
Polyneuropathy Associated with IgM Monoclonal Gammopathy; Advances in Genetics and Treatment, Focusing on Anti-MAG Antibodies
 , and
, and
Abstract
:1. Introduction
2. Methods
3. Discussion
3.1. Pathophysiology of IgM Gammopathies
3.2. Genetic Mutations
3.3. Polyneuropathy in IgM Gammopathies
3.3.1. IgM M-Protein Related Polyneuropathies and Corresponding Phenotypes
3.3.2. IgM Anti-MAG Antibody Polyneuropathy
3.4. Current Therapy for IgM Gammopathy Mediated Polyneuropathy
3.5. Novel Therapy Options
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. A Long-Term Study of Prognosis in Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Mailankody, S.; Landgren, O. Monoclonal gammopathy of undetermined significance and Waldenström’s macroglobulinemia. Best Pract. Res. Clin. Haematol. 2016, 29, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Eisele, L.; Dürig, J.; Hüttmann, A.; Dührsen, U.; Assert, R.; Bokhof, B.; Erbel, R.; Mann, K.; Jöckel, K.-H.; Moebus, S. Prevalence and progression of monoclonal gammopathy of undetermined significance and light-chain MGUS in Germany. Ann. Hematol. 2012, 91, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, D.; Kumar, S.K.; Kyle, R.A.; Dispenzieri, A.; Dasari, S.; Larson, D.R.; Vachon, C.; Cerhan, J.R.; Rajkumar, S.V. Detection and prevalence of monoclonal gammopathy of undetermined significance: A study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin, C.; Reddy-Kolanu, V.; Drayson, M.T.; Sapey, E.; Richter, A.G. The prevalence and significance of monoclonal gammopathy of undetermined significance in acute medical admissions. Br. J. Haematol. 2020, 189, 1127–1135. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Rögnvaldsson, S.; Thorsteinsdottir, S.; Reed, E.R.; Oskarsson, J.T.T.; Petursdottir, I.; Sigurdardottir, G.A.; Vidarsson, B.; Onundarson, P.T.; Agnarsson, B.A.; et al. Screening for Monoclonal Gammopathy of Undetermined Significance: A Population-Based Randomized Clinical Trial. First Results from the Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM) Study. Blood 2021, 138 (Suppl. 1), 156. [Google Scholar] [CrossRef]
- Fermand, J.P.; Bridoux, F.; Dispenzieri, A.; Jaccard, A.; Kyle, R.A.; Leung, N.; Merlini, G. Monoclonal gammopathy of clinical significance: A novel concept with therapeutic implications. Blood 2018, 132, 1478–1485. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, J.; D’Sa, S.; Minnema, M.C.; Kersten, M.J.; Wechalekar, A.; Vos, J.M. IgM monoclonal gammopathies of clinical significance: Diagnosis and management. Haematologica 2022, 107, 2037–2050. [Google Scholar] [CrossRef]
- Castillo, J.J.; Callander, N.S.; Baljevic, M.; Sborov, D.W.; Kumar, S. The evaluation and management of monoclonal gammopathy of renal significance and monoclonal gammopathy of neurological significance. Am. J. Hematol. 2021, 96, 846–853. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Remstein, E.D.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003, 102, 3759–3764. [Google Scholar] [CrossRef]
- Hanewinckel, R.; van Oijen, M.; Ikram, M.A.; van Doorn, P.A. The epidemiology and risk factors of chronic polyneuropathy. Eur. J. Epidemiol. 2016, 31, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Baldereschi, M.; Inzitari, M.; di Carlo, A.; Farchi, G.; Scafato, E.; Inzitari, D. Epidemiology of distal symmetrical neuropathies in the Italian elderly. Neurology 2007, 68, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Nobile-Orazio, E.; Barbieri, S.; Baldini, L.; Marmiroli, P.; Carpo, M.; Premoselli, S.; Manfredini, E.; Scarlato, G. Peripheral neuropathy in monoclonal gammopathy of undetermined significance: Prevalence and immunopathogenetic studies. Acta Neurol. Scand. 1992, 85, 383–390. [Google Scholar] [CrossRef]
- Vrethem, M.; Cruz, M.; Wen-Xin, H.; Malm, C.; Holmgren, H.; Ernerudh, J. Clinical, neurophysiological and immunological evidence of polyneuropathy in patients with monoclonal gammopathies. J. Neurol. Sci. 1993, 114, 193–199. [Google Scholar] [CrossRef]
- Steiner, N.; Schwärzler, A.; Göbel, G.; Löscher, W.; Wanschitz, J.; Gunsilius, E. Are neurological complications of monoclonal gammopathy of undetermined significance underestimated? Oncotarget 2017, 8, 5081–5091. [Google Scholar] [CrossRef] [Green Version]
- Rögnvaldsson, S.; Steingrímsson, V.; Turesson, I.; Björkholm, M.; Landgren, O.; Kristinsson, S.Y. Peripheral neuropathy and monoclonal gammopathy of undetermined significance: A population-based study including 15,351 cases and 58,619 matched controls. Haematologica 2020, 105, 2679–2681. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.J.; Kyle, R.A.; O’Brien, P.C.; Dyck, P.J. Prevalence of monoclonal protein in peripheral neuropathy. Neurology 1981, 31, 1480. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, S.; Kyle, R.A.; Dyck, P.J. Neuropathy associated with monoclonal gammopathies of undetermined significance. Ann. Neurol. 1991, 30, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Baldini, L.; Nobile-Orazio, E.; Guffanti, A.; Barbieri, S.; Carpo, M.; Cro, L.; Cesana, B.; Damilano, I.; Maiolo, A.T. Peripheral neuropathy in IgM monoclonal gammopathy and Wäldenstrom’s macroglobulinemia: A frequent complication in elderly males with low MAG-reactive serum monoclonal component. Am. J. Hematol. 1994, 45, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Latov, N.; Sherman, W.H.; Nemni, R.; Galassi, G.; Shyong, J.S.; Penn, A.S.; Chess, L.; Olarte, M.R.; Rowland, L.P.; Osserman, E.F. Plasma-cell dyscrasia and peripheral neuropathy with a monoclonal antibody to peripheral-nerve myelin. N. Engl. J. Med. 1980, 303, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Lunn, M.P. Neuropathies and paraproteins. Curr. Opin. Neurol. 2019, 32, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Marks, D.; Raza, S. Diagnosis and management of neuropathies associated with plasma cell dyscrasias. Hematol. Oncol. 2018, 36, 3–14. [Google Scholar] [CrossRef]
- Vallat, J.M.; Duchesne, M.; Corcia, P.; Richard, L.; Ghorab, K.; Magy, L.; Mathis, S. The Wide Spectrum of Pathophysiologic Mechanisms of Paraproteinemic Neuropathy. Neurology 2021, 96, 214–225. [Google Scholar] [CrossRef]
- Latov, N. Antibody testing in neuropathy associated with anti-Myelin-Associated Glycoprotein antibodies: Where we are after 40 years. Curr. Opin. Neurol. 2021, 34, 625–630. [Google Scholar] [CrossRef]
- Dalakas, M.C. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617746640. [Google Scholar] [CrossRef] [Green Version]
- Steck, A.J. Anti-MAG neuropathy: From biology to clinical management. J. Neuroimmunol. 2021, 361, 577725. [Google Scholar] [CrossRef]
- Varettoni, M.; Zibellini, S.; Capello, D.; Arcaini, L.; Rossi, D.; Pascutto, C.; Rattotti, S.; Mangiacavalli, S.; Pochintesta, L.; Gotti, M.; et al. Clues to pathogenesis of Waldenström macroglobulinemia and immunoglobulin M monoclonal gammopathy of undetermined significance provided by analysis of immunoglobulin heavy chain gene rearrangement and clustering of B-cell receptors. Leuk. Lymphoma 2013, 54, 2485–2489. [Google Scholar] [CrossRef]
- Paiva, B.; Corchete, L.A.; Vidriales, M.B.; García-Sanz, R.; Perez, J.J.; Aires-Mejia, I.; Sanchez, M.-L.; Barcena, P.; Alignani, D.; Jimenez, C.; et al. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood 2015, 125, 2370–2380. [Google Scholar] [CrossRef]
- García-Sanz, R.; Dogliotti, I.; Zaccaria, G.M.; Ocio, E.M.; Rubio, A.; Murillo, I.; Escalante, F.; Aguilera, C.; García-Mateo, A.; de Coca, A.G.; et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br. J. Haematol. 2021, 192, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Hunter, Z.R.; Yang, G.; Zhou, Y.; Cao, Y.; Liu, X.; Morra, E.; Trojani, A.; Greco, A.; Arcaini, L.; et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013, 121, 2051–2058. [Google Scholar] [CrossRef]
- Cao, X.X.; Meng, Q.; Cai, H.; He, T.H.; Zhang, C.-L.; Su, W.; Sun, J.; Li, Y.; Xu, W.; Zhou, D.-B.; et al. Detection of MYD88 L265P and WHIM-like CXCR4 mutation in patients with IgM monoclonal gammopathy related disease. Ann. Hematol. 2017, 96, 971–976. [Google Scholar] [CrossRef]
- Ferrante, M.; Furlan, D.; Zibellini, S.; Borriero, M.; Candido, C.; Sahnane, N.; Uccella, S.; Genuardi, E.; Alessandria, B.; Bianchi, B.; et al. MYD88L265P Detection in IgM Monoclonal Gammopathies: Methodological Considerations for Routine Implementation. Diagnostics 2021, 11, 779. [Google Scholar] [CrossRef]
- Drandi, D.; Decruyenaere, P.; Ferrante, M.; Offner, F.; Vandesompele, J.; Ferrero, S. Nucleic Acid Biomarkers in Waldenström Macroglobulinemia and IgM-MGUS: Current Insights and Clinical Relevance. Diagnostics 2022, 12, 969. [Google Scholar] [CrossRef]
- Jiménez, C.; Sebastián, E.; Chillón, M.C.; Giraldo, P.; Mariano Hernández, J.; Escalante, F.; González-López, T.J.; Aguilera, C.; de Coca, A.G.; Murillo, I.; et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia 2013, 27, 1722–1728. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Kuzu, I.; Dogan, A.; Dirnhofer, S.; Chan, J.K.C.; Sander, B.; Ott, G.; Xerri, L.; Quintanilla-Martinez, L.; Campo, E. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2016, 468, 259–275. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An Oligomeric Signaling Platform Formed by the Toll-like Receptor Signal Transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [Green Version]
- Balka, K.R.; Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Visentin, A.; Pravato, S.; Castellani, F.; Campagnolo, M.; Angotzi, F.; Cavarretta, C.A.; Cellini, A.; Ruocco, V.; Salvalaggio, A.; Tedeschi, A.; et al. From Biology to Treatment of Monoclonal Gammopathies of Neurological Significance. Cancers 2022, 14, 1562. [Google Scholar] [CrossRef]
- Panwalkar, A.; Verstovsek, S.; Giles, F. Nuclear factor-KappaB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004, 100, 1578–1589. [Google Scholar] [CrossRef]
- Leleu, X.; Eeckhoute, J.; Jia, X.; Roccaro, A.M.; Moreau, A.S.; Farag, M.; Sacco, A.; Ngo, H.T.; Runnels, J.; Melhem, M.R.; et al. Targeting NF-κB in Waldenstrom macroglobulinemia. Blood 2008, 111, 5068–5077. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Buhrlage, S.J.; Tan, L.; Liu, X.; Chen, J.; Xu, L.; Tsakmaklis, N.; Chen, J.G.; Patterson, C.J.; Brown, J.R.; et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood 2016, 127, 3237–3252. [Google Scholar] [CrossRef] [Green Version]
- Pontoriero, M.; Fiume, G.; Vecchio, E.; de Laurentiis, A.; Albano, F.; Iaccino, E.; Mimmi, S.; Pisano, A.; Agosti, V.; Giovannone, E.; et al. Activation of NF-κB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IκB-α. J. Mol. Med. 2019, 97, 675–690. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol.-Neuroimmunol. Neuroinflammat. 2020, 7, e720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.J.; Xu, L.; Gustine, J.N.; Keezer, A.; Meid, K.; Dubeau, T.E.; Liu, X.; Demos, M.G.; Kofides, A.; Tsakmaklis, N.; et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br. J. Haematol. 2019, 187, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Allain, J.S.; Thonier, F.; Pihan, M.; Boulland, M.L.; de Guibert, S.; Launay, V.; Doncker, A.-V.; Ganard, M.; Aliouat, A.; Pangault, C.; et al. IGHV segment utilization in immunoglobulin gene rearrangement differentiates patients with anti-myelin-associated glycoprotein neuropathy from others immunoglobulin M-gammopathies. Haematologica 2018, 103, e207–e210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E.; Mezzapelle, R. The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Kryczek, I.; Wei, S.; Keller, E.; Liu, R.; Zou, W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am. J. Physiol.-Cell Physiol. 2007, 292, C987–C995. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Hunter, Z.R.; Tsakmaklis, N.; Cao, Y.; Yang, G.; Chen, J.; Liu, X.; Kanan, S.; Castillo, J.J.; Tai, Y.-T.; et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström Macroglobulinaemia. Br. J. Haematol. 2016, 172, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Hunter, Z.R.; Liu, X.; Xu, L.; Yang, G.; Chen, J.; Patterson, C.J.; Tsakmaklis, N.; Kanan, S.; Rodig, S.; et al. The WHIM-like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia 2015, 29, 169–176. [Google Scholar] [CrossRef]
- Garbay, B.; Heape, A.M.; Sargueil, F.; Cassagne, C. Myelin synthesis in the peripheral nervous system. Prog. Neurobiol. 2000, 61, 267–304. [Google Scholar] [CrossRef]
- Kidd, G.J.; Ohno, N.; Trapp, B.D. Chapter 5-Biology of Schwann cells. In Handbook of Clinical Neurology; Said, G., Krarup, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 55–79. [Google Scholar]
- Saher, G.; Simons, M. Cholesterol and Myelin Biogenesis. In Cholesterol Binding and Cholesterol Transport Proteins: Structure and Function in Health and Disease; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 489–508. [Google Scholar]
- Patzig, J.; Jahn, O.; Tenzer, S.; Wichert, S.P.; de Monasterio-Schrader, P.; Rosfa, S.; Kuharev, J.; Yan, K.; Bormuth, I.; Bremer, J.; et al. Quantitative and Integrative Proteome Analysis of Peripheral Nerve Myelin Identifies Novel Myelin Proteins and Candidate Neuropathy Loci. J. Neurosci. 2011, 31, 16369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzer, J.L. Polarized Domains of Myelinated Axons. Neuron 2003, 40, 297–318. [Google Scholar] [CrossRef] [Green Version]
- D’Sa, S.; Kersten, M.J.; Castillo, J.J.; Dimopoulos, M.; Kastritis, E.; Laane, E.; Leblond, V.; Merlini, G.; Treon, S.P.; Vos, J.M.; et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: Recommendations from the IWWM-8 consensus panel. Br. J. Haematol. 2017, 176, 728–742. [Google Scholar] [CrossRef]
- Dellagi, K.; Dupouey, P.; Brouet, J.C.; Billecocq, A.; Gomez, D.; Clauvel, J.P.; Seligmann, M. Waldenström’s macroglobulinemia and peripheral neuropathy: A clinical and immunologic study of 25 patients. Blood 1983, 62, 280–285. [Google Scholar] [CrossRef] [Green Version]
- Braun, P.E.; Frail, D.E.; Latov, N. Myelin-associated glycoprotein is the antigen for a monoclonal IgM in polyneuropathy. J. Neurochem. 1982, 39, 1261–1265. [Google Scholar] [CrossRef]
- Leibowitz, S.; Gregson, N.A.; Kennedy, M.; Kahn, S.N. IgM paraproteins with immunological specificity for a Schwann cell component and peripheral nerve myelin in patients with polyneuropathy. J. Neurol. Sci. 1983, 59, 153–165. [Google Scholar] [CrossRef]
- Kahn, S.N.; Smith, I.A.; Eames, R.A.; Thomas, P.K.; Lacey, B.W. IgM paraproteinemia and autoimmune peripheral neuropathy. N. Engl. J. Med. 1981, 304, 1430–1431. [Google Scholar]
- Ilyas, A.A.; Quarles, R.H.; Dalakas, M.C.; Brady, R.O. Polyneuropathy with monoclonal gammopathy: Glycolipids are frequently antigens for IgM paraproteins. Proc. Natl. Acad. Sci. USA 1985, 82, 6697–6700. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, A.A.; Li, S.C.; Chou, D.K.; Li, Y.T.; Jungalwala, F.B.; Dalakas, M.C.; Quarles, R.H. Gangliosides GM2, IV4GalNAcGM1b, and IV4GalNAcGC1a as antigens for monoclonal immunoglobulin M in neuropathy associated with gammopathy. J. Biol. Chem. 1988, 263, 4369–4373. [Google Scholar] [CrossRef]
- Kelly, J.J.; Adelman, L.S.; Berkman, E.; Bhan, I. Polyneuropathies associated with IgM monoclonal gammopathies. Arch. Neurol. 1988, 45, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A. POEMS syndrome: 2021 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2021, 96, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Warren, J.D.; Jacobs, J.M.; Groves, M.J.; Yong, K.; Honan, W.P.; Thomas, P.K.; Reilly, A.M.M. Microvasculitic paraproteinaemic polyneuropathy and B-cell lymphoma. J. Peripher. Nerv. Syst. 2003, 8, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Acute hyperviscosity: Syndromes and management. Blood 2018, 132, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, A.A.; Quarles, R.H.; MacIntosh, T.D.; Dobersen, M.J.; Trapp, B.D.; Dalakas, M.C.; Brady, R.O. IgM in a human neuropathy related to paraproteinemia binds to a carbohydrate determinant in the myelin-associated glycoprotein and to a ganglioside. Proc. Natl. Acad. Sci. USA 1984, 81, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Quarles, R.H.; Ilyas, A.A.; Willison, H.J. Antibodies to glycolipids in demyelinating diseases of the human peripheral nervous system. Chem. Phys. Lipids 1986, 42, 235–248. [Google Scholar] [CrossRef]
- Burger, D.; Simon, M.; Perruisseau, G.; Steck, A.J. The epitope(s) recognized by HNK-1 antibody and IgM paraprotein in neuropathy is present on several N-linked oligosaccharide structures on human P0 and myelin-associated glycoprotein. J. Neurochem. 1990, 54, 1569–1575. [Google Scholar] [CrossRef]
- Willison, H.J.; O’Leary, C.P.; Veitch, J.; Blumhardt, L.D.; Busby, M.; Donaghy, M.; Fuhr, P.; Ford, H.; Hahn, A.; Renaud, S.; et al. The clinical and laboratory features of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies. Brain 2001, 124 Pt 10, 1968–1977. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, A.A.; Quarles, R.H.; Dalakas, M.C.; Fishman, P.H.; Brady, R.O. Monoclonal IgM in a patient with paraproteinemic polyneuropathy binds to gangliosides containing disialosyl groups. Ann. Neurol. 1985, 18, 655–659. [Google Scholar] [CrossRef]
- Carpo, M.; Meucci, N.; Allaria, S.; Marmiroli, P.; Monaco, S.; Toscano, A.; Simonetti, S.; Scarlato, G.; Nobile-Orazio, E. Anti-sulfatide IgM antibodies in peripheral neuropathy. J. Neurol. Sci. 2000, 176, 144–150. [Google Scholar] [CrossRef]
- Pestronk, A.; Li, F.; Griffin, J.; Feldman, E.L.; Cornblath, D.; Trotter, J.; Zhu, S.; Yee, W.C.; Phillips, D.; Peeples, D.M.; et al. Polyneuropathy syndromes associated with serum antibodies to sulfatide and myelin-associated glycoprotein. Neurology 1991, 41, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Dabby, R.; Weimer, L.H.; Hays, A.P.; Olarte, M.; Latov, N. Antisulfatide antibodies in neuropathy: Clinical and electrophysiologic correlates. Neurology 2000, 54, 1448–1452. [Google Scholar] [CrossRef] [PubMed]
- Petratos, S.; Turnbull, V.J.; Papadopoulos, R.; Ayers, M.; Gonzales, M.F. High-titre IgM anti-sulfatide antibodies in individuals with IgM paraproteinaemia and associated peripheral neuropathy. Immunol. Cell Biol. 2000, 78, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Boso, F.; Ruggero, S.; Giannotta, C.; Benedetti, L.; Marfia, G.A.; Ermani, M.; Campagnolo, M.; Salvalaggio, A.; Gallia, F.; De Michelis, C.; et al. Anti-sulfatide/galactocerebroside antibodies in immunoglobulin M paraproteinemic neuropathies. Eur. J. Neurol. 2017, 24, 1334–1340. [Google Scholar] [CrossRef]
- Willison, H.J. Anti-ganglioside Antibodies in Peripheral Nerve Pathology. In Gangliosides: Methods and Protocols; Sonnino, S., Prinetti, A., Eds.; Springer: New York, NY, USA, 2018; pp. 173–188. [Google Scholar]
- Briani, C.; Berger, J.S.; Latov, N. Antibodies to chondroitin sulfate C: A new detection assay and correlations with neurological diseases. J. Neuroimmunol. 1998, 84, 117–121. [Google Scholar] [CrossRef]
- Matà, S.; Torricelli, S.; Barilaro, A.; Grippo, A.; Forleo, P.; del Mastio, M.; Sorbi, S. Polyneuropathy and monoclonal gammopathy of undetermined significance (MGUS); update of a clinical experience. J. Neurol. Sci. 2021, 423, 117335. [Google Scholar] [CrossRef]
- Nobile-Orazio, E.; Manfredini, E.; Carpo, M.; Meucci, N.; Monaco, S.; Ferrari, S.; Bonetti, B.; Cavaletti, G.; Gemignani, F.; Durelli, L.; et al. Frequency and clinical correlates of anti-neural IgM antibodies in neuropathy associated with IgM monoclonal gammopathy. Ann. Neurol. 1994, 36, 416–424. [Google Scholar] [CrossRef]
- Ellie, E.; Vital, A.; Steck, A.; Boiron, J.M.; Vital, C.; Julien, J. Neuropathy associated with “benign” anti-myelin-associated glycoprotein IgM gammopathy: Clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J. Neurol. 1996, 243, 34–43. [Google Scholar] [CrossRef]
- Chassande, B.; Léger, J.M.; Younes-Chennoufi, A.B.; Bengoufa, D.; Maisonobe, T.; Bouche, P.; Baumann, N. Peripheral neuropathy associated with IgM monoclonal gammopathy: Correlations between M-protein antibody activity and clinical/electrophysiological features in 40 cases. Muscle Nerve 1998, 21, 55–62. [Google Scholar] [CrossRef]
- Katz, J.S.; Saperstein, D.S.; Gronseth, G.; Amato, A.A.; Barohn, R.J. Distal acquired demyelinating symmetric neuropathy. Neurology 2000, 54, 615–620. [Google Scholar] [CrossRef]
- Bardel, B.; Molinier-Frenkel, V.; le Bras, F.; Ayache, S.S.; Nordine, T.; Lefaucheur, J.P.; Planté-Bordeneuve, V. Revisiting the spectrum of IgM-related neuropathies in a large cohort of IgM monoclonal gammopathy. J. Neurol. 2022, 269, 4955–4960. [Google Scholar] [CrossRef] [PubMed]
- Nobile-Orazio, E.; Francomano, E.; Daverio, R.; Barbieri, S.; Marmiroli, P.; Manfredini, E.; Carpo, M.; Moggio, M.; Legname, G.; Baldini, L.; et al. Anti-myelin-associated glycoprotein IgM antibody titers in neuropathy associated with macroglobulinemia. Ann. Neurol. 1989, 26, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Kuijf, M.L.; Eurelings, M.; Tio-Gillen, A.P.; van Doorn, P.A.; van den Berg, L.H.; Hooijkaas, H.; Stork, J.; Notermans, N.C.; Jacobs, B.C. Detection of anti-MAG antibodies in polyneuropathy associated with IgM monoclonal gammopathy. Neurology 2009, 73, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Jaskowski, T.D.; Martins, T.B.; Litwin, C.M.; Hill, H.R. Immunoglobulin (Ig) M antibody against myelin associated glycoprotein (MAG): A comparison of methods. J. Clin. Lab. Anal. 2004, 18, 247–250. [Google Scholar] [CrossRef]
- Jaskowski, T.D.; Prince, H.E.; Greer, R.W.; Litwin, C.M.; Hill, H.R. Further comparisons of assays for detecting MAG IgM autoantibodies. J. Neuroimmunol. 2007, 187, 175–178. [Google Scholar] [CrossRef]
- Matà, S.; Ambrosini, S.; Saccomanno, D.; Biagioli, T.; Carpo, M.; Amantini, A.; Giannini, F.; Barilaro, A.; Toscani, L.; Del Mastio, M.; et al. Anti-MAG IgM: Differences in antibody tests and correlation with clinical findings. Neurol. Sci. 2020, 41, 365–372. [Google Scholar] [CrossRef]
- Liberatore, G.; Giannotta, C.; Sajeev, B.P.; Morenghi, E.; Terenghi, F.; Gallia, F.; Doneddu, P.E.; Manganelli, F.; Cocito, D.; Filosto, M.; et al. Sensitivity and specificity of a commercial ELISA test for anti-MAG antibodies in patients with neuropathy. J. Neuroimmunol. 2020, 345, 577288. [Google Scholar] [CrossRef]
- Svahn, J.; Petiot, P.; Antoine, J.C.; Vial, C.; Delmont, E.; Viala, K.; Steck, A.J.; Magot, A.; Cauquil, C.; Zarea, A.; et al. Anti-MAG antibodies in 202 patients: Clinicopathological and therapeutic features. J. Neurol. Neurosurg. Psychiatry 2018, 89, 499–505. [Google Scholar] [CrossRef]
- Magy, L.; Kaboré, R.; Mathis, S.; Lebeau, P.; Ghorab, K.; Caudie, C.; Vallat, J.-M. Heterogeneity of Polyneuropathy Associated with Anti-MAG Antibodies. J. Immunol. Res. 2015, 2015, 450391. [Google Scholar] [CrossRef]
- Hafler, D.A.; Johnson, D.; Kelly, J.J.; Panitch, H.; Kyle, R.; Weiner, H.L. Monoclonal gammopathy and neuropathy: Myelin-associated glycoprotein reactivity and clinical characteristics. Neurology 1986, 36, 75–78. [Google Scholar] [CrossRef]
- Smith, I.S. The natural history of chronic demyelinating neuropathy associated with benign IgM paraproteinaemia. A clinical and neurophysiological study. Brain 1994, 117 Pt 5, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Galassi, G.; Tondelli, M.; Ariatti, A.; Benuzzi, F.; Nichelli, P.; Valzania, F. Long-term disability and prognostic factors in polyneuropathy associated with anti-myelin-associated glycoprotein (MAG) antibodies. Int. J. Neurosci. 2017, 127, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Saifee, T.A.; Schwingenschuh, P.; Reilly, M.M.; Lunn, M.P.; Katschnig, P.; Kassavetis, P.; Pareés, I.; Manji, H.; Bhatia, K.; Rothwell, J.C.; et al. Tremor in inflammatory neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1282–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlskog, M.C.; Kumar, N.; Mauermann, M.L.; Klein, C.J. IgM-monoclonal gammopathy neuropathy and tremor: A first epidemiologic case control study. Park. Relat. Disord. 2012, 18, 748–752. [Google Scholar] [CrossRef] [Green Version]
- Bain, P.G.; Britton, T.C.; Jenkins, I.H.; Thompson, P.D.; Rothwell, J.C.; Thomas, P.K.; Brooks, D.; Marsden, C.D. Tremor associated with benign IgM paraproteinaemic neuropathy. Brain 1996, 119 Pt 3, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Nobile-Orazio, E.; Meucci, N.; Baldini, L.; di Troia, A.; Scarlato, G. Long-term prognosis of neuropathy associated with anti-MAG IgM M-proteins and its relationship to immune therapies. Brain 2000, 123 Pt 4, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Joint Task Force of the EFNS and The PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of multifocal motor neuropathy. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society-first revision. J. Peripher. Nerv. Syst. 2010, 15, 295–301. [Google Scholar] [CrossRef]
- Kelly, J.J., Jr. The electrodiagnostic findings in polyneuropathies associated with IgM monoclonal gammopathies. Muscle Nerve 1990, 13, 1113–1117. [Google Scholar] [CrossRef]
- Goedee, H.S.; Notermans, N.C.; Visser, L.H.; van Asseldonk, J.H.; Franssen, H.; Vrancken, A.; Nikolakopoulos, S.; Berg, L.H.V.D.; Pol, W.L. Neuropathy associated with immunoglobulin M monoclonal gammopathy: A combined sonographic and nerve conduction study. Muscle Nerve 2019, 60, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Franssen, H.; Notermans, N.C. Length dependence in polyneuropathy associated with IgM gammopathy. Ann. Neurol. 2006, 59, 365–371. [Google Scholar] [CrossRef]
- Kaku, D.A.; England, J.D.; Sumner, A.J. Distal accentuation of conduction slowing in polyneuropathy associated with antibodies to myelin-associated glycoprotein and sulphated glucuronyl paragloboside. Brain 1994, 117, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Lozeron, P.; Ribrag, V.; Adams, D.; Brisset, M.; Vignon, M.; Baron, M.; Malphettes, M.; Theaudin, M.; Arnulf, B.; Kubis, N. Is distal motor and/or sensory demyelination a distinctive feature of anti-MAG neuropathy? J. Neurol. 2016, 263, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Notermans, N.C.; Wokke, J.H.J.; Franssen, H. Entrapment in anti myelin-associated glycoprotein neuropathy. J. Neurol. 2009, 256, 620. [Google Scholar] [CrossRef] [PubMed]
- Bourque, P.R.; Masson-Roy, J.; Warman-Chardon, J.; Massie, R.; Melanson, M.; Brooks, J.; Breiner, A. Temporal evolution of nerve conduction study abnormalities in anti-myelin-associated glycoprotein neuropathy. Muscle Nerve 2021, 63, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Quarles, R.H. Presence of the myelin-associated glycoprotein correlates with alterations in the periodicity of peripheral myelin. J. Cell Biol. 1982, 92, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Erb, M.; Flueck, B.; Kern, F.; Erne, B.; Steck, A.J.; Schaeren-Wiemers, N. Unraveling the differential expression of the two isoforms of myelin-associated glycoprotein in a mouse expressing GFP-tagged S-MAG specifically regulated and targeted into the different myelin compartments. Mol. Cell. Neurosci. 2006, 31, 613–627. [Google Scholar] [CrossRef]
- Lopez, P.H.H. Role of Myelin-Associated Glycoprotein (Siglec-4a) in the Nervous System. In Glycobiology of the Nervous System; Yu, R.K., Schengrund, C.L., Eds.; Springer: New York, NY, USA, 2014; pp. 245–262. [Google Scholar]
- Yin, X.; Crawford, T.O.; Griffin, J.W.; Tu, P.H.; Lee, V.M.Y.; Li, C.; Order, J.; Trapp, B.D. Myelin-Associated Glycoprotein Is a Myelin Signal that Modulates the Caliber of Myelinated Axons. J. Neurosci. 1998, 18, 1953. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Mehta, N.R.; Conant, K.; Kim, K.J.; Jones, M.; Calabresi, P.A.; Melli, G.; Hoke, A.; Schnaar, R.; Ming, G.-L.; et al. Axonal Protective Effects of the Myelin-Associated Glycoprotein. J. Neurosci. 2009, 29, 630. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.D.; Luciano, C.A.; Quarles, R.H. Nerve conduction abnormalities in aging mice deficient for myelin-associated glycoprotein. Muscle Nerve 2001, 24, 1380–1387. [Google Scholar] [CrossRef]
- Quarles, R.H. Myelin-associated glycoprotein (MAG): Past, present and beyond. J. Neurochem. 2007, 100, 1431–1448. [Google Scholar] [CrossRef]
- Kelm, S.; Pelz, A.; Schauer, R.; Filbin, M.T.; Tang, S.; de Bellard, M.E.; Schnaar, R.L.; Mahoney, J.A.; Hartnell, A.; Bradfield, P.; et al. Sialoadhesin, myelin-associated glycoprotein and CD22 define a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily. Curr. Biol. 1994, 4, 965–972. [Google Scholar] [CrossRef]
- Crocker, P.R.; Clark, E.A.; Filbin, M.; Gordon, S.; Jones, Y.; Kehrl, J.H.; Kelm, S.; Le Douarin, N.; Powell, L.; Roder, J.; et al. Siglecs: A family of sialic-acid binding lectins. Glycobiology 1998, 8, 5–6. [Google Scholar]
- Chester, M.A. IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN) Nomenclature of glycolipids. Eur. J. Biochem. 1998, 257, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Pronker, M.F.; Lemstra, S.; Snijder, J.; Heck, A.J.R.; Thies-Weesie, D.M.E.; Pasterkamp, R.J.; Janssen, B.J.C. Structural basis of myelin-associated glycoprotein adhesion and signalling. Nat. Commun. 2016, 7, 13584. [Google Scholar] [CrossRef]
- Takatsu, M.; Hays, A.P.; Latov, N.; Abrams, G.M.; Nemni, R.; Sherman, W.H.; Nobile-Orazio, E.; Saito, T.; Freddo, L. Immunofluorescence study of patients with neuropathy and IgM M proteins. Ann. Neurol. 1985, 18, 173–181. [Google Scholar] [CrossRef]
- Vital, A.; Vital, C.; Julien, J.; Baquey, A.; Steck, A.J. Polyneuropathy associated with IgM monoclonal gammopathy. Acta Neuropathol. 1989, 79, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.M.; Erne, B.; Bernasconi, L.; Tosi, C.; Probst, A.; Landmann, L.; Steck, A.J. Confocal microscopic localization of anti-myelin-associated glycoprotein autoantibodies in a patient with peripheral neuropathy initially lacking a detectable IgM gammopathy. Acta Neuropathol. 1998, 95, 540–546. [Google Scholar] [CrossRef]
- Kawagashira, Y.; Koike, H.; Tomita, M.; Morozumi, S.; Iijima, M.; Nakamura, T.; Katsuno, M.; Tanaka, F.; Sobue, G. Morphological Progression of Myelin Abnormalities in IgM-Monoclonal Gammopathy of Undetermined Significance Anti-Myelin-Associated Glycoprotein Neuropathy. J. Neuropathol. Exp. Neurol. 2010, 69, 1143–1157. [Google Scholar] [CrossRef] [Green Version]
- Vital, A.; Lagueny, A.; Julien, J.; Ferrer, X.; Barat, M.; Hermosilla, E.; Rouanet-Larrivière, M.; Henry, P.; Bredin, A.; Louiset, P.; et al. Chronic inflammatory demyelinating polyneuropathy associated with dysglobulinemia: A peripheral nerve biopsy study in 18 cases. Acta Neuropathol. 2000, 100, 63–68. [Google Scholar] [CrossRef]
- Monaco, S.; Bonetti, B.; Ferrari, S.; Moretto, G.; Nardelli, E.; Tedesco, F.; Mollnes, T.E.; Nobile-Orazio, E.; Manfredini, E.; Bonazzi, L.; et al. Complement-mediated demyelination in patients with IgM monoclonal gammopathy and polyneuropathy. N. Engl. J. Med. 1990, 322, 649–652. [Google Scholar] [CrossRef]
- Lunn, M.P.; Crawford, T.O.; Hughes, R.A.; Griffin, J.W.; Sheikh, K.A. Anti-myelin-associated glycoprotein antibodies alter neurofilament spacing. Brain 2002, 125 Pt 4, 904–911. [Google Scholar] [PubMed] [Green Version]
- Madrid, R.; Bradley, W.G. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): Studies on the formation of the abnormal myelin sheath. J. Neurol. Sci. 1975, 25, 415–448. [Google Scholar] [CrossRef]
- Kawagashira, Y.; Koike, H.; Takahashi, M.; Ohyama, K.; Iijima, M.; Katsuno, M.; Niwa, J.-I.; Doyu, M.; Sobue, G. Aberrant Expression of Nodal and Paranodal Molecules in Neuropathy Associated With IgM Monoclonal Gammopathy With Anti-Myelin-Associated Glycoprotein Antibodies. J. Neuropathol. Exp. Neurol. 2020, 79, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Erne, B.; Lauria, G.; Pareyson, D.; Borgna, M.; Morbin, M.; Arnold, A.; Czaplinski, A.; Fuhr, P.; Schaeren-Wiemers, N.; et al. IgM deposits on skin nerves in anti-myelin-associated glycoprotein neuropathy. Ann. Neurol. 2005, 57, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Tatum, A.H. Experimental paraprotein neuropathy, demyelination by passive transfer of human IgM anti-myelin-associated glycoprotein. Ann. Neurol. 1993, 33, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J.; Trapp, B.D.; Bacher, J.D.; Dalakas, M.C.; Griffin, J.W.; Quarles, R.H. Demyelination induced by intraneural injection of human antimyelin-associated glycoprotein antibodies. Muscle Nerve 1988, 11, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, A.A.; Gu, Y.; Dalakas, M.C.; Quarles, R.H.; Bhatt, S. Induction of experimental ataxic sensory neuronopathy in cats by immunization with purified SGPG. J. Neuroimmunol. 2008, 193, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Bigbee, J.W.; Maeda, R.; Miyatani, N.; Kalb, R.G.; Yu, R.K. Induction of demyelination by intraneural injection of antibodies against sulfoglucuronyl paragloboside. Exp. Neurol. 1991, 113, 221–225. [Google Scholar] [CrossRef]
- Ritz, M.F.; Erne, B.; Ferracin, F.; Vital, A.; Vital, C.; Steck, A.J. Anti-MAG IgM penetration into myelinated fibers correlates with the extent of myelin widening. Muscle Nerve 1999, 22, 1030–1037. [Google Scholar] [CrossRef]
- Hays, A.P.; Lee, S.S.L.; Latov, N. Immune reactive C3d on the surface of myelin sheaths in neuropathy. J. Neuroimmunol. 1988, 18, 231–244. [Google Scholar] [CrossRef]
- Stork, A.C.; Cats, E.A.; Vlam, L.; Heezius, E.; Rooijakkers, S.; Herpers, B.; De Jong, B.A.; Rijkers, G.; Van Strijp, J.; Notermans, N.C.; et al. Classical and lectin complement pathway activity in polyneuropathy associated with IgM monoclonal gammopathy. J. Neuroimmunol. 2016, 290, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Amaador, K.; Wieske, L.; Koel-Simmelink, M.J.A.; Kamp, A.; Jongerius, I.; de Heer, K.; Teunissen, C.E.; Minnema, M.C.; Notermans, N.C.; Eftimov, F.; et al. Serum neurofilament light chain, contactin-1 and complement activation in anti-MAG IgM paraprotein-related peripheral neuropathy. J. Neurol. 2022, 269, 3700–3705. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.M.; Notermans, N.C.; D’Sa, S.; Lunn, M.P.; van der Pol, W.L.; Kraan, W.; Reilly, M.M.; Chalker, J.; Gupta, R.; Kersten, M.-J.; et al. High prevalence of the MYD88 L265P mutation in IgM anti-MAG paraprotein-associated peripheral neuropathy. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1007–1009. [Google Scholar] [CrossRef]
- Hänggi, P.; Aliu, B.; Martin, K.; Herrendorff, R.; Steck, A.J. Decrease in Serum Anti-MAG Autoantibodies Is Associated With Therapy Response in Patients With Anti-MAG Neuropathy: Retrospective Study. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1109. [Google Scholar] [CrossRef] [PubMed]
- Niermeijer, J.M.; Fischer, K.; Eurelings, M.; Franssen, H.; Wokke, J.H.; Notermans, N.C. Prognosis of polyneuropathy due to IgM monoclonal gammopathy: A prospective cohort study. Neurology 2010, 74, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Low, P.A.; Windebank, A.J.; Jaradeh, S.S.; Gosselin, S.; Bourque, P.; Smith, B.E.; Kratz, K.M.; Karnes, J.L.; Evans, B.A.; et al. Plasma exchange in polyneuropathy associated with monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 1991, 325, 1482–1486. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Chevret, S.; Léger, J.M.; Louboutin, J.P.; Bussel, A.; Brouet, J.C. Plasma exchange and chlorambucil in polyneuropathy associated with monoclonal IgM gammopathy. IgM-associated Polyneuropathy Study Group. J. Neurol. Neurosurg. Psychiatry 1995, 59, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Gorson, K.C.; Ropper, A.H.; Weinberg, D.H.; Weinstein, R. Treatment experience in patients with anti–myelin-associated glycoprotein neuropathy. Muscle Nerve 2001, 24, 778–786. [Google Scholar] [CrossRef]
- Baron, M.; Lozeron, P.; Harel, S.; Bengoufa, D.; Vignon, M.; Asli, B.; Malphettes, M.; Parquet, N.; Brignier, A.; Fermand, J.P.; et al. Plasma exchanges for severe acute neurological deterioration in patients with IgM anti-myelin-associated glycoprotein (anti-MAG) neuropathy. J. Neurol. 2017, 264, 1132–1135. [Google Scholar] [CrossRef]
- Siciliano, G.; Moriconi, L.; Gianni, G.; Richieri, E.; Vignocchi, M.G.; Rossi, B. Selective techniques of apheresis in polyneuropathy associated with monoclonal gammopathy of undetermined significance. Acta Neurol. Scand. 1994, 89, 117–122. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti–myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Léger, J.M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Campagnolo, M.; Zambello, R.; Nobile-Orazio, E.; Benedetti, L.; Marfia, G.A.; Riva, N.; Castellani, F.; Bianco, M.; Salvalaggio, A.; Garnero, M.; et al. IgM MGUS and Waldenstrom-associated anti-MAG neuropathies display similar response to rituximab therapy. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1094–1097. [Google Scholar] [CrossRef]
- Niermeijer, J.M.; Eurelings, M.; van der Linden, M.W.; Lokhorst, H.M.; Franssen, H.; Fischer, K.; Teunissen, L.L.; Berg, L.H.V.D.; Schobben, F.; Wokke, J.H.J.; et al. Intermittent cyclophosphamide with prednisone versus placebo for polyneuropathy with IgM monoclonal gammopathy. Neurology 2007, 69, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Hamidou, M.A.; Belizna, C.; Wiertlewsky, S.; Audrain, M.; Biron, C.; Grolleau, J.Y.; Mussini, J.-M. Intravenous cyclophosphamide in refractory polyneuropathy associated with IgM monoclonal gammopathy: An uncontrolled open trial. Am. J. Med. 2005, 118, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.C.; Lunn, M.P.T.; Schey, S.; Hughes, R.A.C. Successful treatment of IgM paraproteinaemic neuropathy with fludarabine. J. Neurol. Neurosurg. Psychiatry 1999, 66, 575. [Google Scholar] [CrossRef] [Green Version]
- Niermeijer, J.M.; Eurelings, M.; Lokhorst, H.; Franssen, H.; Fijnheer, R.; Wokke, J.H.; Notermans, N.C.; Halperin, J.J.; Landis, D.M.; Kleinman, G.M. Neurologic and hematologic response to fludarabine treatment in IgM MGUS polyneuropathy. Neurology 2006, 67, 2076–2079. [Google Scholar] [CrossRef]
- Ghosh, A.; Littlewood, T.; Donaghy, M. Cladribine in the treatment of IgM paraproteinemic polyneuropathy. Neurology 2002, 59, 1290. [Google Scholar] [CrossRef]
- Ernerudh, J.H.; Vrethem, M.; Andersen, O.; Lindberg, C.; Berlin, G. Immunochemical and clinical effects of immunosuppressive treatment in monoclonal IgM neuropathy. J. Neurol. Neurosurg. Psychiatry 1992, 55, 930–934. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C.; Quarles, R.H.; Farrer, R.G.; Dambrosia, J.; Soueidan, S.; Stein, D.P.; Cupler, E.; Sekul, E.A.; Otero, C. A controlled study of intravenous immunoglobulin in demyelinating neuropathy with IgM gammopathy. Ann. Neurol. 1996, 40, 792–795. [Google Scholar] [CrossRef]
- Comi, G.; Roveri, L.; Swan, A.; Willison, H.; Bojar, M.; Illa, I.; Karageorgiou, C.; Nobile-Orazio, E.; Bergh, P.V.D.; Hughes, R.; et al. A randomised controlled trial of intravenous immunoglobulin in IgM paraprotein associated demyelinating neuropathy. J. Neurol. 2002, 249, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Brouet, J.C.; Chevret, S.; Léger, J.M.; Clavelou, P.; Pouget, J.; Vallat, J.; Vial, C. A randomised double blind trial versus placebo does not confirm the benefit of α-interferon in polyneuropathy associated with monoclonal IgM. J. Neurol. Neurosurg. Psychiatry 2000, 69, 279. [Google Scholar] [CrossRef] [PubMed]
- Notermans, N.C.; Vermeulen, M.; Lokhorst, H.M.; van Doorn, P.A.; van den Berg, L.H.; Teunissen, L.L. Pulsed high-dose dexamethasone treatment of polyneuropathy associated with monoclonal gammopathy. J. Neurol. 1997, 244, 462–463. [Google Scholar] [CrossRef] [Green Version]
- Gorson, K.C.; Amato, A.A.; Ropper, A.H. Efficacy of mycophenolate mofetil in patients with chronic immune demyelinating polyneuropathy. Neurology 2004, 63, 715–717. [Google Scholar] [CrossRef]
- Lunn, M.P.; Nobile-Orazio, E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst. Rev. 2016, 10, Cd002827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruppers, M.H.J.; Merkies, I.S.J.; Notermans, N.C. Recent advances in outcome measures in IgM-anti-MAG+ neuropathies. Curr. Opin. Neurol. 2015, 28, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Pruppers, M.H.J.; Merkies, I.S.J.; Lunn, M.P.T.; Notermans, N.C.; van den Bergh, P.; Blomkwist-Markens, P. 230th ENMC International Workshop: Improving future assessment and research in IgM anti-MAG peripheral neuropathy: A consensus collaborative effort, Naarden, The Netherlands, 24–26 February 2017. Neuromuscul. Disord. 2017, 27, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.; Sherman, W.; Olarte, M.R.; Lovelace, R.E. Peripheral neuropathy associated with plasma cell dyscrasia: A clinical and electrophysiological follow-up study. Acta Neurol. Scand. 1987, 75, 244–248. [Google Scholar] [CrossRef]
- van Meerten, T.; Hagenbeek, A. CD20-Targeted Therapy: The Next Generation of Antibodies. Semin. Hematol. 2010, 47, 199–210. [Google Scholar] [CrossRef]
- Leget, G.A.; Czuczman, M.S. Use of rituximab, the new FDA-approved antibody. Curr. Opin. Oncol. 1998, 10, 548–551. [Google Scholar] [CrossRef]
- van Sorge, N.M.; van der Pol, W.L.; van de Winkel, J.G.J. FcγR polymorphisms: Implications for function, disease susceptibility and immunotherapy. Tissue Antigens 2003, 61, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.; Pestronk, A.; Florence, J.; Al-Lozi, M.T.; Lopate, G.; Miller, T.; Ramneantu, I.; Waheed, W.; Stambuk, M.; Stone, M.J.; et al. Peripheral neuropathies in Waldenström’s macroglobulinaemia. J. Neurol. Neurosurg. Psychiatry 2006, 77, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M. Rituximab in anti-MAG neuropathy: More evidence for efficacy and more predictive factors. J. Neurol. Sci. 2017, 377, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, L.; Briani, C.; Grandis, M.; Vigo, T.; Gobbi, M.; Ghiglione, E.; Carpo, M.; Cocito, D.; Caporale, C.M.; Sormani, M.P.; et al. Predictors of response to rituximab in patients with neuropathy and anti–myelin associated glycoprotein immunoglobulin M. J. Peripher. Nerv. Syst. 2007, 12, 102–107. [Google Scholar] [CrossRef]
- Gazzola, S.; Delmont, E.; Franques, J.; Boucraut, J.; Salort-Campana, E.; Verschueren, A.; Sagui, E.; Hubert, A.-M.; Pouget, J.; Attarian, S. Predictive factors of efficacy of rituximab in patients with anti-MAG neuropathy. J. Neurol. Sci. 2017, 377, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Kawagashira, Y.; Koike, H.; Ohyama, K.; Hashimoto, R.; Iijima, M.; Adachi, H.; Katsuno, M.; Chapman, M.; Lunn, M.; Sobue, G. Axonal loss influences the response to rituximab treatment in neuropathy associated with IgM monoclonal gammopathy with anti-myelin-associated glycoprotein antibody. J. Neurol. Sci. 2015, 348, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Stork, A.C.; Notermans, N.C.; van den Berg, L.H.; Schellevis, R.D.; Niermeijer, J.M.; Nederend, M.; Leusen, J.H.W.; van der Pol, W. Fcγ receptor IIIA genotype is associated with rituximab response in antimyelin-associated glycoprotein neuropathy. J. Neurol. Neurosurg. Psychiatry 2014, 85, 918–920. [Google Scholar] [CrossRef]
- Sala, E.; Robert-Varvat, F.; Paul, S.; Camdessanché, J.P.; Antoine, J.C. Acute neurological worsening after Rituximab treatment in patients with anti-MAG neuropathy. J. Neurol. Sci. 2014, 345, 224–227. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Zervas, C.; Zomas, A.; Kiamouris, C.; Viniou, N.A.; Grigoraki, V.; Karkantaris, C.; Mitsouli, C.; Gika, D.; Christakis, J.; et al. Treatment of Waldenström’s Macroglobulinemia With Rituximab. J. Clin. Oncol. 2002, 20, 2327–2333. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Fonseca, R.; Greipp, P.R.; Blood, E.; Rue, M.; Vesole, D.H.; Gertz, M.A. Initial immunoglobulin M ‘flare’ after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia. Cancer 2004, 101, 2593–2598. [Google Scholar] [CrossRef]
- Vo, M.L.; Martin, P.; Latov, N. Correlation of Changes in Gait Parameters, With Phenotype, Outcome Measures, and Electrodiagnostic Abnormalities in a Patient With Anti-MAG Neuropathy After Exacerbation and Improvement. J. Clin. Neuromuscul. Dis. 2015, 17, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Advani, R.H.; Branagan, A.R.; Buske, C.; Dimopoulos, M.A.; D’Sa, S.; Kersten, M.J.; Leblond, V.; Minnema, M.C.; Owen, R.G.; et al. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020, 7, e827–e837. [Google Scholar] [CrossRef]
- Colchester, N.T.H.; Allen, D.; Katifi, H.A.; Burt, T.; Lown, R.N.; Pinto, A.A.; Duncombe, A.S. Chemoimmunotherapy with rituximab, cyclophosphamide and prednisolone in IgM paraproteinaemic neuropathy: Evidence of sustained improvement in electrophysiological, serological and functional outcomes. Haematologica 2021, 106, 302–305. [Google Scholar] [CrossRef]
- Gruson, B.; Ghomari, K.; Beaumont, M.; Garidi, R.; Just, A.; Merle, P.; Merlusca, L.; Marolleau, J.P.; Royer, B. Long-term response to rituximab and fludarabine combination in IgM anti-myelin-associated glycoprotein neuropathy. J. Peripher. Nerv. Syst. 2011, 16, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Massa, F.; Zuppa, A.; Pesce, G.; Demichelis, C.; Bergamaschi, M.; Garnero, M.; Briani, C.; Ferrari, S.; Schenone, A.; Benedetti, L. Bendamustine-rituximab (BR) combined therapy for treatment of immuno-mediated neuropathies associated with hematologic malignancy. J. Neurol. Sci. 2020, 413, 116777. [Google Scholar] [CrossRef]
- Hospital, M.A.; Viala, K.; Dragomir, S.; Levy, V.; Cohen-Aubart, F.; Neil, J.; Musset, L.; Choquet, S.; Leger, J.-M.; Leblond, V. Immunotherapy-based regimen in anti-MAG neuropathy: Results in 45 patients. Haematologica 2013, 98, e155–e157. [Google Scholar] [CrossRef]
- Nivet, T.; Baptiste, A.; Belin, L.; Ghillani-Dalbin, P.; Algrin, C.; Choquet, S.; Lamy, T.; Morel, V.; Musset, L.; Roos-Weil, D.; et al. Immunochemotherapy versus rituximab in anti-myelin-associated glycoprotein neuropathy: A report of 64 patients. Br. J. Haematol. 2022, 198, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.; El-Sharkawi, D.; Kothari, J.; D’Sa, S.; Auer, R.; McCarthy, H.; Krishna, R.; Miles, O.; Kyriakou, C.; Owen, R. Diagnosis and management of Waldenström macroglobulinaemia—A British Society for Haematology guideline. Br. J. Haematol. 2022, 197, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Leblond, V.; Dimopoulos, M.A.; Kimby, E.; Staber, P.; Kersten, M.J.; Tedeschi, A.; Buske, C. Waldenström’s macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv41–iv50. [Google Scholar] [CrossRef]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Minnema, M.C.; Vos, J.; Eftimov, F.; Vrancken, A. P-034: MAGNAZ trial-A prospective phase II study in patients with monoclonal gammopathy of unknown significance (MGUS) and anti-Myelin Associated Glycoprotein (MAG) Neuropathy and Zanubrutinib Treatment. Clin. Lymphoma Myeloma Leuk. 2021, 21, S57. [Google Scholar] [CrossRef]
- Castillo, J.J.; Allan, J.N.; Siddiqi, T.; Advani, R.H.; Meid, K.; Leventoff, C.; White, T.P.; Flynn, C.A.; Sarosiek, S.; Branagan, A.R.; et al. Venetoclax in Previously Treated Waldenström Macroglobulinemia. J. Clin. Oncol. 2021, 40, 63–71. [Google Scholar] [CrossRef]
- Treon, S.P.; Meid, K.; Hunter, Z.R.; Flynn, C.A.; Sarosiek, S.R.; Leventoff, C.R.; White, T.P.; Cao, Y.; Roccaro, A.M.; Sacco, A.; et al. Phase 1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4-mutated Waldenström macroglobulinemia. Blood 2021, 138, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Dimopoulos, M.A. Proteasome Inhibitors in Waldenström Macroglobulinemia. Hematol. Oncol. Clin. North Am. 2018, 32, 829–840. [Google Scholar] [CrossRef]
- Casan, J.M.L.; Wong, J.; Northcott, M.J.; Opat, S. Anti-CD20 monoclonal antibodies: Reviewing a revolution. Hum. Vaccines Immunother. 2018, 14, 2820–2841. [Google Scholar] [CrossRef]
- Tobinai, K.; Klein, C.; Oya, N.; Fingerle-Rowson, G. A Review of Obinutuzumab (GA101), a Novel Type II Anti-CD20 Monoclonal Antibody, for the Treatment of Patients with B-Cell Malignancies. Adv. Ther. 2017, 34, 324–356. [Google Scholar] [CrossRef] [Green Version]
- Rakocevic, G.; Martinez-Outschoorn, U.; Dalakas, M.C. Obinutuzumab, a potent anti–B-cell agent, for rituximab-unresponsive IgM anti-MAG neuropathy. Neurol.-Neuroimmunol. Neuroinflammat. 2018, 5, e460. [Google Scholar] [CrossRef] [Green Version]
- Briani, C.; Visentin, A.; Salvalaggio, A.; Cacciavillani, M.; Trentin, L. Obinutuzumab, a new anti-CD20 antibody, and chlorambucil are active and effective in anti-myelin-associated glycoprotein antibody polyneuropathy. Eur. J. Neurol. 2019, 26, 371–375. [Google Scholar] [CrossRef]
- Briani, C.; Visentin, A.; Castellani, F.; Cacciavillani, M.; Trentin, L. The BCL2 Inhibitor Venetoclax Plus Rituximab Is Active in MYD88 Wild-Type Polyneuropathy With Anti-MAG Antibodies. Neurol.-Neuroimmunol. Neuroinflammation 2022, 9, e1181. [Google Scholar] [CrossRef]
- Howard, J.F., Jr. Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 2018, 1412, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Querol, L.A.; Hartung, H.P.; Lewis, R.A.; van Doorn, P.A.; Hammond, T.R.; Atassi, N.; Alonso-Alonso, M.; Dalakas, M.C. The Role of the Complement System in Chronic Inflammatory Demyelinating Polyneuropathy: Implications for Complement-Targeted Therapies. Neurotherapeutics 2022, 19, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Vlam, L.; Cats, E.A.; Harschnitz, O.; Jansen, M.D.; Piepers, S.; Veldink, J.H.; Franssen, H.; Stork, A.C.; Heezius, E.; Rooijakkers, S.H.; et al. Complement activity is associated with disease severity in multifocal motor neuropathy. Neurol.-Neuroimmunol. Neuroinflammation 2015, 2, e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, R.; Giardino, G.; Cirillo, E.; Prencipe, R.; Pignata, C. Complement system network in cell physiology and in human diseases. Int. Rev. Immunol. 2021, 40, 159–170. [Google Scholar] [CrossRef]
- Varga, L.; Farkas, H. rhC1INH: A new drug for the treatment of attacks in hereditary angioedema caused by C1-inhibitor deficiency. Expert Rev. Clin. Immunol. 2011, 7, 143–153. [Google Scholar] [CrossRef]
- van de Walle, I.; Silence, K.; Budding, K.; van de Ven, L.; Dijkxhoorn, K.; de Zeeuw, E.; Yildiz, C.; Gabriels, S.; Percier, J.-M.; Wildemann, J.; et al. ARGX-117, a therapeutic complement inhibiting antibody targeting C2. J. Allergy Clin. Immunol. 2021, 147, 1420–1429. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Yancopoulou, D.; Kokkinos, P.; Huber-Lang, M.; Hajishengallis, G.; Biglarnia, A.R.; Lupu, F.; Nilsson, B.; Risitano, A.M.; Ricklin, D.; et al. Compstatin: A C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur. J. Clin. Investig. 2015, 45, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Patriquin, C.J.; Kuo, K.H.M. Eculizumab and Beyond: The Past, Present, and Future of Complement Therapeutics. Transfus. Med. Rev. 2019, 33, 256–265. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef]
- Cardozo, T.; Cardozo, L.; Boutjdir, M. Autoantibody:Autoantigen Competitor Decoys: Application to Cardiac Phenotypes. Front. Immunol. 2022, 13, 812649. [Google Scholar] [CrossRef] [PubMed]
- Herrendorff, R.; Hänggi, P.; Pfister, H.; Yang, F.; Demeestere, D.; Hunziker, F.; Frey, S.; Schaeren-Wiemers, N.; Steck, A.J.; Ernst, B. Selective in vivo removal of pathogenic anti-MAG autoantibodies, an antigen-specific treatment option for anti-MAG neuropathy. Proc. Natl. Acad. Sci. USA 2017, 114, E3689–E3698. [Google Scholar] [CrossRef] [PubMed]
- Aliu, B.; Demeestere, D.; Seydoux, E.; Boucraut, J.; Delmont, E.; Brodovitch, A.; Oberholzer, T.; Attarian, S.; Théaudin, M.; Tsouni, P.; et al. Selective inhibition of anti-MAG IgM autoantibody binding to myelin by an antigen-specific glycopolymer. J. Neurochem. 2020, 154, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef] [PubMed]


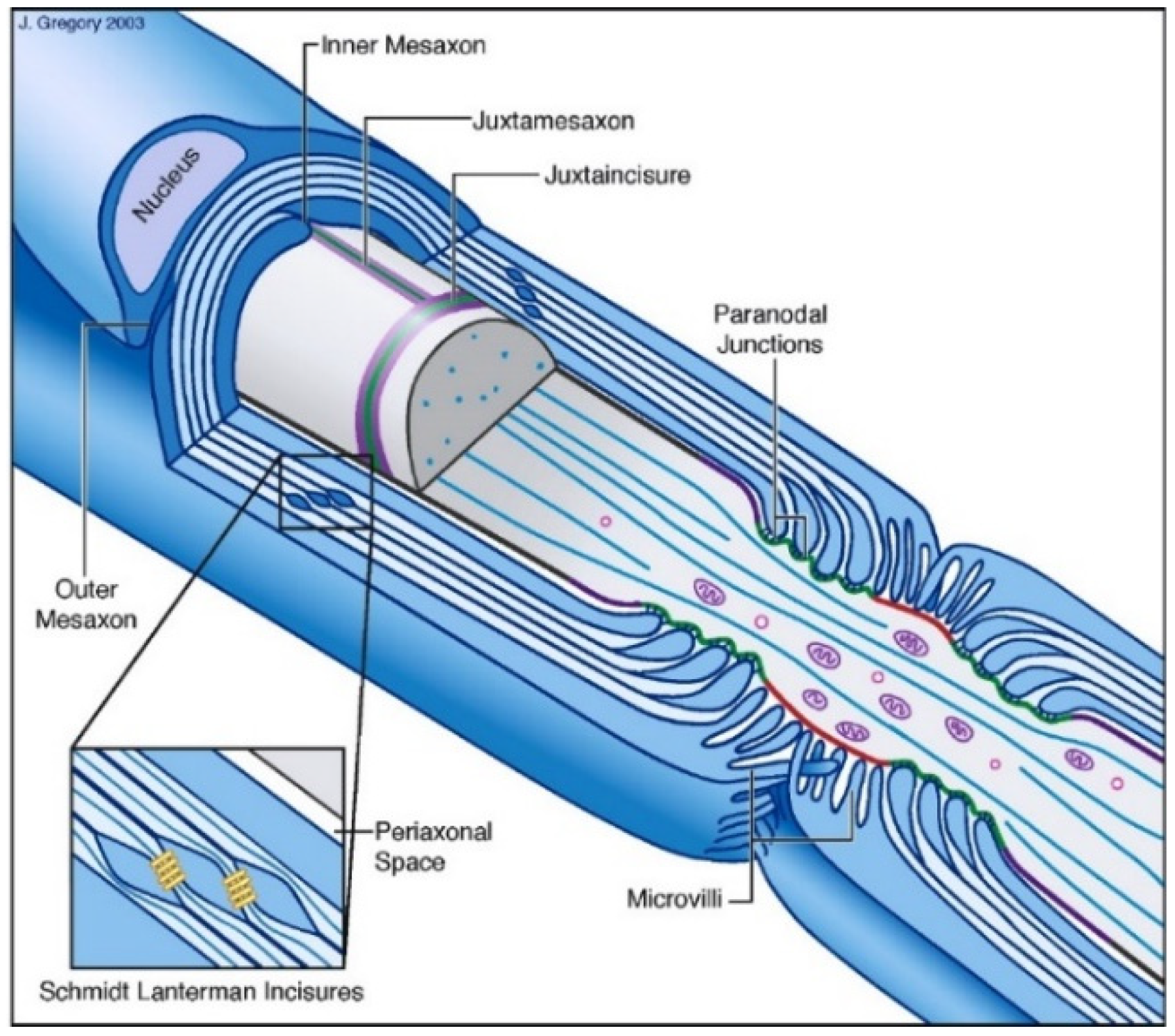{kind=link}
| Target Molecule | Associated PNP | References |
|---|---|---|
| HNK1 (present on SGPG, MAG, MP0 and other glycoproteins) | Anti-MAG PNP | [77,78,79] |
| Disialosyl epitope | B-series anti-ganglioside PNP (GD1b, GD3, GT1b and GQ1b) | [80,81] |
| Terminal Galβ(1–3)GalNAc structure | Anti-GM1/-GD1b/-asialo-GM1 ganglioside PNP | [82,83] |
| Sulfatide | Anti-sulfatide PNP | [82,83,84,85] |
| Combination sulfatide/galactocerebroside | Anti-sulfatide PNP | [86] |
| Internal sialylated epitope | Anti-GM1/-GM2 PNP | [87] |
| Chondroitin sulfate C | Anti-chondroitin sulfate C PNP | [88] |
| Technique | Intervention | Design | Study Arms | Number of Patients | Outcome | Authors |
|---|---|---|---|---|---|---|
| Scavenging M-protein | Plasma exchange (PE) | Prospective double-blind RCT. Follow-up 3 weeks | PE vs. sham exchange | 19 (11 IgM) vs. 20 (10 IgM). No information on MAG status | Significant improvement in motor score in IgG and IgA paraprotein patients, but not for IgM paraprotein patients | Dyck et al., 1991. [150] |
| Prospective, open-label RCT. Follow-up 12 months | PE + chlorambucil vs. chlorambucil | 22 vs. 23 (16 & 17 antimyelin Abs, respectively) | No difference in clinical outcome | Oksenhendler et al., 1995. [151] | ||
| Retrospective case series | PE | 6 (all anti-MAG) | 1 patient subjective improvement, 1 patient objective improvement but became unresponsive to PE later in disease course | Ellie et al., 1996. [91] | ||
| Retrospective case series | PE PE + chlorambucil | 5 2 (all anti-MAG) | 2/5 patients with PE alone improved, 2/2 patients with PE + chlorambucil improved on functional impairment scale. Effect duration not mentioned | Nobile-Orazio et al., 2000. [109] | ||
| Retrospective case series | PE | 20 (all anti-MAG) | 8 patients had at least temporal neurological improvement on MRC sum scale and neurological impairment scale | Gorson et al., 2001. [152] | ||
| Pro- and retrospective, uncontrolled open-label case series. Follow-up 6–12 months | PE | 24 (all anti-MAG) | 4 patients had a significant clinical improvement (ONLS and/or modified functional impairment scale score) after 6 months, 1 after 12 months | Svahn et al., 2018. [101] | ||
| Retrospective case series | PE (in acute worsening) | 4 (all anti-MAG) | 4 patients improved on ONLS score and subjectively after 3 to 6 PEs | Baron et al., 2017. [153] | ||
| Selective apheresis | Prospective case series. Follow-up 12 months | Selective apheresis | 1 IgM (2 IgG). No information on MAG status. | Sensory and motor response | Siciliano et al., 1994. [154] | |
| Decreasing M-protein production by targeting B-cell | Rituximab (RTx) | Prospective, double-blind RCT. Follow-up 8 months | RTx vs. placebo | 13 vs. 13 (all anti-MAG) | No significant improvement in primary outcome, but significant improvement in secondary outcome measures (e.g., 10 m walk test) | Dalakas et al., 2009. [155] |
| Prospective, double-blind RCT. Follow-up 12 months | RTx vs. placebo | 26 vs. 28 (all anti-MAG) | No significant improvement in primary outcome, but significant improvement in secondary outcome measures (e.g., INCAT disability scale) | Léger et al., 2013. [156] | ||
| Pro- and retrospective, uncontrolled open-label case series. Follow-up 6–12 months | RTx | 92 (all anti-MAG) | 29 patients had a significant clinical improvement (ONLS and/or modified functional impairment scale score), 27 remained stable | Svahn et al., 2018. [101] | ||
| Retrospective case series | RTx | 25 IgM MGUS (23 anti-MAG), 8 WM (all anti-MAG) | 18 of 33 patients clinically significant improvement (INCAT, INCAT sensory sum score, MRC sum score). MGUS statistically significant, WM not reached | Campagnolo et al., 2017. [157] | ||
| Cyclo-phosphamide | Prospective, double-blind RCT, 2nd phase cross-over study. Follow-up 1st phase 6 months, 2nd phase 24 months | Cyclophosphamide + prednisone vs. placebo | 16 vs. 19 (all anti-MAG) | No difference in primary outcome (Rivermead mobility index). Some secondary outcomes (e.g., MRC sum score, sensory sum score) improved significantly more than placebo after 24 months | Niermeijer et al. 2007. [158] | |
| Prospective, uncontrolled open-label case series. Follow-up mean 28 months (range 18–30) | Cyclophosphamide | 9 (all anti-MAG) | 7 patients significantly improved in muscle strength and modified ranking scale, 2 remained stable | Hamidou et al., 2005. [159] | ||
| Fludarabine | Prospective uncontrolled open-label case series. Follow-up mean 14.5 months (range 4–28) | Fludarabine | 4 (2 anti-MAG) | 4 patients significantly improved in motor and sensory neurological functioning and MRC sum score increase | Wilson et al., 1999. [160] | |
| Prospective uncontrolled open label case series. Follow-up 12 months | Fludarabine | 16 (6 anti-MAG) | 5 patients (all anti-MAG) significantly improved (1 point on median ranking scale), others were stable. Similar findings with Rivermead mobility index and MRC sum score | Niermeijer et al., 2006. [161] | ||
| Cladribine | Case report | Cladribine + IVIG | 1 (anti-MAG-positive) | Significant motor and sensory improvement (albeit after second IVIG trial) | Ghosh et al., 2002. [162] | |
| Chlorambucil | Retrospective case series | Chlorambucil | 2 (both anti-MAG) | No significant difference in clinical outcome | Gorson et al., 2001. [152] | |
| Pro- and retrospective, uncontrolled open-label case series. Follow-up 6–12 months | Chlorambucil | 33 (all anti-MAG) | 1 patient had a clinically significant improvement (ONLS and/or modified functional impairment scale score), 8 remained stable | Svahn et al., 2018. [101] | ||
| Melphalan | Prospective, uncontrolled open-label case series. Follow-up unknown | Melphalan + chlorambucil | 1 (anti-peripheral nerve myelin Ab’s) | No significant difference in clinical outcome | Ernerudh et al., 1992. [163] | |
| Decreasing damage done by M-protein by immunosuppression or -modulation | Intravenous immunoglobulins (IVIG) | Randomized double-blind crossover trial. Follow-up 6 months (3 months for 1st treatment, 3 months for 2nd treatment) | IVIG vs. placebo | 11 vs. 11 (9 anti-MAG) | 2 patients had a short (<3 months) clinically significant improvement in neurological functions | Dalakas et al., 1996. [164] |
| Randomized double-blind crossover trial. Follow-up 4 weeks | IVIG vs. placebo | 11 vs. 11 (11 anti-MAG) | Significant clinical improvement after 4 weeks (INCAT score and 10 m walk test). Secondary outcomes significantly increased as well. No percentages of patients given. No data after follow-up | Comi et al., 2002. [165] | ||
| Pro- and retrospective, uncontrolled open-label case series. Follow-up 6–12 months | IVIG | 68 (all anti-MAG) | Significant clinical improvement 19 after 6 months, 3 after 12 months | Svahn et al., 2018. [101] | ||
| Interferon alpha-2a (IFNα-2a) | Prospective, double-blind RCT. Follow-up 6 months | IFNα-2a vs. placebo | 12 vs. 12 (all anti-MAG) | No significant difference in clinical outcome | Mariette et al., 2000. [166] | |
| Dexamethasone (pulsed high dose) | Prospective, uncontrolled open-label case series, follow-up mean 19 months | Dexamethasone | 6 (5 IgM anti-MAG, 1 IgG) | 2 patients improved significantly, 4 had serious adverse events | Notermans et al., 1997. [167] | |
| Prednisone | Retrospective case series | Prednisone | 6 (all anti-MAG) | No significant difference in clinical outcome | Nobile-Orazio et al., 2000. [109] | |
| Retrospective case series | Prednisone | 8 (all anti-MAG) | No significant difference in clinical outcome | Gorson et al., 2001. [152] | ||
| Pro- and retrospective, uncontrolled open-label case series. Follow-up 6–12 months | Oral prednisone IV prednisone | 14 7 (all anti-MAG) | No significant clinical difference after 6 months, 3 patients significantly improved 12 months after oral prednisone | Svahn et al., 2018. [101] | ||
| Azathioprine | Retrospective case series | Azathioprine | 2 (all anti-MAG) | No significant difference in clinical outcome | Gorson et al., 2001. [152] | |
| Mycophenolate | Retrospective case series | Mycophenolate | 8 (4 anti-MAG) | No significant difference in clinical outcome | Gorson et al., 2004. [168] |
| Technique | Treatment | Anti-MAG Literature | Effect on Anti-MAG |
|---|---|---|---|
| Decreasing M-protein production by targeting B-cell | BTK inhibitors (e.g., ibrutinib, zanubrutinib, acalabrutinib) | Castellani et al., 2020. Prospective case series, follow-up 9–12 months [52] | Clinical improvement of 3/3 WM anti-MAG patients (sensory, motor and ataxia scales) |
| BCL2 inhibitors (Venetoclax) | Briani et al., 2022. Case report [204] | Clinical improvement in CLL anti-MAG patient. | |
| Anti-BCL2 monoclonal antibody (ulocuplumab) | |||
| Next-generation anti-CD20 monoclonal antibodies (e.g., ofatumumab and obinutuzumab) | Rakocevic et al., 2018. Retrospective IgM MGUS anti-MAG case series (N = 2) [202] Briani et al., 2019. Retrospective CLL IgM anti-MAG case series (N = 2) [203] | Rakocevic et al. reported no clinical improvement, but decrease in total IgM and anti-MAG titer Briani et al. reported both | |
| Proteasome inhibitors (e.g., carfilzomib and ixazomib) | |||
| Decreasing damage by M-protein by immunosuppression or -modulation | Complement inhibitors (e.g., Cinryze, ARGX-117, compstatin family C3 inhibitors, and eculizumab) | ||
| Preventing the M-protein from binding/attacking target antigen | Autoantibody competitor decoys or antigen mimics (e.g., PPSGG) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van de Mortel, J.P.M.; D’Sa, S.; Vrancken, A.F.J.E.; Notermans, N.C.; Vos, J.M.I.; Minnema, M.C. Polyneuropathy Associated with IgM Monoclonal Gammopathy; Advances in Genetics and Treatment, Focusing on Anti-MAG Antibodies. Hemato 2022, 3, 663-688. https://doi.org/10.3390/hemato3040045
van de Mortel JPM, D’Sa S, Vrancken AFJE, Notermans NC, Vos JMI, Minnema MC. Polyneuropathy Associated with IgM Monoclonal Gammopathy; Advances in Genetics and Treatment, Focusing on Anti-MAG Antibodies. Hemato. 2022; 3(4):663-688. https://doi.org/10.3390/hemato3040045
Chicago/Turabian Stylevan de Mortel, Johannes P. M., Shirley D’Sa, Alexander F. J. E. Vrancken, Nicolette C. Notermans, Josephine M. I. Vos, and Monique C. Minnema. 2022. "Polyneuropathy Associated with IgM Monoclonal Gammopathy; Advances in Genetics and Treatment, Focusing on Anti-MAG Antibodies" Hemato 3, no. 4: 663-688. https://doi.org/10.3390/hemato3040045