
Cold Agglutinin Disease: A Distinct Clonal B-Cell Lymphoproliferative Disorder of the Bone Marrow


Abstract
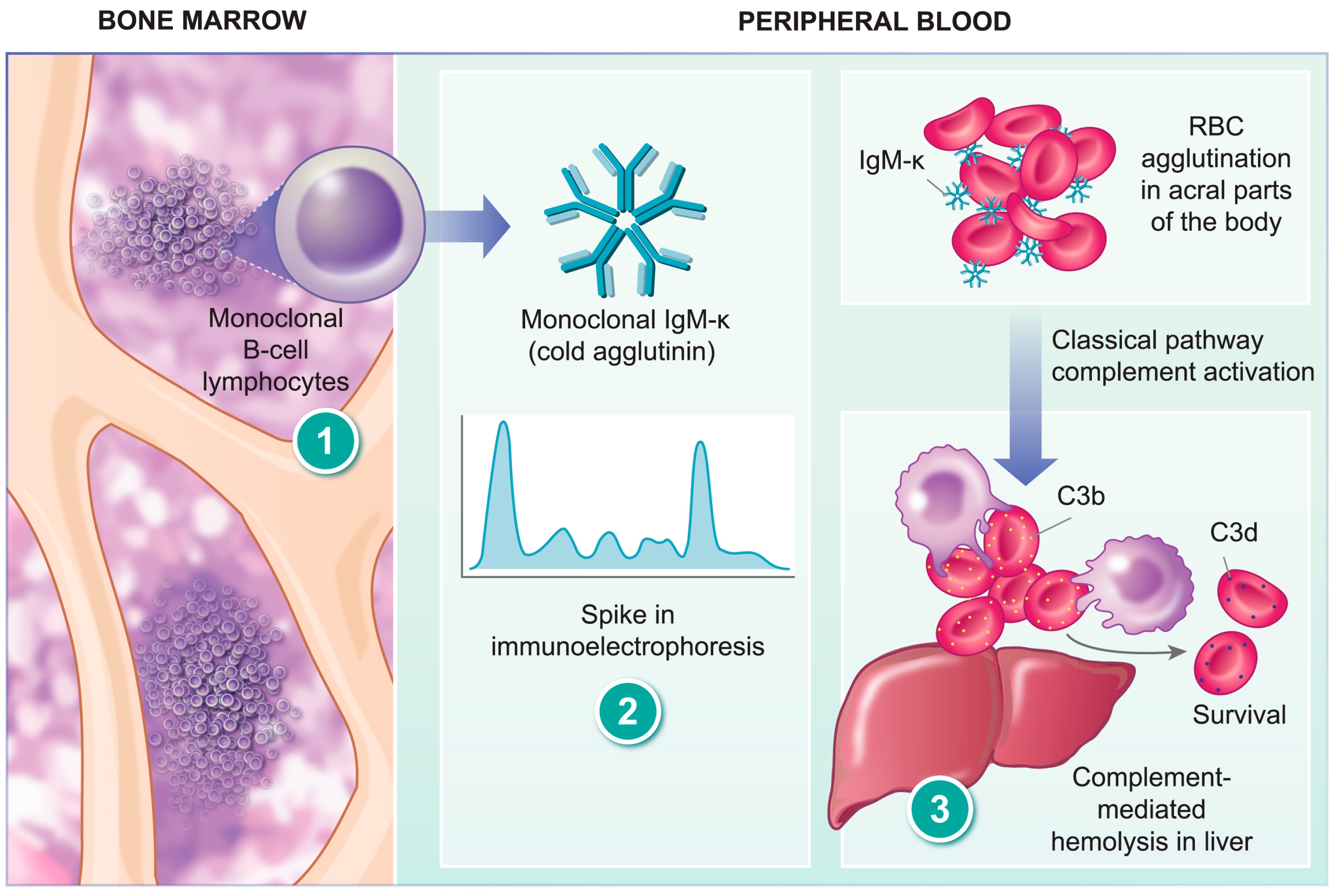
:1. History
2. Definition of Cold Agglutinin Disease (CAD)
3. Epidemiology
4. Pathogenesis of CAD
5. Clinical Presentation
5.1. Fatigue
5.2. Anemia
5.3. Cold-Induced Symptoms
5.4. Thromboembolic Complications
6. Diagnosis
6.1. Principles of DAT
6.2. Methods of Performing DAT
6.3. Results of DAT in CAD and Further Characterization
7. Treatment
7.1. Fatigue and Anemia
7.2. Cold-Induced Symptoms and TE Complications
7.3. Emergency Situations
7.4. Current Recommendations
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mack, P.; Freedman, J. Autoimmune hemolytic anemia: A history. Transfus. Med. Rev. 2000, 14, 223–233. [Google Scholar] [CrossRef]
- Petz, L.D.; Garratty, G. Historical concepts of immune hemolytic anemias. In Immune Hemolytic Anemias, 2nd ed.; Petz, L.D., Garratty, G., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2004; Volume 1, pp. 1–31. [Google Scholar]
- Coombs, R.R. Historical note: Past, present and future of the antiglobulin test. Vox Sang. 1998, 74, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Miller, G.; Christian, R.M. Clinical and laboratory observations on autoimmune hemolytic disease. Ann. Intern. Med. 1951, 35, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Schubothe, H. The cold hemagglutinin disease. Semin. Hematol. 1966, 3, 27–47. [Google Scholar] [PubMed]
- Wiener, A.S.; Unger, L.J.; Cohen, L.; Feldman, J. Type-specific cold auto-antibodies as a cause of acquired hemolytic anemia and hemolytic transfusion reactions: Biologic test with bovine red cells. Ann. Intern. Med. 1956, 44, 221–240. [Google Scholar] [CrossRef]
- Swiecicki, P.L.; Hegerova, L.T.; Gertz, M.A. Cold agglutinin disease. Blood 2013, 122, 1114–1121. [Google Scholar] [CrossRef]
- Hill, Q.A.; Hill, A.; Berentsen, S. Defining autoimmune hemolytic anemia: A systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019, 3, 1897–1906. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, S.; Barcellini, W. Autoimmune Hemolytic Anemias. N. Engl. J. Med. 2021, 385, 1407–1419. [Google Scholar] [CrossRef]
- Berentsen, S. New Insights in the Pathogenesis and Therapy of Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia. Front. Immunol. 2020, 11, 590. [Google Scholar] [CrossRef]
- Berentsen, S.; Ulvestad, E.; Langholm, R.; Beiske, K.; Hjorth-Hansen, H.; Ghanima, W.; Sorbo, J.H.; Tjonnfjord, G.E. Primary chronic cold agglutinin disease: A population based clinical study of 86 patients. Haematologica 2006, 91, 460–466. [Google Scholar]
- Berentsen, S.; Barcellini, W.; D’Sa, S.; Randen, U.; Tvedt, T.H.A.; Fattizzo, B.; Haukas, E.; Kell, M.; Brudevold, R.; Dahm, A.E.A.; et al. Cold agglutinin disease revisited: A multinational, observational study of 232 patients. Blood 2020, 136, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Petz, L.D.; Garratty, G. Classification and clinical characteristics of autoimmune hemolytic anemias. In Immune Hemolytic Anemias, 2nd ed.; Petz, L.D., Garratty, G., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2004; Volume 1, pp. 61–131. [Google Scholar]
- Campbell, A.; Podbury, B.; Yue, M.; Mollee, P.; Bird, R.; Hapgood, G. The Role of a Routine Bone Marrow Biopsy in Autoimmune Hemolytic Anemia for the Detection of an Underlying Lymphoproliferative Disorder. Hemasphere 2022, 6, e674. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S. Centralized review offers promise for the clinician, the pathologist, and the patient with newly diagnosed lymphoma. J. Clin. Oncol. 2011, 29, 1398–1399. [Google Scholar] [CrossRef] [PubMed]
- Randen, U.; Troen, G.; Tierens, A.; Steen, C.; Warsame, A.; Beiske, K.; Tjonnfjord, G.E.; Berentsen, S.; Delabie, J. Primary cold agglutinin-associated lymphoproliferative disease: A B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica 2014, 99, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Reyero, J.; Martinez Magunacelaya, N.; Gonzalez de Villambrosia, S.; Gomez Mediavilla, A.; Urquieta Lam, M.; Insunza, A.; Tonda, R.; Beltran, S.; Gut, M.; Gonzalez, A.; et al. Diagnostic value of bone marrow core biopsy patterns in lymphoplasmacytic lymphoma/Waldenstrom macroglobulinaemia and description of its mutational profiles by targeted NGS. J. Clin. Pathol. 2020, 73, 571–577. [Google Scholar] [CrossRef]
- Malecka, A.; Delabie, J.; Ostlie, I.; Tierens, A.; Randen, U.; Berentsen, S.; Tjonnfjord, G.E.; Troen, G. Cold agglutinin-associated B-cell lymphoproliferative disease shows highly recurrent gains of chromosome 3 and 12 or 18. Blood Adv. 2020, 4, 993–996. [Google Scholar] [CrossRef]
- Bertoni, F.; Rossi, D.; Zucca, E. Recent advances in understanding the biology of marginal zone lymphoma. F1000Research 2018, 7, 406. [Google Scholar] [CrossRef] [Green Version]
- Van den Brand, M.; van Krieken, J.H. Recognizing nodal marginal zone lymphoma: Recent advances and pitfalls. A systematic review. Haematologica 2013, 98, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Krugmann, J.; Tzankov, A.; Dirnhofer, S.; Fend, F.; Wolf, D.; Siebert, R.; Probst, P.; Erdel, M. Complete or partial trisomy 3 in gastro-intestinal MALT lymphomas co-occurs with aberrations at 18q21 and correlates with advanced disease stage: A study on 25 cases. World J. Gastroenterol. 2005, 11, 7384–7385. [Google Scholar] [CrossRef]
- Malecka, A.; Troen, G.; Delabie, J.; Malecki, J.; Ostlie, I.; Tierens, A.; Randen, U.; Berentsen, S.; Tjonnfjord, G.E. The mutational landscape of cold agglutinin disease: CARD11 and CXCR4 mutations are correlated with lower hemoglobin levels. Am. J. Hematol. 2021, 96, E279–E283. [Google Scholar] [CrossRef]
- Kaiser, L.M.; Hunter, Z.R.; Treon, S.P.; Buske, C. CXCR4 in Waldenstrom’s Macroglobulinema: Chances and challenges. Leukemia 2021, 35, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Malecka, A.; Troen, G.; Tierens, A.; Ostlie, I.; Malecki, J.; Randen, U.; Berentsen, S.; Tjonnfjord, G.E.; Delabie, J.M. Immunoglobulin heavy and light chain gene features are correlated with primary cold agglutinin disease onset and activity. Haematologica 2016, 101, e361–e364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, A.; Barcellini, W.; D’Sa, S.; Miyakawa, Y.; Broome, C.M.; Michel, M.; Kuter, D.J.; Jilma, B.; Tvedt, T.H.A.; Fruebis, J.; et al. Sutimlimab in Cold Agglutinin Disease. N. Engl. J. Med. 2021, 384, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Fryzek, J.; Jiang, X.; Reichert, H.; Patel, P.; Su, J.; Morales Arias, J.; Broome, C.M. Complement-mediated hemolysis persists year round in patients with cold agglutinin disease. Transfusion 2021, 62, 51–59. [Google Scholar] [CrossRef]
- Linz, W.J.; Tauscher, C.; Winters, J.L.; Gastineau, D.A.; Moore, B. Transfusion medicine illustrated: Cold agglutinin disease. Transfusion 2003, 43, 1185. [Google Scholar] [CrossRef]
- Ungprasert, P.; Tanratana, P.; Srivali, N. Autoimmune hemolytic anemia and venous thromboembolism: A systematic review and meta-analysis. Thromb. Res. 2015, 136, 1013–1017. [Google Scholar] [CrossRef]
- Bylsma, L.C.; Gulbech Ording, A.; Rosenthal, A.; Ozturk, B.; Fryzek, J.P.; Arias, J.M.; Roth, A.; Berentsen, S. Occurrence, thromboembolic risk, and mortality in Danish patients with cold agglutinin disease. Blood Adv. 2019, 3, 2980–2985. [Google Scholar] [CrossRef]
- Broome, C.M.; Cunningham, J.M.; Mullins, M.; Jiang, X.; Bylsma, L.C.; Fryzek, J.P.; Rosenthal, A. Increased risk of thrombotic events in cold agglutinin disease: A 10-year retrospective analysis. Res. Pr. Thromb. Haemost. 2020, 4, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Cid, J.; Ortin, X.; Beltran, V.; Escoda, L.; Contreras, E.; Elies, E.; Martin-Vega, C. The direct antiglobulin test in a hospital setting. Immunohematology 2003, 19, 16–18. [Google Scholar] [CrossRef]
- Berentsen, S.; Roth, A.; Randen, U.; Jilma, B.; Tjonnfjord, G.E. Cold agglutinin disease: Current challenges and future prospects. J. Blood Med. 2019, 10, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Petz, L.D.; Garratty, G. The diagnosis of hemolytic anemia. In Immune Hemolytic Anemias, 2nd ed.; Petz, L.D., Garratty, G., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2004; Volume 1, pp. 33–60. [Google Scholar]
- Kaplan, H.S.; Garratty, G. Predictive value of direct antiglobulin test results. Diagn. Med. 1985, 8, 29–33. [Google Scholar]
- Kamesaki, T.; Kajii, E. A Comprehensive Diagnostic Algorithm for Direct Antiglobulin Test-Negative Autoimmune Hemolytic Anemia Reveals the Relative Ratio of Three Mechanisms in a Single Laboratory. Acta Haematol. 2018, 140, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Revelli, N.; Imperiali, F.G.; Villa, M.A.; Manera, M.C.; Paccapelo, C.; Zaninoni, A.; Zanella, A. Comparison of traditional methods and mitogen-stimulated direct antiglobulin test for detection of anti-red blood cell autoimmunity. Int. J. Hematol. 2010, 91, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Kamesaki, T. Diagnostic algorithm for classification and characterization of direct antiglobulin test-negative autoimmune hemolytic anemia with 1-year clinical follow-up. Transfusion 2021, 62, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Rives, S.; Montoto, S.; Sanz, C.; Pereira, A. Relative sensitivity of direct antiglobulin test, antibody’s elution and flow cytometry in the serologic diagnosis of immune hemolytic transfusion reactions. Haematologica 2000, 85, 186–188. [Google Scholar]
- Kamesaki, T.; Oyamada, T.; Omine, M.; Ozawa, K.; Kajii, E. Cut-off value of red-blood-cell-bound IgG for the diagnosis of Coombs-negative autoimmune hemolytic anemia. Am. J. Hematol. 2009, 84, 98–101. [Google Scholar] [CrossRef]
- Jager, U.; Barcellini, W.; Broome, C.M.; Gertz, M.A.; Hill, A.; Hill, Q.A.; Jilma, B.; Kuter, D.J.; Michel, M.; Montillo, M.; et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2020, 41, 100648. [Google Scholar] [CrossRef]
- Berentsen, S.; Hill, A.; Hill, Q.A.; Tvedt, T.H.A.; Michel, M. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther. Adv. Hematol. 2019, 10, 2040620719873321. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, S. How I treat cold agglutinin disease. Blood 2021, 137, 1295–1303. [Google Scholar] [CrossRef]
- Reeves, H.M.; Winters, J.L. The mechanisms of action of plasma exchange. Br. J. Haematol. 2014, 164, 342–351. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354. [Google Scholar] [CrossRef] [PubMed]
- Rovira, J.; Cid, J.; Gutierrez-Garcia, G.; Pereira, A.; Fernandez-Aviles, F.; Rosinol, L.; Martinez, C.; Carreras, E.; Urbano, A.; Rovira, M.; et al. Fatal immune hemolytic anemia following allogeneic stem cell transplantation: Report of 2 cases and review of literature. Transfus. Med. Rev. 2013, 27, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, I.; Cid, J.; Palomino, A.; Gine, E.; Alvarez-Larran, A.; Cibeira, M.T.; Lozano, M. Role of therapeutic plasma exchanges in refractory severe warm autoimmune hemolytic anemia: Presentation of two case reports. Transfusion 2020, 60, 2753–2757. [Google Scholar] [CrossRef] [PubMed]
- Pons-Estel, G.J.; Salerni, G.E.; Serrano, R.M.; Gomez-Puerta, J.A.; Plasin, M.A.; Aldasoro, E.; Lozano, M.; Cid, J.; Cervera, R.; Espinosa, G. Therapeutic plasma exchange for the management of refractory systemic autoimmune diseases: Report of 31 cases and review of the literature. Autoimmun. Rev. 2011, 10, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Cid, J.; Carbasse, G.; Andreu, B.; Baltanas, A.; Garcia-Carulla, A.; Lozano, M. Efficacy and safety of plasma exchange: An 11-year single-center experience of 2730 procedures in 317 patients. Transfus. Apher. Sci. 2014, 51, 209–214. [Google Scholar] [CrossRef]
- Cid, J.; Perez-Valencia, A.I.; Torrente, M.A.; Avarez-Larran, A.; Diaz-Ricart, M.; Esteve, J.; Lozano, M. Successful management of three patients with autoimmune thrombotic thrombocytopenic purpura with paradigm-changing therapy: Caplacizumab, steroids, plasma exchange, rituximab, and intravenous immunoglobulins (CASPERI). Transfus. Apher. Sci. 2021, 60, 103011. [Google Scholar] [CrossRef]
- Deng, J.; Zhou, F.; Wong, C.Y.; Huang, E.; Zheng, E. Efficacy of therapeutic plasma exchange for treatment of autoimmune hemolytic anemia: A systematic review and meta-analysis of randomized controlled trials. J. Clin. Apher. 2020, 35, 294–306. [Google Scholar] [CrossRef]
- Berentsen, S.; Ulvestad, E.; Gjertsen, B.T.; Hjorth-Hansen, H.; Langholm, R.; Knutsen, H.; Ghanima, W.; Shammas, F.V.; Tjonnfjord, G.E. Rituximab for primary chronic cold agglutinin disease: A prospective study of 37 courses of therapy in 27 patients. Blood 2004, 103, 2925–2928. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, S.; Randen, U.; Oksman, M.; Birgens, H.; Tvedt, T.H.A.; Dalgaard, J.; Galteland, E.; Haukas, E.; Brudevold, R.; Sorbo, J.H.; et al. Bendamustine plus rituximab for chronic cold agglutinin disease: Results of a Nordic prospective multicenter trial. Blood 2017, 130, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, S.; Randen, U.; Vagan, A.M.; Hjorth-Hansen, H.; Vik, A.; Dalgaard, J.; Jacobsen, E.M.; Thoresen, A.S.; Beiske, K.; Tjonnfjord, G.E. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood 2010, 116, 3180–3184. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Gramegna, D.; Paoloni, F.; Fattizzo, B.; Binda, F.; D’Adda, M.; Farina, M.; Lucchini, E.; Mauro, F.R.; Salvi, F.; et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: A phase 2 prospective GIMEMA study. Blood 2018, 132, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Jalink, M.; Berentsen, S.; Castillo, J.J.; Treon, S.P.; Cruijsen, M.; Fattizzo, B.; Cassin, R.; Fotiou, D.; Kastritis, E.; De Haas, M.; et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood 2021, 138, 2002–2005. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Warm AIHA | Cold AIHA | Atypical AIHA |
|---|---|---|
| Idiopathic | Primary: cold agglutinin disease (CAD) | Warm and cold |
| Secondary | Secondary: cold agglutinin syndrome (CAS)Paroxysmal cold hemoglobinuria (PCH) | DAT-negative AIHA |
| Characteristic | CA-Associated LPD | LPL | MZL |
|---|---|---|---|
| Histology | Intraparenchymal nodules | Interstitial, nodular, paratrabecular, and intrasinusoidal infiltrates | Intraparenchymal nodules and/or intrasinusoidal infiltrates (splenic) |
| Cytology | Small lymphoid cells, plasma cells | Small lymphocytes, lymphoplasmacytoid cells, and plasma cells | Small lymphocytes with abundant, pale cytoplasm and few admixed plasma cells |
| Cytogenetics | +3, +12, +18 | del(6q), gain(6p), +18 | +3, +12, +18 |
| IGHV gene | IGHV4-34 | IGHV3, IGHV3-23, IGHV3-7 | IGHV1-2 (splenic), IGHV3-4 (nodal) |
| MYD88 L265P | Absent | Present (>90% cases) | Present (10% cases) |
| KMT2D | 67% | Absent | 34% |
| CARD11 | 33% | Absent | 8% |
| CXCR4 | 28% | 40% | Absent |
| Transformation to large B-cell lymphoma | Rare (3.4%) | Yes (5–13%) | Yes (15%) |
| Study | Results |
|---|---|
| Direct antiglobulin test | Positive for C3 (negative or weakly positive for IgG) |
| Titration | ≥64 |
| Specificity | I, i |
| Thermal amplitude | >4 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Climent, F.; Cid, J.; Sureda, A. Cold Agglutinin Disease: A Distinct Clonal B-Cell Lymphoproliferative Disorder of the Bone Marrow. Hemato 2022, 3, 163-173. https://doi.org/10.3390/hemato3010014
Climent F, Cid J, Sureda A. Cold Agglutinin Disease: A Distinct Clonal B-Cell Lymphoproliferative Disorder of the Bone Marrow. Hemato. 2022; 3(1):163-173. https://doi.org/10.3390/hemato3010014
Chicago/Turabian StyleCliment, Fina, Joan Cid, and Anna Sureda. 2022. "Cold Agglutinin Disease: A Distinct Clonal B-Cell Lymphoproliferative Disorder of the Bone Marrow" Hemato 3, no. 1: 163-173. https://doi.org/10.3390/hemato3010014