | 1.07 (2) | 134 (1) | 54(1) | 53 (1) | α 2 | 1.19 (2) | 11 (1) | 99(1) | 121(1) |
| α 3 | −0.47 (3) | 104 (1) | 133(1) | 40 (1) | α 3 | −2.28 (3) | 87 (1) | 42(1) | 132(1) |
| α p | 3.1 | α p | 3.3 | ||||||
| Microcline 6434 Or62.4 This work | 20–1000 | Microcline 71134 Or100 Openshaw et | 20–1005 | ||||||
| α 1 | 23.7 (0.8) | 17 (1) | 88 (1) | 99 (1) | α 1 | 19.1 (0.5) | 23 (1) | 94(1) | 94 (1) |
| α 2 | 1.4 (0.5) | 101 (4) | 40(17) | 52(17) | α 2 | 0.3 (0.4) | 113 (1) | 109(2) | 18 (8) |
| α 3 | −0.01 (6) | 103 (1) | 130(17) | 40(17) | α 3 | −2.2(0.4) | 91 (3) | 91 (3) | 72 (8) |
| α p | α p | ||||||||
| RbAlSi3O8 This work | 20–1000 | RbGaSi3O8 This work | 20–1000 | ||||||
| α 1 | 16.9 (0.5) | 34 (1) | 90 | 82 (1) | α 1 | 16.6 (0.3) | 34 (1) | 90 | 82 (1) |
| α 2 | 0.4 (0.4) | 124(4) | 90 | 8 (1) | α 2 | 1.4 (0.3) | 123 (1) | 90 | 6 (1) |
| α 3 | −2.5 (0.4) | 90 | 0 | 90 | α 3 | −2.7 (0.2) | 90 | 0 | 90 |
| α p | 14.8 (0.8) | α p | 15.3 (0.5) | ||||||
| SrAl2Si2O8 this work | 20 −1273 | SrAl2Si2O8 Benna and Bruno | 20–679 | ||||||
| α 1 | 7.2 (0.2) | 30 (3) | 90 | 86 (3) | α 1 | 6.9 (5) | 32 | 90 | 83 |
| α 2 | 4.7 (0.2) | 120 (3) | 90 | 4 (3) | α 2 | 3.2 (3) | 122 | 90 | 7 |
| α 3 | 1.8 (0.2) | 90 | 0 | 90 | α 3 | 0.9 (4) | 90 | 0 | 90 |
| α p | 13.7 (0.3) | α p | 11.0 | ||||||
| CaAl2Si2O8 Grundy and Brown | 20–850 | PbAl2Si2O8 Benna et al. | 20–700 | ||||||
| α 1 | 8.9 (0.6) | 23 (1) | 94 (1) | 94 (1) | α 1 | 5.9 (3) | 19 | 90 | 96 |
| α 2 | 2.2 (0.5) | 113 (1) | 109(2) | 18 (8) | α 2 | 1.6 (3) | 109 | 90 | 6 |
| α 3 | −2.4 (0.4) | 91 (3) | 91 (3) | 72 (8) | α 3 | 1.1 (2) | 90 | 0 | 90 |
| α p | 8.8 (0.9) | α p | 8.6 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henderson, C.M.B. Composition, Thermal Expansion and Phase Transitions in Framework Silicates: Revisitation and Review of Natural and Synthetic Analogues of Nepheline-, Feldspar- and Leucite-Mineral Groups. Solids 2021, 2, 1-49. https://doi.org/10.3390/solids2010001
Henderson CMB. Composition, Thermal Expansion and Phase Transitions in Framework Silicates: Revisitation and Review of Natural and Synthetic Analogues of Nepheline-, Feldspar- and Leucite-Mineral Groups. Solids. 2021; 2(1):1-49. https://doi.org/10.3390/solids2010001
Chicago/Turabian StyleHenderson, C. Michael B. 2021. "Composition, Thermal Expansion and Phase Transitions in Framework Silicates: Revisitation and Review of Natural and Synthetic Analogues of Nepheline-, Feldspar- and Leucite-Mineral Groups" Solids 2, no. 1: 1-49. https://doi.org/10.3390/solids2010001