
Aetiology of MDS: With a Focus on Hereditary Predisposition


Department of Haematology, Leeds Teaching Hospitals, Leeds LS9 7TF, UK
*
Author to whom correspondence should be addressed.
Hemato 2022, 3(1), 17-37; https://doi.org/10.3390/hemato3010003
Submission received: 17 June 2021
/
Revised: 14 October 2021
/
Accepted: 3 November 2021
/
Published: 24 December 2021
(This article belongs to the Special Issue Challenges in the Treatment of Myelodysplastic Syndrome)
Abstract
:Myelodysplastic syndromes affect an older age group with a median age at onset in the eighth decade of life. As such, there is a relationship between the pathogenesis of MDS and age-related processes affecting haematopoietic stem/progenitor cells and/or the bone marrow microenvironment. MDS with an onset in younger people may be associated with recognised hereditary myeloid malignancy syndromes, and ‘forme fruste’ presentations of inherited syndromes in later life are now increasingly recognised such as germline mutations in DDX41. The considerable clinical and research interest in hereditary disorders is reflected in the relative emphasis within our manuscript. Prior chemo/radiotherapy is a clear cause of MDS but the predisposition factors for therapy-related MDS remain unclear. Clonal haematopoiesis is common in older people and may evolve to MDS, although once again, the biological factors driving this evolution are largely unknown. Finally, environmental exposure to genotoxic agents is likely to play only a minor role in the contemporary occupational/recreational setting.
1. Introduction
Despite abundant epidemiological research activity over the past 30 years, for the majority of newly presenting patients with MDS, the aetiology remains unclear. Considerable progress in the description of molecular/cytogenetic abnormalities is leading to a better definition of biological subtypes. In parallel, the emerging understanding of the molecular basis of hereditary myeloid malignancy syndromes provides further descriptive data. There is increasing evidence that ‘inflammation’ is associated with some subtypes of MDS, but the chicken vs. egg argument is only beginning. The recently described clonal haematopoiesis of indeterminate prognosis (CHIP) requires an etiological explanation, as do the mechanistic drivers that promote the evolution from CHIP to MDS. The role of environmental factors, such as putative occupational or recreational carcinogens remains uncertain, but at this stage appear to be only a minor contributor in the multistep ontogeny towards the clinical presentation of MDS. We will discuss these concepts in turn. Other contributors to this MDS monograph series will expand on many of these concepts. In this review we will attempt to integrate these concepts where relevant to aetiology (Figure 1).
Demographics
MDS is more common in older individuals, typically presenting in the eighth decade of life [1]. As such, there is a relationship between ageing bone marrow (haematopoiesis and microenvironment) and the development of MDS, which is nevertheless still a rare disease. In common with most malignant diseases, MDS is more common in males, except in MDS with del (5q), which has a striking female predominance. Therefore, why do those few patients develop MDS when the majority of older people do not, and why is del (5q) so strikingly female predominant, for example?
Finally, there are few studies exploring demographic differences between Western (predominantly Caucasian) MDS populations and other ethnic groups, and none with direct relevance to putative etiological variation. In general, south-east Asian MDS patients appear to be younger than Western MDS populations with some differences in cytogenetic profile [2].
MDS Is Heterogeneous; Etiology Must Also Be So
Although classified under a single term of Myelodysplastic Syndromes, morphological, clinical and biological features fragment MDS into an increasing number of subtypes [3,4]. As such, the etiological basis for these individual subtypes will inevitably be different. The aetiology of MDS with ring sideroblasts and isolated SF3B1 mutation [5] must be vastly different from that resulting in MDS-EB2 with monosomal karyotype and dual-hit TP53 aberration [6]. This creates an obvious challenge for researchers, namely, to create well-annotated large datasets with sufficient power to answer etiological questions within each biological subtype.
2. Environmental Epidemiology
The older, extensive literature (pre-2013) on the role of occupational and environmental carcinogens in the aetiology of MDS has been thoroughly reviewed elsewhere [7]. A recent systematic review of relevant case-control studies on relatively small patient and control cohorts published since 2001, indicates an increased Odds/Hazard ratio (OR/HR) for MDS in patients with high BMI, smokers, and with coexistent autoimmune disease. Other associations with MDS such as anaemia, community-acquired infections and anti-tuberculosis drugs are more likely not etiological [8]. Similarly, a meta-analysis comprising a larger cohort (1942 MDS vs. 5359 controls) describes exposure to pesticides as a risk factor for MDS [9]. However, in both of these papers, most reported OR/HR were <2.0, which, given older literature indicating inconsistent associations, does not provide strong and reliable evidence for clinically important association/etiological factors.
The conclusions continue to be that there is some evidence for an etiological role of environmental exposure and that it is plausible that a combination of low penetrance inherited predisposition and exposure to selected carcinogens may contribute to the aetiology of MDS in some patients, but to only a modest degree. Other recent and relevant ideas can be summarised as follows:
Carcinogens; Benzene as the Paradigm
For more than 50 years it has been known that exposure to high concentrations of benzene (>>5 ppm) can cause bone marrow failure, typically aplastic anaemia, sometimes transforming to AML [10]. There is evidence linking low-level benzene exposure (<0.5 ppm-years) to the development of MDS, both in the workplace and in the ambient community setting [11,12,13]. However, the in vitro and in vivo biological data do not provide a convincing and cogent mechanistic explanation, with, for example, cytogenetic abnormalities in lymphocytes exposed in vitro to benzene not consistent with those seen in typical MDS [14,15,16]. Odds ratios for exposure in MDS cohorts compared with controls are relatively low, typically 1.0–5.0, indicating a statistically significant relationship but of debatable clinical relevance for the majority of MDS patients.
Exposure to tobacco is more often observed in MDS cohorts compared with controls, a consistent finding but again with low odds ratio/relative risk (typically < 3.0) [17]. The putative carcinogenic constituents of tobacco smoke include benzene.
Thus far only limited studies of the influence of benzene exposure on the marrow microenvironment are available. The potential interaction of intrinsic inflammation and environmental carcinogen exposure, which together may dysregulate the bone marrow niche is worthy of attention, particularly in the context of inflammation related to clonal hematopoiesis [18].
3. Inherited Quantitative Trait Loci
Blood cell numbers vary between individuals and the reference range for ‘normality’ is correspondingly wide for most blood cells. Recent single cell analyses suggest that up to 15% of heritability of blood counts can be explained by inheritance of specific genomic loci [19]. This principle is supported by other in vitro techniques [20].
Animal models also indicate some heritability of stem cell numbers and cell cycling status, either as traits inherent to the HSC or to microenvironmental cells [21]. Given that the mutation rate in human tissue is linked to the number of cell divisions, it is plausible that individuals with a greater inherited number/cycling of HSCs could be more likely to develop stochastic critical genomic damage as a component of the multistep pathogenesis of myeloid malignancy.
4. ‘Inflammation’ as an Indirect Etiological Factor
The association between MDS and systemic autoimmune disorders has long been recognised but emerging data now also imply a link with autoinflammatory states. The recent description of VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome creates a direct link between bone marrow dysplasia and clinical autoinflammatory manifestations [22]. In addition, low-risk MDS is often associated with dysregulation of the NLRP3 inflammasome pathway [23]. An inflammatory environment may promote the evolution of cytopenias, may be permissive for clonal expansion and/or may be genotoxic, promoting disease progression.
This is reviewed in more detail in an accompanying article (Mekinian and Fain).
5. Clonal Haematopoiesis
Clonal haematopoiesis describes identification of a population of hematopoietic cells that share a distinct molecular aberration, typically a gene mutation, found in haematological malignancies. (CH) detected by high sensitivity molecular analysis may be almost universal in people aged > 50 years. The term Clonal Haematopoiesis of Indeterminate Potential (CHIP) describes people with CH and a normal blood count [24], in whom the clonal size is <2% hematopoietic cells. Whilst the portfolio of genes with acquired mutations is well established for myeloid malignancy, mutations in three genes dominate CHIP, namely DNMT3A, TET2 and ASXL1 (so-called DTA group) in that order of mutation frequency. Recent analyses of large datasets (>50,000 individuals) purport to define both fitness and mutation rate of specific CH mutations. In general terms, mutations in spliceosome genes have greater fitness (competitive expansion advantage compared to wild-type cells). In contrast, the inferred mutation rate per year is higher for DNMT3A mutations [25]. This would be consistent with a higher likelihood to develop MDS for patients with CHIP and isolated spliceosome mutations compared with isolated DTA mutations [26]. However, only a small proportion of people with CHIP will progress to MDS, and curiously the cardiovascular consequences of some CHIP variants may be of considerably greater clinical relevance [24]. Whilst CHIP may represent one step of the multistep pathogenesis for some subtypes of MDS, this may not be a universal MDS pathogenetic necessity.
6. Therapy-Related MDS
Chemo-radiotherapy is a well-established risk factor for the development of myeloid malignancy. There is an extensive literature review on this subject [27], but we will discuss new concepts that may explain at least a component of the predisposition. Previously, most pathogenetic hypotheses focussed on DNA damage induced by chemotherapeutic agents. These pathogenetic mechanisms still apply within the framework of ‘new’ concepts. Indeed, low penetrance predisposition factors may yet contribute, such as polymorphisms in genes encoding enzymes that metabolise chemotherapeutic agents or protect cells from damage, including DNA repair or antioxidant pathways reviewed in [28].
Firstly, an inherited predisposition to malignancy may result in multiple malignancies within the same individual. In the context of myeloid malignancy manifesting after a solid tumour, this may be interpreted as therapy-related, but equally plausible is that both malignancies have the same predisposition factors and that therapy per se may not be the cause.
Secondly, the role of CH as a predisposition factor is emerging. Recent data suggest that chemotherapy may induce clonal selection of specific CH mutations in specific therapeutic contexts. CH of genes encoding proteins in the DNA damage response pathways such as TP53, PPM1D and CHEK2 are selected in patients treated with radiotherapy, topoisomerase-2 inhibitors and platinum, for example [29]. This clonal selection creates an increased likelihood of evolution to therapy-related myeloid malignancy.
7. Familial Predisposition to Myeloid Malignancy
Recently, there has been increasing recognition of the role of germline genetic mutations associated with MDS, particularly but not exclusively presenting in children and younger adults. This subgroup has now been formally recognised within the recent revision of the World Health Organisation Classification as myeloid neoplasms with germline predisposition [3]. In contrast to somatic MDS, where only one known mutation (SF3B1) can be used as a diagnostic criterion, germline mutations in specific genes are sufficient for subclassification in the context of a myeloid neoplasm [3].
Historically, these disorders have been associated with well-defined non-haematological phenotypic changes, particularly those associated with bone marrow failure syndromes. It is becoming apparent that the majority of patients with a genetic predisposition to MDS have no phenotypically characteristic features and can be diagnosed only by genetic screening. Furthermore, these predisposition syndromes may present in patients above the age of 40, particularly those with DDX41 mutations. As a whole, germline predisposition is associated with at least 5% of MDS cases, and prevalence will only increase as new susceptibility genes are discovered [30].
Diagnosis and management of germline predisposition differs from somatic MDS in terms of substantial implications for the patient and the wider family. Many such disorders are associated with other medical conditions, including susceptibility to complications such as infection or secondary malignancies. These patients may require alterations in the recommended treatment such as haematopoietic stem cell transplantation.
Although these mutations are considered rare, in light of these implications, prompt recognition of germline associations and careful discussion of prognosis with the patient and their family is essential. In this review, we will discuss the subset of mutations specifically predisposing to MDS and chronic myelomonocytic leukaemia (CMML). We will not cover germline mutations predisposing to acute myeloid leukaemia (AML) alone, such as biallelic CEBPA, or lymphoid malignancies.
As a whole, causative mutations fall into at least three groups: (1) ubiquitous transcription factors critical for haematopoiesis such as RUNX1 and GATA2, (2) mutations associated with bone marrow failure and fundamental cellular processes such as ribosome biogenesis, telomere maintenance and DNA repair, and (3) newly discovered variants involved in innate immunity and antiviral responses, such as DDX41 and SAMD9/SAMD9L. An extended gene list is provided in Table 1.
Myeloid Neoplasms with Germline RUNX1 Mutation
RUNX1 encodes a master regulator of haematopoiesis highly expressed in haematopoietic stem cells. Located on the long arm of chromosome 21, it comprises three major isoforms, expressed differentially throughout haematopoietic differentiation (1A and 1B) or just at the time of stem cell emergence (1C). All isoforms carry a runt-homology domain (RHD), whereas a transactivation domain (TAD) containing activating and inhibitory domains is present only in isoforms 1B and 1C. This DNA-binding subunit forms a heterodimer with its partner CBFβ to activate transcription of a variety of key target genes involved in haematopoietic differentiation, cell cycle regulation and ribosome biogenesis [35]. One of the most common subtypes of AML with a relatively favourable prognosis results from the t (8; 21) translocation, causing fusion of the RUNX1 DNA binding domain to the RUNX1T1 (ETO) protein and blockade of haematopoietic differentiation. RUNX1 is involved in a host of other chromosomal translocations, including some responsible for therapy-related MDS and acute lymphoblastic leukaemia (ALL). RUNX1 was amongst the first genes to be identified in association with familial myeloid malignancy [36]. In contrast to the majority of the genes defining known Hereditary Myeloid Malignancy Syndromes (HMMS), somatic mutations in RUNX1 are commonly seen in sporadic myeloid malignancies with poor prognosis, including 10% of MDS cases, posing a particular diagnostic and clinical challenge for the identification and management of germline RUNX1 variants [37].
Familial platelet disorder with predisposition to myeloid malignancy (FPD-MM) is a rare autosomal dominant condition presenting with thrombocytopenia, abnormal platelet function and preserved platelet size. Eczema, psoriasis and arthritis are prominent in some families [38]. Affected family members may lack a bleeding tendency and present later in life. Opportunities for diagnosis from blood counts and platelet function testing, which raise the possibility of germline carriage, may not be available in childhood or young adulthood. However, genetic anticipation is also reported. Two-thirds of samples from asymptomatic young RUNX1 carriers demonstrate clonal haematopoiesis at high variant allele frequencies, resulting in a lifetime risk of 44% for the development of myeloid malignancy [30,39]. The average age at onset is 29 years; however, the age range spans a full lifespan, and includes a predilection for other leukaemias, including T-ALL, CMML and hairy cell leukaemia [40]. The majority of germline mutations cluster in the RHD and transactivation (TAD) domains, with unique mutations seen in each family pedigree. The range of variants seen, from missense and nonsense variants with dominant-negative effects, to deletions and frameshift mutations causing haploinsufficiency, result in considerable phenotypic heterogeneity [35].
The most frequent somatic partners in germline RUNX1-mutated MDS/AML cases are variants affecting the second RUNX1 allele (40%), most commonly duplication of the germline allele from partial trisomy or uniparental disomy of chromosome 21. Clonal haematopoiesis-associated mutations such as DNMT3A and TET2 are also frequently found, but mutations in ASXL1 were extremely rare, in marked contrast to strong co-association in sporadic AML [38,41]. In further contrast to sporadic RUNX1-mutated AML cohorts, germline cases are more likely to exhibit somatic variants in the second RUNX1 allele and GATA2, as a likely later event, suggesting that further reduction in wild-type RUNX1 levels may be more leukaemogenic [42]. Functional studies are consistent with both a haploinsufficient tumour suppressor model where only one mutation in RUNX1 is required, and the classic second-hit model requiring two mutations.
The range of variants and phenotypes exclusive to distinct kindreds precludes definitive conclusions, but several observations are noteworthy. Firstly, somatic mutations in RUNX1 and DNMT3A do not co-occur with germline RUNX1 lesions, suggesting germline RUNX1 haploinsufficiency combined with alterations in epigenetic states is sufficient for the development of malignancy. Secondly, the range of mutational partners differs between germline and sporadic disease. In sporadic disease, epigenetic and spliceosomal modifiers are commonly initiating lesions with subclonal RUNX1 mutations manifesting at an intermediate point in the disease course [4]. In contrast, familial disease has a preference for co-occurrence of tumour suppressors and transcription factors such as GATA2, PHF6, BCOR and WT1, presumably relating to the differential effects of early RUNX1 loss of function [38]. One potential differentiating role of early RUNX1 haploinsufficiency is the alteration of stem cell and progenitor bone marrow niche residency, due to RUNX1 effects upon adherence and motility genes [43]. RUNX1-deficient stem cells have reduced levels of apoptosis and p53, leading to resistance to genotoxic stress and a long term survival advantage. Coupled with increased levels of genomic instability, this may be important for increased rates of clonal haematopoiesis in asymptomatic carriers and subsequent transforming events [42,44,45].
These data suggest that pre-existing RUNX1 mutations may set the scene for altered evolution of pre-leukaemic stem cells, precluding sporadic mutation models from being applicable to germline disease. The true prognosis for FPD-MM, therefore, cannot be extrapolated from somatic disease and awaits further work on familial variants. Early identification of affected families is key, not least because transplantation from carrier siblings is associated with very poor outcomes, including engraftment failure and early relapse [46]. Encouragingly, inhibition of RUNX1 degradation has been demonstrated to restore RUNX1 levels and improve megakaryocytic differentiation in vitro. These and other potential future treatment pathways, which may prevent mutation of the wild-type allele, are particularly suitable for germline carriers and highlight the importance of early detection [47,48]. Molecular diagnosis by sequencing of all coding exons, including copy number analysis for deletions, is suggested for families with young MDS/AML patients.
GATA2-Spectrum Disorders
The role of GATA2 within haematopoiesis has many similarities with RUNX1. The gene encodes a transcription factor critical for multilineage haematopoiesis, stem cell homeostasis and lymphatic development [49]. Haploinsufficiency of GATA2 depletes haematopoietic stem cells [50] and select differentiated immune subsets [51,52] causing immune senescence. This leads to a broad spectrum of phenotypic changes, including the MonoMac and DCML syndromes (monocytopenia, B, NK and dendritic cell deficiencies associated with nontuberculous mycobacterial and fungal infections, human papillomavirus-associated warts and pulmonary alveolar proteinosis (PAP)) in up to 50% of patients [53,54]. Other manifestations include Emberger syndrome (primary lymphoedema), hearing loss, chronic neutropenia and autoimmune disorders [55,56,57].
Heterozygous germline mutations in GATA2 are the most common cause of paediatric and young adult MDS, particularly associated with monosomy 7, trisomy 8, AML and aplastic anaemia. Myeloid malignancy penetrance is extremely high at 75% [58], with a median age of 19 at leukaemic onset. In paediatric and adolescent MDS, GATA deficiency accounts for 7% of all MDS in this age-group, rising to 15% of advanced cases, and 37% of monosomy 7 patients, including two-thirds of adolescents with monosomy 7 [54]. Germline mutations are particularly associated with truncating or missense mutations affecting the second zinc finger (ZF2), as opposed to zinc finger 1 (ZF1) missense mutations seen mostly in somatic disease. Studies carefully assessing non-coding GATA2 regulatory regions and synonymous mutations suggest that the true prevalence could be higher than currently described [54,59].
Although initial kindreds demonstrated an autosomal dominant inheritance pattern, de novo loss of function variants are most common in paediatric MDS. Less than 30% of patients with germline GATA2 mutations have an affected family member [54].
Somatic ASXL1 variants frequently co-occur in up to 29% of patients and associate with transformation to proliferative CMML in young women [60]. Seen in isolation, the prognosis is poorer than for GATA2WT patients but similar when MDS subtype and karyotypic risk are accounted for [54]. Outcomes post-chemotherapy are poor [61], and the optimal timing for transplant may be in the late hypocellular phase before progression to advanced disease or upon progressive organ manifestations, as life-threatening complications such as PAP respond rapidly to stem cell transplantation. Monitoring serum Flt3 ligand may be useful to detect early stress upon haematopoiesis and clinical progression [51]. Favourable outcomes are seen post-transplantation in young patients; however, conditioning regimes may need to be tailored to reduce toxicity and incorporate prophylaxis against opportunistic infections [32,62,63,64].
Fanconi Anaemia
First described a century ago, the combination of congenital physical anomalies and cytopenias, now known as Fanconi anaemia (FA), is well described in association with bone marrow failure and MDS/AML. This diverse syndrome is caused by defective DNA repair and diagnosed traditionally by chromosome hypersensitivity to DNA crosslinking agents. Germline mutations in 23 different genes are responsible for increased DNA breakage and very high cancer susceptibility. The FA genes code for proteins that form the core complex responding to DNA damage. They perform critical roles in removing interstrand crosslinks preventing DNA replication and transcription, as well as other roles in replication fork stability and telomere maintenance [65]. Almost all mutations are autosomal recessive and mutations in FANCA, FANCC and FANCG are responsible for 90% of cases, whilst other mutations cause similar syndromes without confirmed MDS predisposition. Rare biallelic mutations in BRCA2 (also known as FANCD1) and PALB2 (FANCN) cause MDS/AML and solid tumours exceptionally early in childhood [66].
FA is the most common inherited cause of BM failure, with a median age of 7 at onset. Up to 30% of patients meet VACTERL-H criteria for anomalies (vertebral, anal, cardiac, tracheo-oesophageal fistula, oesophageal atresia, renal, upper limb and hydrocephalus); however, 25% of patients lack any characteristic physical findings such as short stature, failure to thrive and café au lait spots, or subtle limb abnormalities [65]. Presentations can include macrocytosis and a hypocellular marrow: baseline dyserythropoiesis is widely seen and does not constitute sufficient grounds for an MDS diagnosis. Progressive proliferation-induced stress leads to stem cell depletion, chronic inflammation and clonal evolution, culminating in a 40% cumulative incidence of MDS by the age of 50 [67,68]. Chromosomal gains of 1q are seen in almost half of cases at all stages of disease; however, gain 3q (in 40% of cases), loss of 7/7q and cryptic RUNX1 abnormalities are seen only in high-grade MDS/AML [69].
The curative option of transplantation must be specifically tailored to prevent the toxicity of radiation and alkylating agent-susceptibility, highlighting the importance of careful screening and timing of transplantation. Graft failure and solid malignancies, including squamous cell carcinomas associated with chronic GVHD, are more frequently seen post-transplantation [70]. A specific feature of the genetic instability seen in FA blood lymphocytes is somatic mosaicism, whereby one of the mutated alleles spontaneously reverts to functionally normal status, providing a growth advantage and clinical blood count improvement but persisting risks of haematological malignancy [71]. Somatic reversion causes false-negative peripheral blood testing and reinforces the requirement for skin fibroblast testing at diagnosis.
Ribosome Disorders
Diamond–Blackfan anaemia (DBA) was the first ribosomopathy to be identified in humans [72]. A congenital hypoplastic anaemia associated with increased red blood cell erythrocyte deaminase, half of the patients carry physical abnormalities, including craniofacial, genitourinary and thumb anomalies. Infants present at a median age of 8 weeks with macrocytic anaemia and reticulocytopenia, with typical features of red cell aplasia on BM assessment. DBA is unique amongst the inherited bone marrow failure (IBMF) syndromes in manifesting a specific defect in erythropoiesis, although other progenitors can be affected. The relative risk for MDS is amplified 300-fold, with a median age at onset of 13, a notably lower incidence of AML compared to FA reaching 5% by the fifth decade, and increases in solid tumours such as osteogenic sarcoma and colon cancer [73,74]. Most cases are due to haploinsufficiency of ribosomal proteins (RPs), leading to defects in rRNA maturation and paralleling erythroid hypoplasia seen in the acquired RPS14 haploinsufficiency of del (5q) MDS [75,76]. Causative heterozygous mutations coding for ribosomal subunits have been identified in 20 genes, although mutations in six genes account for 70% of all cases, most commonly RPS19 in 25% of cases. Half of all mutations arise de novo and unresolved questions surround the connection between impaired ribosomal processing and a block in erythroid differentiation. Reduction in haematopoietic ribosomes selectively reduces translation of a subset of transcripts, which may affect the erythroid lineage to a greater extent due to extremely high rates of protein synthesis [77]. Disruption of ribosome biogenesis leads to activation of p53, increased autophagy and heme toxicity causing excess cell death [78,79,80]. Somatic RP heterozygosity is strongly linked to inactivating TP53 mutations; however, specific molecular features associated with MDS aetiology in DBA have not yet been identified [76].
Shwachman–Diamond syndrome (SDS) is a rare autosomal recessive disorder caused in 90% of cases by compound heterozygous mutations in the SBDS gene, located on 7q. The protein product functions as an essential cofactor for the GTPase elongation factor 1 (EFL1), catalysing removal of the assembly protein eukaryotic initiation factor 6 (eIF6) to enable ribosomal maturation [81]. Uncoupling of this process leads to a multi-system disease encompassing bone marrow failure, exocrine pancreatic insufficiency and impaired bone metabolism [82]. Patients commonly present with neutropenia and infection and exhibit malabsorption, cognitive impairment and impaired neutrophil and monocyte chemotaxis. The bone marrow is hypocellular and MDS evolves in one in three patients by the age of 30 [83]. Similar phenotypes have been reported for mutations in related proteins (EFL1, DNAJC21) and signal recognition particle 54 (SRP54), an essential component of the protein translation machinery [84].
Associated somatic mutations in EIF6 are common and benign, acting similarly to somatic reversion seen in Fanconi anaemia to enhance clonal fitness by compensating for the ribosomal defect and alleviating cellular stress [85]. In contrast, clonal haematopoiesis due to TP53 mutations is seen in 50% of paediatric SDS patients [86] preceding frank transformation to MDS/AML by several years. SDS is likely under-diagnosed and associated with poor survival even in the context of allogeneic stem cell transplantation [32,87]. Ribosomal stress leads to mTOR/STAT3 and p53 pathway hyperactivation and consequent growth inhibition of stem cells. This process selects for the development of multiple independent TP53-mutated clones due to an evolutionary growth advantage, which overcomes normal tumour suppressor checkpoints without correcting the ribosomal stress defect. The frequency of TP53 alterations increases with age, and 80% of patients over 10 years of age carry at least one TP53 mutation. The lack of chemo-sensitivity of biallelic TP53 disease highlights the importance of close disease monitoring, which may in the future encompass single cell DNA sequencing, and the need for novel targeted treatments such as pharmacological inactivation of EIF6 [6,85].
Disorders of Telomere Maintenance
Telomere biology disorders (TBDs) are intimately linked to the processes underlying haematopoiesis: stem cell renewal, cellular ageing and the effects of replicative stress. Telomeres are specialised repetitive structures protecting chromosomal ends from fusion and replenishing terminal DNA sequences [88]. Telomeres shorten naturally with age, and physiological telomere loss is a protective process to halt cell division in normal somatic cells with a long proliferative history once a critically short telomere length is reached. Haematopoietic stem cells avert senescence by expression of telomerase, a reverse transcriptase encoded by TERT, which synthesises telomeric repeats to prevent shortening using an RNA template encoded by TERC. Although short leucocyte telomeres correlating with advanced disease were first described in acquired aplastic anaemia patients [89], germline mutations in 13 genes coding for components of the telomerase complex and associated proteins have now been identified as causing a heterogeneous spectrum of overlapping disorders.
Dyskeratosis congenita (DKC), caused by mutations in DKC1, classically presents with the mucocutaneous triad of skin hypopigmentation, nail dystrophy and oral leucoplakia, reflecting organ-specific high cellular turnover and senescence of dermal stem cells [90]. The gene codes for dyskerin, which maintains the stability of telomerase, and when severe presents early in life with high penetrance. Hoyeraal–Hreidarsson syndrome is a severe form associated with cerebellar hypoplasia. At the other end of the spectrum, forme fruste variants may present in adulthood with variable penetrance of BM failure, pulmonary fibrosis and cryptogenic liver cirrhosis associated with heterozygous mutations in TERT, TERC and RTEL1 [91]. Although there are similarities with FA in a 500-fold greater incidence of MDS, lifetime cumulative risk is far lower at 2% and disorders present at an older median age of 31, and there are also significant increases in squamous cell carcinoma risk. In young patients < 40, customised sequencing platforms covering non-coding regions identified pathogenic or likely pathogenic mutations affecting telomere biology in 4% of MDS cohorts and 8% of the aplastic anaemia population [92,93]. Whole exome sequencing can enhance the yield for a causal or likely germline telomeropathy to 16% when testing is restricted to a highly selective undiagnosed cytopenic cohort comprising young patients with defined physical signs, family history or infants ≤ 2 [94].
Screening for these variants should be considered in patients presenting with longstanding cytopenia, BM failure or hypoplastic MDS/AML at younger ages than expected [91]. Physical examination is relevant to assess for the presence of subtle signs such as premature hair greying and extensive dental caries. Leucocyte telomere length (LTL) < 1st percentile for age as measured by flow-FISH is highly suggestive of a TBD, although accuracy is diminished in older patients and those lacking classical features [91]. Screening is best performed as close to diagnosis as possible, given that immunosuppressive agents, chemotherapy and BM failure induced-replicative stress all cause telomere shortening. Clarifying genotype-phenotype heterogeneity is further confounded by shortened telomere lengths in patients with low-risk MDS lacking identified pathogenic telomere mutations, and AA patients with somatic myeloid mutations or monosomy 7, presumably relating to haematopoietic stress [95,96,97].
Blood counts and telomere length often respond directly to androgens, possibly due to upregulation of TERT expression, suggesting mitigating strategies to prevent telomere attrition and chromosomal instability [98]. Curative transplantation regimes may require tailored reduced intensity protocols to prevent excess organ toxicity. Novel regimes aiming to exploit a competitive disadvantage in DKC stem cells by omitting alkylating agents and radiotherapy are under investigation [99]. MDS patients with shorter telomere lengths pre-transplant have independently higher levels of non-relapse mortality following allogeneic stem cell transplant, likely due to replicative exhaustion from cellular stresses induced by infection, graft-versus-host-disease and immune reconstitution, and analogous to post-transplant toxicities seen in DKC patients [100]. The possibility that a proportion of these cases may relate to undiagnosed TBDs is suggested by associations between LTL and pre-transplant pulmonary and hepatic dysfunction rather than blast percentage [101].
Li-Fraumeni Syndrome
TP53 encodes a transcription factor critical for cellular protection and activates in response to a wide variety of stress signals, including DNA damage, oncogene activation and hypoxia, with a broad range of target downstream effects, including apoptosis, cell cycle arrest, senescence, metabolic regulation and DNA repair [102,103]. As the most frequently mutated gene in human cancer, autosomal dominant germline mutations in TP53 are highly penetrant for early ‘core’ malignancies, including sarcomas, adrenocortical, brain and premenopausal breast cancer, and almost half of children with low hypodiploid ALL [104,105]. Missense mutations are most commonly seen, resulting in dominant negative or loss of function effects [106]. Detection in the de novo MDS setting is very rare, but the yield in therapy-related disease appears higher as alkylating agent and ionising radiation-induced stress enable mutant p53 to promote clonal haematopoietic stem cell expansion and subsequent karyotypic complexity with dire long-term outcomes [6,107,108,109,110]. The germline yield is low at 7–8% even when investigations are limited to MDS/AML [111] tumour panel variants with a VAF > 0.4 [112] or therapy-related myeloid neoplasms [111], suggesting the choice of whom to test should be further filtered based upon refinement of the classical Chompret criteria [113,114].
Myeloid Neoplasms with Germline DDX41 Mutation
Germline mutations in the DEAD-box helicase 41 gene (DDX41) were recently identified in a number of families associated with MDS/AML and less frequently CMML and MPN [115]. The gene product acts as a DNA sensor mediating the innate immune type 1 interferon response via the stimulator of the interferon gene (STING) pathway [116]. Defects in DDX41 lead to altered pre-mRNA splicing and RNA processing via spliceosomal interactions and may disrupt putative tumour suppressor function; however, the precise mechanism underlying the development of MDS remains unknown. Contrary to previous assumptions associating heritable risk with early disease onset, these mutations define a unique cluster predisposing to late-onset MDS/AML, with an average age at diagnosis of over 60 years [117]. The majority of affected individuals are male (79%), and up to half have a history of cytopenia for years prior to diagnosis. The majority of patients have a normal karyotype, and only 27% have a family history of haematological malignancies, which also includes a predisposition to lymphoid malignancy.
The frequency of germline DDX41 in unselected cohorts is estimated to be at least 2–3%, suggesting this is likely to form the largest contributor to HMMS yet discovered, and should be incorporated within routine diagnostic testing. The majority of DDX41-associated AML cases arise from antecedent MDS, suggesting opportunities for early intervention [115,117,118]. Germline variants strongly predispose to somatic mutations in the unaffected allele, in 50–80% of cases [115,117]. Germline variants include start codon loss, frameshift, missense or nonsense mutations, while somatic lesions are almost always missense and the majority involve the amino acid substitution R525H, causing loss of RNA helicase activity [118]. DDX41 is located at the distal end of chromosome 5, and corresponding deletions on 5q35.5 in a small proportion of cases, including a quarter of MDS and secondary AML del (5q) cases, result in reduced DDX41 mRNA levels. Co-occurrence of germline and somatic mutations in this pattern suggests that DDX41 haploinsufficiency is sufficient to cause disease in the context of epigenetic or spliceosomal modifiers; however, hypomorphism of the second allele enhances clonal advantage in a ‘second-hit’ model analogous to other HMMS such as CEBPA. In contrast to these lesions, isolated somatic DDX41 variants are rarely found in MDS/AML, and the substantially later age of onset suggests lower potency for leukemogenicity. The true penetrance remains unclear and is strongly influenced by gender. The range of other somatic mutations associated with DDX41-driven MDS/AML raises the possibility that these lesions induce pre-leukaemic stem cells analogous to those previously characterised in MDS lacking known germline predisposition, promoting the risk of leukaemia development in later life due to unknown factors [119].
Patients with DDX41 mutations or low DDX41 mRNA expression treated with lenalidomide experienced significantly improved response rates compared to DDX41 wild-type patients [115,120]. The largest series of patients thus far published indicates a relatively favourable outcome for germline DDX41 mutations, including excellent responses to intensive chemotherapy for high-risk patients with a 100% response rate and median overall survival exceeding 5 years. Patients were bridged to transplant without obvious excess toxicity, and the finding of mutated DDX41 has been used to risk-stratify older AML patients into a low risk subgroup, also including patients with GATA2 mutations, who may be candidates for reduced toxicity approaches [117,121].
SAMD9/SAMD9LMutations
The phenotype of somatic reversion is also seen in children with mutations in SAMD9/SAMD9L genes located on 7q22 and implicated in interferon-dependent control of cellular proliferation. These rare heterozygous gain-of-function mutations, variously associated with neurological dysfunction and cytopenia (SAMD9L) or the multi-system MIRAGE syndrome (SAMD9) may present at a young age with monosomy 7/del (7q) MDS or bone marrow failure [122]. Considerable phenotypic variation is seen: correction by way of loss of function mutations on the alternate allele or uniparental disomy can induce complete recovery of blood counts. The alternative evolutionary strategy to improve cell turnover involves the selection pressure of emergency haematopoiesis, perhaps in the context of viral infection. This results in loss of the mutant allele via deletion of 7/7q, overcoming the bottleneck in cell turnover but also resulting in presumptive loss of other resident tumour suppressors such as EZH2 and the potential development of myeloid malignancy. This phenomenon has also been suggested as a potential mechanism for reported transient monosomy 7 syndrome [123,124]. Given the loss of the mutant allele by the time of malignant presentation, non-haematopoietic germline screening is the only approach to detection in suspected cases.
Germline SAMD9/SAMD9L mutation has not been widely studied in adult MDS; the only series reported to date identified ‘germline’ mutations in 3% adult MDS cases, although the source of germline tissue in this study was CD3+ lymphocytes with the possibility of malignant cell contamination. Mutations were not in similar exons compared with paediatric cases and no corresponding somatic reversion events were evident, raising the possibility that these were somatic events [125].
Practicalities in the Clinic
A very common question from patients is how did this happen to me, and why me? For the vast majority of patients, there is no simple answer to this, other than this remains unknown.
The consultation history for newly diagnosed patients should always enquire about:
- Prior chemo/radiotherapy,
- Family history of myeloid malignancy, other cancers,
- Family history of attendance at haematology clinics (thrombocytopenia, macrocytosis, vitamin B12/folate supplementation),
- Pulmonary or hepatic disease (typically fibrosis), or other non-haematological features described below.
We would no longer recommend a routine discussion of exposure to occupational and environmental carcinogens, as the evidence for their etiological role is weak.
Consideration should be given to the laboratory features; for example, hypocellular MDS may alert to the possibility of germline DDX41 or telomeropathies, Fanconi anaemia and other inherited bone marrow failure syndromes.
When to Consider HMMS in the Clinical Consultation?
All patients presenting with MDS, particularly younger patients, should receive a focussed clinical history and examination, with specific reference to personal or family history of neoplasms and clinical features as in Table 2. The expected yield for positive germline testing varies from almost 30% for patients with two or more close relatives with MDS/AML [30], particularly in the presence of indicative features such as idiopathic pulmonary fibrosis or lymphoedema, to 13–19% in young MDS cohorts up to the age of 45 [92,93]. In contrast, the yield is low in the case of families associated with non-Hodgkin’s lymphoma or myeloproliferative neoplasms (MPN), and single gene associations for these diseases are lacking, despite particularly strong evidence of a heritable component for MPN risk. Population-level epidemiological studies suggest that the risks of being diagnosed with MDS given a first degree relative with the same condition are amongst the highest known for cancers, at 7-fold higher than the general population. The same magnitude of MDS-specific risk is present for first degree relatives of a patient with myelofibrosis; however, absolute risks remain low in the absence of specific gene associations [126]. Given the presumed rarity of currently known single-gene associations, the mechanisms underlying much of this heritable risk remain unknown. In a young adult MDS cohort aged between 18 and 40, almost 40% of patients with pathogenic or likely pathogenic germline variants had neither family history nor any phenotypic features [92]. The phenomenon of anticipation seen within familial myeloid malignancies means that younger generations within a family may present earlier than older generations. The majority of known hereditary myeloid malignancy syndromes (HMMS) are incompletely penetrant, such that family members with pathogenic variants may remain fit and well, and 50–70% of HMMS patients have no family history [93,117]. Considering that up to 12% of MDS patients report a first-degree relative with haematological malignancy [127], family history alone is of limited use as a predictive tool and many patients are likely to require more detailed assessment.
Diagnosis
Suspicion for HMMS in an individual has historically been raised by two routes: a strong family history of haematological malignancy, or specific clinical features fitting aspects of a syndromic presentation. More recently, with the advent of genetic panels screening for somatic mutations at diagnosis of MDS, a new and important development is the question of how to approach the finding of genes associated with HMMS. Targeted somatic gene mutation panels cannot be used to diagnose germline mutations, as these panels may not cover all key sites of interest within a given gene and fail to detect gene duplications or deletions. Some gene variants are predominantly associated with acquired variants in the older population, such as RUNX1, and routine germline testing in this setting would have a low yield. In order to avoid excessive testing in the absence of standardised germline testing at diagnosis, further germline testing should be considered in a targeted approach for young patients with variant allele frequency (VAF) for a relevant mutation ≥ 40%, or other features detailed in Table 1 [112,129].
Patients with suspected HMMS should ideally undergo genetic counselling prior to germline testing. Important points to consider include:
- Discussion of the limitations of current testing, including the possibility of unexpected or difficult-to-interpret results such as private variants of unknown significance unique to specific families. These may require functional testing to prove a pathogenic role, which may not be available at the time of the test.
- Possible implications for the patient and family members should be discussed, including the importance of sharing relevant results to guide further testing.
- The possibility that tests may not yield any significant findings should be raised, and the fact that this would not exclude a germline predisposition, given the ongoing discovery of novel variants as knowledge and technology progress over time.
The gold standard for germline testing on constitutional DNA is culture of skin fibroblasts. To avoid contamination from somatic variants in the blood, nail, saliva and buccal samples are usually avoided [130]. The turnaround time for this procedure may exceed 12 weeks, and in cases where a more rapid result may be required, such as work-up for related donor allogeneic stem cell transplantation, alternative testing sources may be required, with consideration of the potential for false-positive results.
Outcomes
Management principles following diagnosis of HMMS are outlined in Table 3. Poor outcomes have been seen with use of related donors for allogenic stem cell transplantation where the donor has later been to carry a pathogenic germline variant in genes such as RUNX1, GATA2, DDX41 or CEBPA [46,131,132,133]. These complications include poor donor stem cell mobilisation, delayed engraftment, poor immune function and early relapse or donor-derived leukaemias. Avoidance of donors is advised where a family history or syndromic presentation is present or suspected, even in the absence of positive testing, due to the possibility of novel variants. Related donors with mild anaemia, neutropenia, thrombocytopenia or lymphopenia should be excluded from donation [134]. Patients with normal full blood counts may also carry a deleterious variant and management may need to be individualised, particularly where a suitably matched unrelated donor is not present.
8. Future Directions
Increasing variant discovery and phenotypic heterogeneity have combined with the issue of variable family history and the need to make rapid and far-reaching decisions on management, including surveillance, allogeneic transplantation timing and donor source to create difficult diagnostic and treatment decisions. The ability to accurately diagnose germline predisposition syndromes has the potential to improve patient prognosis through targeted surveillance and pre-emptive treatment. Set against this is the spectre of over-investigation, patient and familial anxiety, and difficulties in variant interpretation. These challenges highlight the need for considered investigation of patients. Depending on the context, this may involve tailored large-scale somatic NGS panels encompassing germline variants, expert-guided variant curation and standardised germline testing at baseline to minimise therapeutic delay and guide important decisions such as allogeneic donor selection. The recent roll-out in the United Kingdom of whole genome sequencing coupled with paired germline analysis for newly diagnosed acute leukaemias, and allied to centralised expert-led variant interpretation services, is an exemplar of the way forward.
Increasing interest in the role of inflammation in CHIP, and in clonal dysplasias such as CCUS, coupled with an interrogation of the bone marrow microenvironment and immune regulation of haematopoiesis has the potential to further inform the etiological processes leading to the majority of MDS cases that are not associated with inherited predisposition.
Author Contributions
Both authors contributed equally to all elements of this document. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Roman, E.; Smith, A.; Appleton, S.; Crouch, S.; Kelly, R.; Kinsey, S.; Cargo, C.; Patmore, R. Myeloid malignancies in the real-world: Occurrence, progression and survival in the UK’s population-based Haematological Malignancy Research Network 2004–15. Cancer Epidemiol. 2016, 42, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Tuechler, H.; Sanz, G.; Schanz, J.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Differing clinical features between Japanese and Caucasian patients with myelodysplastic syndromes: Analysis from the International Working Group for Prognosis of MDS. Leuk. Res. 2018, 73, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Bowen, D.T. Occupational and environmental etiology of MDS. Best Pr. Res. Clin. Haematol. 2013, 26, 319–326. [Google Scholar] [CrossRef]
- Sweeney, M.R.; Applebaum, K.M.; Arem, H.; Braffett, B.H.; Poynter, J.N.; Robien, K. Medical Conditions and Modifiable Risk Factors for Myelodysplastic Syndrome: A Systematic Review. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1502–1517. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Yu, M.; Hu, C.; Ye, L.; Xie, L.; Jin, J.; Chen, F.; Tong, H. Pesticide Exposure as a Risk Factor for Myelodysplastic Syndromes: A Meta-Analysis Based on 1942 Cases and 5359 Controls. PLoS ONE 2014, 9, e110850. [Google Scholar]
- Aksoy, M.; Dincol, K.; Erdem, S.; Dinçol, G.; Dinçol, G.; Dinçol, G. Acute leukemia due to chronic exposure to benzene. Am. J. Med. 1972, 52, 160–166. [Google Scholar] [CrossRef]
- Teras, L.R.; Diver, W.R.; Deubler, E.L.; Krewski, D.; Flowers, C.R.; Switchenko, J.M.; Gapstur, S.M. Residential ambient benzene exposure in the United States and subsequent risk of hematologic malignancies. Int. J. Cancer 2019, 145, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Linet, M.S.; Yin, S.-N.; Gilbert, E.S.; Dores, G.M.; Hayes, R.B.; Vermeulen, R.; Tian, H.-Y.; Lan, Q.; Portengen, L.; Ji, B.-T.; et al. A retrospective cohort study of cause-specific mortality and incidence of hematopoietic malignancies in Chinese benzene-exposed workers. Int. J. Cancer 2015, 137, 2184–2197. [Google Scholar] [CrossRef] [PubMed]
- Schnatter, A.R.; Glass, D.; Tang, G.; Irons, R.D.; Rushton, L. Myelodysplastic Syndrome and Benzene Exposure among Petroleum Workers: An International Pooled Analysis. J. Natl. Cancer Inst. 2012, 104, 1724–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHale, C.M.; Zhang, L.; Smith, M.T. Current understanding of the mechanism of benzene-induced leukemia in humans: Implications for risk assessment. Carcinogenesis 2011, 33, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.T.; Zhang, L.; McHale, C.M.; Skibola, C.F.; Rappaport, S.M. Benzene, the exposome and future investigations of leukemia etiology. Chem. Interact. 2011, 192, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Escobar, P.A.; Smith, M.T.; Vasishta, A.; Hubbard, A.E.; Zhang, L. Leukaemia-specific chromosome damage detected by comet with fluorescence in situ hybridization (comet-FISH). Mutagenesis 2007, 22, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Björk, J.; Albin, M.; Mauritzson, N.; Strömberg, U.; Johansson, B.; Hagmar, L. Smoking and Myelodysplastic Syndromes. Epidemiology 2000, 11, 285–291. [Google Scholar] [CrossRef]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2 -mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Ulirsch, J.C.; Lareau, C.A.; Bao, E.L.; Ludwig, L.S.; Guo, M.H.; Benner, C.; Satpathy, A.T.; Kartha, V.K.; Salem, R.M.; Hirschhorn, J.N.; et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 2019, 51, 683–693. [Google Scholar] [CrossRef]
- Ferreira, M.A.; Hottenga, J.-J.; Warrington, N.M.; Medland, S.E.; Willemsen, G.; Lawrence, R.W.; Gordon, S.; de Geus, E.J.; Henders, A.K.; Smit, J.H.; et al. Sequence Variants in Three Loci Influence Monocyte Counts and Erythrocyte Volume. Am. J. Hum. Genet. 2009, 85, 745–749. [Google Scholar] [CrossRef] [Green Version]
- Waterstrat, A.; Rector, K.; Geiger, H.; Liang, Y. Quantitative trait gene Slit2 positively regulates murine hematopoietic stem cell numbers. Sci. Rep. 2016, 6, 31412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; McLemore, A.; Vincelette, N.D.; Ward, G.A.; Eksioglu, E.A.; Sallman, D.A.; Al Ali, N.; Padron, E.; et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: An observational cohort study. Lancet Haematol. 2018, 5, e393–e402. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Papula, A.L.; Poon, G.Y.P.; Wong, W.H.; Young, A.L.; Druley, T.E.; Fisher, D.S.; Blundell, J.R. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 2020, 367, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Galli’, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Abou Zahr, A.; Kavi, A.M.; Mukherjee, S.; Zeidan, A.M. Therapy-related myelodysplastic syndromes, or are they? Blood Rev. 2017, 31, 119–128. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2021, 52, 1219–1226. [Google Scholar] [CrossRef]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.-L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef] [Green Version]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsley, R.C.; Saber, W.; Mar, B.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Reilly, C.R.; Myllymäki, M.; Redd, R.A.; Padmanaban, S.; Karunakaran, D.; Tesmer, V.M.; Tsai, F.D.; Gibson, C.J.; Rana, H.Q.; Zhong, L.; et al. The clinical and functional effects of TERT variants in myelodysplastic syndrome. Blood 2021, 138, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.R.; Ma, J.; Lamprecht, T.; Walsh, M.; Wang, S.; Bryant, V.; Song, G.; Wu, G.; Easton, J.; Kesserwan, C.; et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat. Commun. 2017, 8, 1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Song, W.-J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 1999, 23, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.L.; Arts, P.; Carmichael, C.; Babic, M.; Dobbins, J.; Chong, C.-E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef] [Green Version]
- Carraway, H.E.; LaFramboise, T. Myeloid neoplasms with germline predisposition: Practical considerations and complications in the search for new susceptibility loci. Best Pr. Res. Clin. Haematol. 2020, 33, 101191. [Google Scholar] [CrossRef]
- Brown, A.L.; Hahn, C.N.; Scott, H.S. Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood 2020, 136, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debré, I.; Duployez, N.; Bucci, M.; Geffroy, S.; Micol, J.-B.; Renneville, A.; Boissel, N.; Dhédin, N.; Rea, D.; Nelken, B.; et al. Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia 2016, 30, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debré, I.; Manchev, V.T.; Balayn, N.; Bluteau, M.; Tomowiak, C.; Legrand, C.; Langlois, T.; Bawa, O.; Tosca, L.; Tachdjian, G.; et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood 2015, 125, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Behrens, K.; Triviai, I.; Schwieger, M.; Tekin, N.; Alawi, M.; Spohn, M.; Indenbirken, D.; Ziegler, M.; Müller, U.; Alexander, W.S.; et al. Runx1 downregulates stem cell and megakaryocytic transcription programs that support niche interactions. Blood 2016, 127, 3369–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, J.; Simpson, K.M.; Escher, R.; Buchet-Poyau, K.; Beissbarth, T.; Carmichael, C.; Ritchie, M.E.; Schütz, F.; Cannon, P.; Liu, M.; et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genom. 2008, 9, 363. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Gao, L.; Teng, L.; Ge, J.; Oo, Z.M.; Kumar, A.R.; Gilliland, D.G.; Mason, P.J.; Tan, K.; Speck, N.A. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 17, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Owen, C.J.; Toze, C.L.; Koochin, A.; Forrest, D.L.; Smith, C.A.; Stevens, J.M.; Jackson, S.C.; Poon, M.; Sinclair, G.D.; Leber, B.; et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood 2008, 112, 4639–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutein, M.C.; Hart, M.R.; Anderson, D.J.; Jeffery, J.; Kotini, A.G.; Dai, J.; Chien, S.; DelPriore, M.; Borst, S.; Maguire, J.A.; et al. Restoring RUNX1 deficiency in RUNX1 familial platelet disorder by inhibiting its degradation. Blood Adv. 2021, 5, 687–699. [Google Scholar] [CrossRef]
- Connelly, J.P.; Kwon, E.M.; Gao, Y.; Trivedi, N.S.; Elkahloun, A.G.; Horwitz, M.S.; Cheng, L.; Liu, P.P. Targeted correction of RUNX1 mutation in FPD patient-specific induced pluripotent stem cells rescues megakaryopoietic defects. Blood 2014, 124, 1926–1930. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.-Y.; Keller, G.; Kuo, F.C.; Weiss, M.; Chen, J.; Rosenblatt, M.; Alt, F.W.; Orkin, S.H. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nat. Cell Biol. 1994, 371, 221–226. [Google Scholar] [CrossRef]
- Rodrigues, N.P.; Janzen, V.; Forkert, R.; Dombkowski, D.M.; Boyd, A.S.; Orkin, S.H.; Enver, T.; Vyas, P.; Scadden, D.T. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood 2005, 106, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, R.E.; Milne, P.; Jardine, L.; Zandi, S.; Swierczek, S.I.; McGovern, N.; Cookson, S.; Ferozepurwalla, Z.; Langridge, A.; Pagan, S.; et al. The evolution of cellular deficiency in GATA2 mutation. Blood 2014, 123, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Nováková, M.; Žaliová, M.; Suková, M.; Wlodarski, M.; Janda, A.; Froňková, E.; Campr, V.; Lejhancová, K.; Zapletal, O.; Pospíšilová, D.; et al. Loss of B cells and their precursors is the most constant feature of GATA-2 deficiency in childhood myelodysplastic syndrome. Haematologica 2016, 101, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.; Sampaio, E.P.; Khan, J.; Calvo, K.; Lemieux, J.E.; Patel, S.; Frucht, D.M.; Vinh, D.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; Van Den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef]
- Ostergaard, P.; Simpson, M.; Connell, F.C.; Steward, C.; Brice, G.; Woollard, W.J.; Dafou, D.; Kilo, T.; Smithson, S.; Lunt, P.; et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 2011, 43, 929–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinner, M.A.; Sanchez, L.; Hsu, A.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, C.; Chong, C.E.; Carmichael, C.; Wilkins, E.J.; Brautigan, P.J.; Li, X.-C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Collin, M.; Horwitz, M.S. GATA2 deficiency and related myeloid neoplasms. Semin. Hematol. 2017, 54, 81–86. [Google Scholar] [CrossRef]
- Kozyra, E.J.; Pastor, V.B.; Lefkopoulos, S.; Sahoo, S.S.; Busch, H.; Voss, R.K.; Erlacher, M.; Lebrecht, D.; Szvetnik, E.A. Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia 2020, 34, 2673–2687. [Google Scholar] [CrossRef]
- West, R.R.; Hsu, A.P.; Holland, S.M.; Cuellar-Rodriguez, J.; Hickstein, D.D. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica 2014, 99, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Donadieu, J.; Lamant, M.; Fieschi, C.; De Fontbrune, F.S.; Caye, A.; Ouachee, M.; Beaupain, B.; Bustamante, J.; Poirel, H.A.; Isidor, B.; et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 2018, 103, 1278–1287. [Google Scholar] [CrossRef] [Green Version]
- Tholouli, E.; Sturgess, K.; Dickinson, R.E.; Gennery, A.; Cant, A.J.; Jackson, G.; Lordan, J.; Hambleton, S.; Slatter, M.A.; Bigley, V.; et al. In vivo T-depleted reduced-intensity transplantation for GATA2-related immune dysfunction. Blood 2018, 131, 1383–1387. [Google Scholar] [CrossRef]
- Bogaert, D.J.; Laureys, G.; Naesens, L.; Mazure, D.; De Bruyne, M.; Hsu, A.P.; Bordon, V.; Wouters, E.; Tavernier, S.J.; Lambrecht, B.N.; et al. GATA2 deficiency and haematopoietic stem cell transplantation: Challenges for the clinical practitioner. Br. J. Haematol. 2020, 188, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parta, M.; Shah, N.N.; Baird, K.; Rafei, H.; Calvo, K.R.; Hughes, T.; Cole, K.; Kenyon, M.; Schuver, B.B.; Cuellar-Rodriguez, J.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency Using a Busulfan-Based Regimen. Biol. Blood Marrow Transplant. 2018, 24, 1250–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Peffault de Latour, R.; Soulier, J. How I treat MDS and AML in Fanconi anemia. Blood 2016, 127, 2971–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaycoechea, J.I.; Patel, K.J. Why does the bone marrow fail in Fanconi anemia? Blood 2014, 123, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pr. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Quentin, S.; Cuccuini, W.; Ceccaldi, R.; Nibourel, O.; Pondarre, C.; Pagès, M.-P.; Vasquez, N.; D’Enghien, C.D.; Larghero, J.; De Latour, R.P.; et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 2011, 117, e161–e170. [Google Scholar] [CrossRef] [Green Version]
- Ebens, C.L.; MacMillan, M.; Wagner, J.E. Hematopoietic cell transplantation in Fanconi anemia: Current evidence, challenges and recommendations. Expert Rev. Hematol. 2017, 10, 81–97. [Google Scholar] [CrossRef]
- Gross, M.; Hanenberg, H.; Lobitz, S.; Friedl, R.; Herterich, S.; Dietrich, R.; Gruhn, B.; Schindler, D.; Hoehn, H. Reverse mosaicism in Fanconi anemia: Natural gene therapy via molecular self-correction. Cytogenet. Genome Res. 2002, 98, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Léger-Silvestre, I.; Caffrey, J.M.; Dawaliby, R.; Alvarez-Arias, D.A.; Gas, N.; Bertolone, S.J.; Gleizes, P.-E.; Ellis, S.R. Specific Role for Yeast Homologs of the Diamond Blackfan Anemia-associated Rps19 Protein in Ribosome Synthesis. J. Biol. Chem. 2005, 280, 38177–38185. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, A.; Rosenberg, P.; Atsidaftos, E.; Alter, B.P.; Lipton, J.M. Incidence of neoplasia in Diamond Blackfan anemia: A report from the Diamond Blackfan Anemia Registry. Blood 2012, 119, 3815–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.; Rosenberg, P. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, L.; Narla, A.; Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef]
- Vlachos, A. Acquired ribosomopathies in leukemia and solid tumors. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 716–719. [Google Scholar] [CrossRef] [Green Version]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra67. [Google Scholar] [CrossRef] [Green Version]
- Le Goff, S.; Boussaid, I.; Floquet, C.; Raimbault, A.; Hatin, I.; Andrieu-Soler, C.; Salma, M.; Leduc, M.; Gautier, E.-F.; Guyot, B.; et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood 2021, 137, 89–102. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Van Wijk, R.; Pereboom, T.C.; Goos, Y.J.; Seinen, C.W.; Van Oirschot, B.A.; Van Dooren, R.; Gastou, M.; Giles, R.H.; Van Solinge, W.; et al. Ribosomal Protein Mutations Induce Autophagy through S6 Kinase Inhibition of the Insulin Pathway. PLoS Genet. 2014, 10, e1004371. [Google Scholar] [CrossRef]
- Finch, A.J.; Hilcenko, C.; Basse, N.; Drynan, L.F.; Goyenechea, B.; Menne, T.F.; González-Fernández, Á.; Simpson, P.; D’Santos, C.S.; Arends, M.J.; et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011, 25, 917–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzerri, V.; Cipolli, M. Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives. Mol. Diagn. Ther. 2019, 23, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.C.; Davies, S.M.; Shimamura, A. Clinical and Molecular Pathophysiology of Shwachman–Diamond Syndrome: An Update. Hematol. Clin. N. Am. 2013, 27, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carapito, R.; Konantz, M.; Paillard, C.; Miao, Z.; Pichot, A.; LeDuc, M.S.; Yang, Y.; Bergstrom, K.L.; Mahoney, D.H.; Shardy, D.L.; et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond–like features. J. Clin. Investig. 2017, 127, 4090–4103. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Myers, K.C.; Bowman, J.; Gibson, C.J.; Camarda, N.D.; Furutani, E.; Muscato, G.M.; Klein, R.H.; Ballotti, K.; Liu, S.; et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat. Commun. 2021, 12, 1334. [Google Scholar] [CrossRef]
- Xia, J.; Miller, C.; Baty, J.; Ramesh, A.; Jotte, M.; Fulton, R.S.; Vogel, T.P.; Cooper, M.; Walkovich, K.J.; Makaryan, V.; et al. Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood 2018, 131, 408–416. [Google Scholar] [CrossRef]
- Myers, K.C.; Furutani, E.; Weller, E.; Siegele, B.; Galvin, A.; Arsenault, V.; Alter, B.P.; Boulad, F.; Bueso-Ramos, C.; Burroughs, L.; et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: A multicentre, retrospective, cohort study. Lancet Haematol. 2020, 7, e238–e246. [Google Scholar] [CrossRef]
- Bertuch, A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016, 13, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Ball, S.E.; Gibson, F.M.; Rizzo, S.; Tooze, J.A.; Marsh, J.C.; Gordon-Smith, E.C. Progressive telomere shortening in aplastic anemia. Blood 1998, 91, 3582–3592. [Google Scholar] [CrossRef]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.J.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef]
- Townsley, D.M.; Dumitriu, B.; Young, N.S. Bone marrow failure and the telomeropathies. Blood 2014, 124, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, S.; Churpek, J.E.; Walsh, T.; Keel, S.; Hakkarainen, M.; Schroeder, T.; Germing, U.; Geyh, S.; Heuser, M.; Thol, F.; et al. Germline variants drive myelodysplastic syndrome in young adults. Leukemia 2021, 35, 2439–2444. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Scott, A.; Sanchez-Bonilla, M.; Ho, P.A.; Gulsuner, S.; Pritchard, C.C.; Abkowitz, J.L.; King, M.-C.; Walsh, T.; Shimamura, A. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 2016, 101, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Bluteau, O.; Sebert, M.; Leblanc, T.; De Latour, R.P.; Quentin, S.; Lainey, E.; Hernandez, L.; Dalle, J.-H.; De Fontbrune, F.S.; Lengline, E.; et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood 2018, 131, 717–732. [Google Scholar] [CrossRef] [Green Version]
- Rollison, D.E.; Epling-Burnette, P.K.; Park, J.Y.; Lee, J.-H.; Park, H.; Jonathan, K.; Cole, A.L.; Painter, J.S.; Guerrier, M.; Meléndez-Santiago, J.; et al. Telomere length in myelodysplastic syndromes. Leuk. Lymphoma 2011, 52, 1528–1536. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Jiang, J.; Smith, A.E.; Mohamedali, A.M.; Mian, S.A.; Gandhi, S.; Gaken, J.; Czepulkowski, B.; Marsh, J.C.W.; Mufti, G.J. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood 2014, 124, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.T.; Cooper, J.N.; Padilla-Nash, H.M.; Sloand, E.M.; Wu, C.O.; Scheinberg, P.; Ried, T.; Young, N.S. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia 2012, 26, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsley, D.M.; Dumitriu, B.; Liu, D.; Biancotto, A.; Weinstein, B.; Chen, C.; Hardy, N.; Mihalek, A.D.; Lingala, S.; Kim, Y.J.; et al. Danazol Treatment for Telomere Diseases. N. Engl. J. Med. 2016, 374, 1922–1931. [Google Scholar] [CrossRef]
- Agarwal, S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis Congenita. Hematol. Clin. N. Am. 2018, 32, 669–685. [Google Scholar] [CrossRef]
- Gadalla, S.M.; Sales-Bonfim, C.; Carreras, J.; Alter, B.P.; Antin, J.H.; Ayas, M.; Bodhi, P.; Davis, J.; Davies, S.M.; Deconinck, E.; et al. Outcomes of Allogeneic Hematopoietic Cell Transplantation in Patients with Dyskeratosis Congenita. Biol. Blood Marrow Transplant. 2013, 19, 1238–1243. [Google Scholar] [CrossRef] [Green Version]
- Myllymäki, M.; Redd, R.A.; Reilly, C.R.; Saber, W.; Spellman, S.R.; Gibson, C.J.; Hu, Z.-H.; Wang, T.; Orr, E.H.; Grenier, J.G.; et al. Short telomere length predicts nonrelapse mortality after stem cell transplantation for myelodysplastic syndrome. Blood 2020, 136, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ Line p53 Mutations in a Familial Syndrome of Breast Cancer, Sarcomas, and Other Neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Hagström-Andersson, A.; Chen, S.-C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 2009, 23, 203–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Zebisch, A.; Lal, R.; Müller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.; Fuchs, D.; Wölfler, A.; Geigl, J.B.; Sill, H. Acute myeloid leukemia with TP53 germ line mutations. Blood 2016, 128, 2270–2272. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Valentin, A.; Ulz, P.; Beham-Schmid, C.; Lind, K.; Rupp, V.; Lackner, H.; Wölfler, A.; Zebisch, A.; Olipitz, W.; et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J. Med Genet. 2012, 49, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Drazer, M.W.; Kadri, S.; Sukhanova, M.; Patil, S.; West, A.H.; Feurstein, S.; Calderon, D.A.; Jones, M.F.; Weipert, C.M.; Daugherty, C.K.; et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Adv. 2018, 2, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Ruijs, M.W.G.; Verhoef, S.; Rookus, M.A.; Pruntel, R.; van der Hout, A.H.; Hogervorst, F.B.L.; Kluijt, I.; Sijmons, R.H.; Aalfs, C.M.; Wagner, A.; et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: Mutation detection rate and relative frequency of cancers in different familial phenotypes. J. Med Genet. 2010, 47, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, M.; Bannon, S.A.; Routbort, M.; Naqvi, K.; Kadia, T.M.; Takahashi, K.; Alvarado, Y.; Ravandi-Kashani, F.; Patel, K.P.; Champlin, R.; et al. Hematologic malignancies and Li–Fraumeni syndrome. Mol. Case Stud. 2019, 5, a003210. [Google Scholar] [CrossRef] [Green Version]
- Polprasert, C.; Schulze, I.; Sekeres, M.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvatiyar, K.; Zhang, Z.; Teles, R.; Ouyang, S.; Jiang, Y.; Iyer, S.S.; Zaver, S.A.; Schenk, M.; Zeng, S.; Zhong, W.; et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 2012, 13, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Sébert, M.; Passet, M.; Raimbault, A.; Rahmé, R.; Raffoux, E.; De Fontbrune, F.S.; Cerrano, M.; Quentin, S.; Vasquez, N.; Da Costa, M.; et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood 2019, 134, 1441–1444. [Google Scholar] [CrossRef]
- Quesada, A.E.; Routbort, M.J.; Dinardo, C.D.; Bueso-Ramos, C.E.; Kanagal-Shamanna, R.; Khoury, J.D.; Thakral, B.; Zuo, Z.; Yin, C.C.; Loghavi, S.; et al. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am. J. Hematol. 2019, 94, 757–766. [Google Scholar] [CrossRef]
- Woll, P.S.; Kjällquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic Syndromes Are Propagated by Rare and Distinct Human Cancer Stem Cells In Vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef] [Green Version]
- Negoro, E.; Radivoyevitch, T.; Polprasert, C.; Adema, V.; Hosono, N.; Makishima, H.; Przychodzen, B.; Hirsch, C.; Clemente, M.J.; Nazha, A.; et al. Molecular predictors of response in patients with myeloid neoplasms treated with lenalidomide. Leukemia 2016, 30, 2405–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdock, H.M.; Kim, H.T.; Hambley, B.; Vachhani, P.; Denlinger, N.; Gier, S.H.; Cho, C.; Perales, M.; Koreth, J.; Soiffer, V.T.; et al. Genetic Alterations at Diagnosis Predict Outcome of AML Patients Age 60 or Older Undergoing Allogeneic Transplantation in First Remission. Blood 2019, 134, 48. [Google Scholar] [CrossRef]
- Buonocore, F.; Kühnen, P.; Suntharalingham, J.P.; Del Valle, I.; Digweed, M.; Stachelscheid, H.; Khajavi, N.; Didi, M.; Brady, A.F.; Blankenstein, O.; et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J. Clin. Investig. 2017, 127, 1700–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, V.B.; Sahoo, S.S.; Boklan, J.; Schwabe, G.C.; Saribeyoglu, E.; Strahm, B.; Lebrecht, D.; Voss, M.; Bryceson, Y.T.; Erlacher, M.; et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica 2018, 103, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Nilsson, A.R.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Narumi, S.; Guan, Y.; Przychodzen, B.P.; Hirsch, C.M.; Makishima, H.; Shima, H.; Aly, M.; Pastor, V.; Kuzmanovic, T.; et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 2018, 132, 2309–2313. [Google Scholar] [CrossRef] [Green Version]
- Sud, A.; Chattopadhyay, S.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Houlston, R.S.; Hemminki, K. Familial risks of acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms. Blood 2018, 132, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Strom, S.S.; Gu, Y.; Gruschkus, S.K.; Pierce, S.; Estey, E.H. Risk factors of myelodysplastic syndromes: A case–control study. Leukemia 2005, 19, 1912–1918. [Google Scholar] [CrossRef]
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- University of Chicago Hematopoietic Malignancies Cancer Risk Team. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood 2016, 128, 1800–1813. [Google Scholar] [CrossRef] [Green Version]
- Padron, E.; Ball, M.C.; Teer, J.K.; Painter, J.S.; Yoder, S.J.; Zhang, C.; Zhang, L.; Moscinski, L.C.; Rollison, D.E.; Gore, S.D.; et al. Germ line tissues for optimal detection of somatic variants in myelodysplastic syndromes. Blood 2018, 131, 2402–2405. [Google Scholar] [CrossRef]
- Kobayashi, S.; Osawa, Y.; Nagao, S.; Takano, K.; Okada, Y.; Tachi, N.; Teramoto, M.; Kawamura, T.; Horiuchi, T.; Kato, S.; et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia 2017, 31, 1020–1022. [Google Scholar] [CrossRef]
- Xiao, H.; Shi, J.; Luo, Y.; Tan, Y.; He, J.; Xie, W.; Zhang, L.; Wang, Y.; Liu, L.; Wu, K.; et al. First report of multiple CEBPA mutations contributing to donor origin of leukemia relapse after allogeneic hematopoietic stem cell transplantation. Blood 2011, 117, 5257–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galera, P.; Hsu, A.P.; Wang, W.; Droll, S.; Chen, R.; Schwartz, J.R.; Klco, J.M.; Arai, S.; Maese, L.; Zerbe, C.; et al. Donor-derived MDS/AML in families with germline GATA2 mutation. Blood 2018, 132, 1994–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churpek, J.E.; Artz, A.; Bishop, M.; Liu, H.; Godley, L.A. Correspondence Regarding the Consensus Statement from the Worldwide Network for Blood and Marrow Transplantation Standing Committee on Donor Issues. Biol. Blood Marrow Transplant. 2016, 22, 183–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Etiological and pathological processes leading to MDS. Graded blue fill—inherited factors. Light blue and dark blue fill—pathological mechanisms.
Figure 1.
Etiological and pathological processes leading to MDS. Graded blue fill—inherited factors. Light blue and dark blue fill—pathological mechanisms.


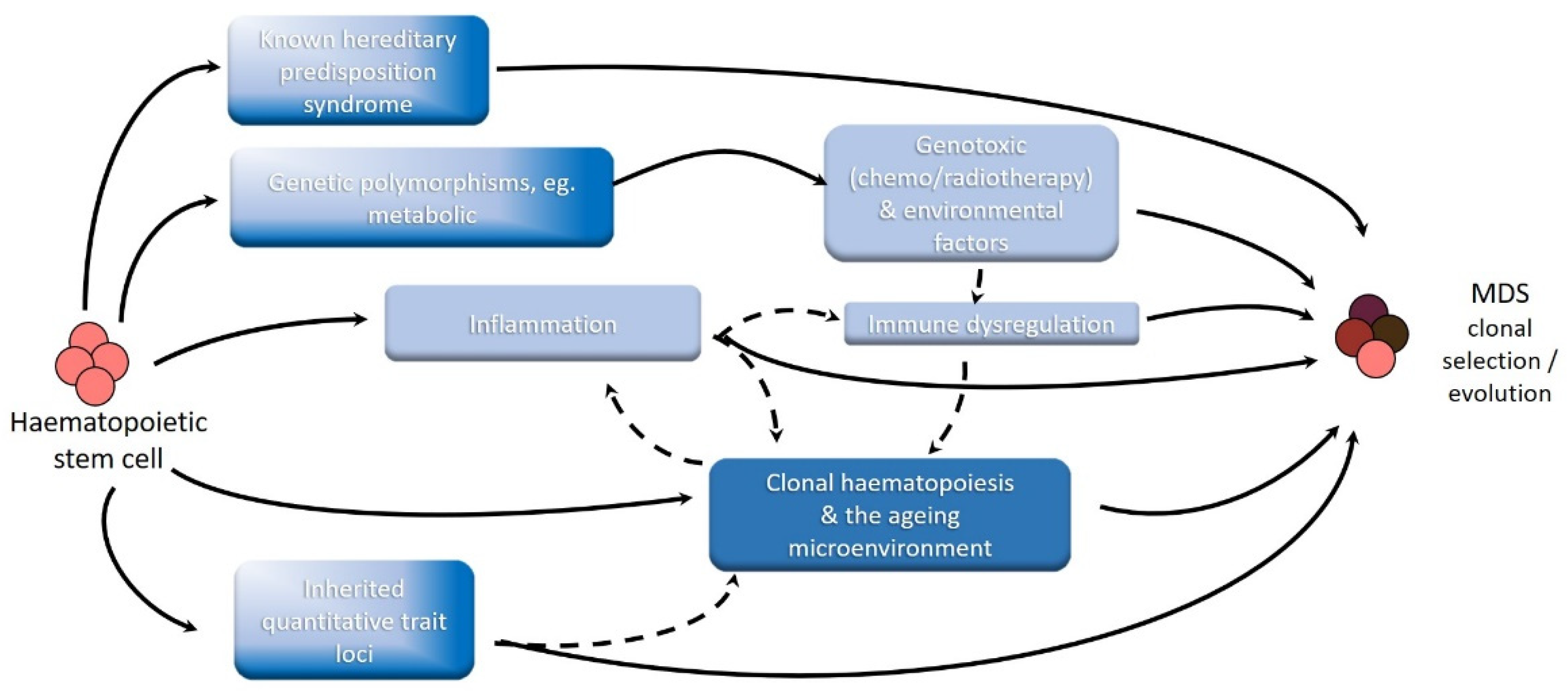{kind=link}
Table 1.
Selected genes involved in germline MDS predisposition and associated phenotypes.
| Cellular Function, Gene | Clinical Features | Frequency |
|---|---|---|
| Transcription factors | ||
| Familial platelet disorder with predisposition to myeloid malignancy RUNX1 | Thrombocytopenia, platelet dysfunction, bleeding phenotype, eczema, T-ALL, hairy cell leukaemia | Rare |
| GATA2-spectrum disorders GATA2 | Emberger syndrome, MonoMAC syndrome, immunodeficiency (DC, monocyte, B and NK-cell deficiency), lymphoedema, warts, atypical mycobacterial infection, hearing loss, CMML, JMML, monosomy 7 | Childhood MDS: 7%; Adult MDS: unknown but likely underdiagnosed |
| Thrombocytopenia 5 ETV6 | Thrombocytopenia, platelet dysfunction, B-ALL, CMML, MM | Rare |
| DNA repair and genome instability syndromes | ||
| Fanconi anaemia FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1/BACH1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4, FANCQ/ERCC4, FANCR/RAD51, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7, FANCW/RFWD3 | Bone marrow failure, short stature, skeletal and facial abnormalities, congenital cardiac, renal and ocular/auditory abnormalities, café-au-lait spots, squamous cell carcinoma (oral, gastrointestinal, genitourinary), breast cancer, positive chromosome breakage testing, elevated HbF | Childhood BM failure: most frequent cause [31] Adult MDS: very rare |
| Mismatch repair disorders MLH1, MSH6, PMS2, MSH2, EPCAM | Colon/ovarian/uterine/CNS cancer, ALL, lymphoma | |
| Bloom syndrome BLM | Short stature, photosensitive rash, pulmonary disease, diabetes, ALL, lymphoma | |
| Ribosome disorders | ||
| Diamond-Blackfan anaemia RPS19, RPL5, RPS26, RPL11, RPL35a, RPS10 | Bone marrow failure, short stature, congenital cardiac, renal, skeletal and facial abnormalities, elevated HbF, sarcoma | Extremely rare |
| Schwachman-Diamond syndrome SBDS | Bone marrow failure, short stature, pancreatic insufficiency, preceding isolated neutropenia, skeletal abnormalities | Formerly thought to be very rare, phenotypically silent cases may represent 4% of young MDS [32] |
| Telomeropathies | ||
| Dyskeratosis congenita and telomere biology disorders DKC1, TERC, TERT, TINF2, RTEL1 | Nail dystrophy, abnormal skin and hair pigmentation, oral leukoplakia, pulmonary and hepatic fibrosis, oral and GI squamous cell carcinoma, telomere lengths < 1st percentile for age | Syndromically rare, however rare TERT variants associated with short telomeres have been recently identified in 3% of MDS transplant recipients [33] |
| Signal transducers (Ras pathway) | ||
| Noonan syndrome PTPN11, SOS1, RAF1, KRAS, NRAS, BRAF, MAP2K1 | Short stature, cardiac and facial abnormalities, coagulopathy, webbed neck, developmental delay, JMML | |
| Noonan-like CBL | JMML | |
| Tumour suppressors Li-Fraumeni syndrome TP53 | Early onset breast malignancy (age < 30), sarcoma, CNS, adrenocortical carcinoma, paediatric hypodiploid ALL, lymphoma, therapy-related leukaemias | Very rare |
| Other DDX41 | Malignancies at older ages (>50), CML, lymphoma | Most frequent HMMS: 2–3% in adult MDS |
| Thrombocytopenia 2 ANKRD26 | Thrombocytopenia, platelet dysfunction, CLL, CML, CMML | |
| MIRAGE Syndrome SAMD9 | Myelodysplasia, infection, restriction of growth, adrenal insufficiency, genital phenotypes and enteropathy, monosomy 7 | Previously thought to be very rare, one study recently identified SAMD9/SAMD9L lesions in 17% of paediatric MDS [34] |
| Ataxia-pancytopenia syndrome SAMD9L | Bone marrow failure, ataxia, monosomy 7 | |
| Neurofibromatosis 1 NF1 | Café au-lait spots, neurofibromas, axillary freckles, Lisch nodules, CNS cancers | |
| SRP72 | Bone marrow failure, sensorineural hearing loss | |
| Severe congenital neutropenia ELANE, CSF3R, HAX1, G6PC3, WAS | Osteopenia (ELANE), neurodevelopmental (HAX1), cardiac (G6PC3), monocytopenia (WAS) |
Table 2.
Individuals in whom inherited predisposition to MDS must be considered, adapted WHO criteria [128].
Table 2.
Individuals in whom inherited predisposition to MDS must be considered, adapted WHO criteria [128].
|
Table 3.
Key management principles for patients with germline MDS predisposition.
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Khan, A.B.; Bowen, D. Aetiology of MDS: With a Focus on Hereditary Predisposition. Hemato 2022, 3, 17-37. https://doi.org/10.3390/hemato3010003
AMA Style
Khan AB, Bowen D. Aetiology of MDS: With a Focus on Hereditary Predisposition. Hemato. 2022; 3(1):17-37. https://doi.org/10.3390/hemato3010003
Chicago/Turabian StyleKhan, Anjum B., and David Bowen. 2022. "Aetiology of MDS: With a Focus on Hereditary Predisposition" Hemato 3, no. 1: 17-37. https://doi.org/10.3390/hemato3010003