
Simple One–Pot Synthesis of Hexakis(2-alkoxy-1,5-phenyleneimine) Macrocycles by Precipitation–Driven Cyclization

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Instruments
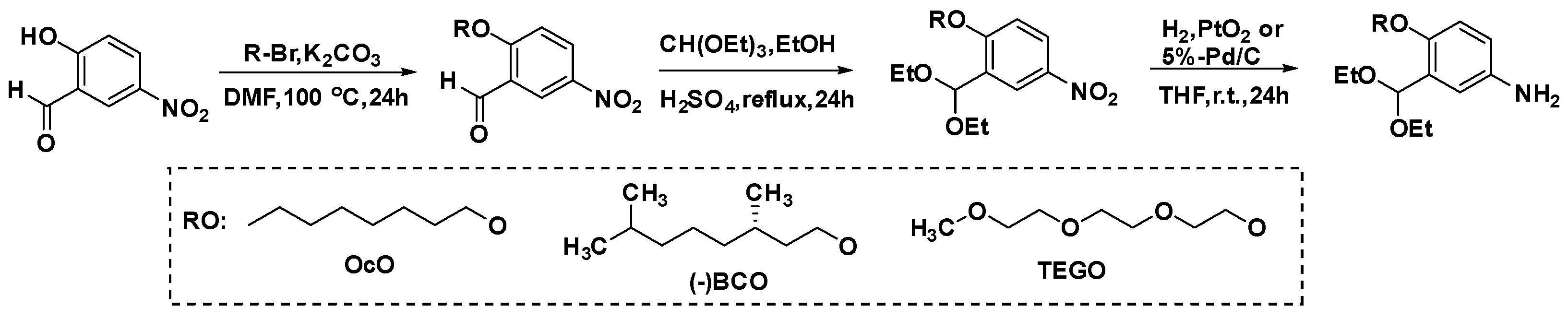
2.2. Preparation of Monomers
2.2.1. 2-Alkoxy-5-nitrobenzaldehydes
2.2.2. 2-Alkoxy-5-nitrobenzaldehyde Diethyl Acetals
2.2.3. 2-Alkoxy-5-aminobenzaldehyde Diethyl Acetals
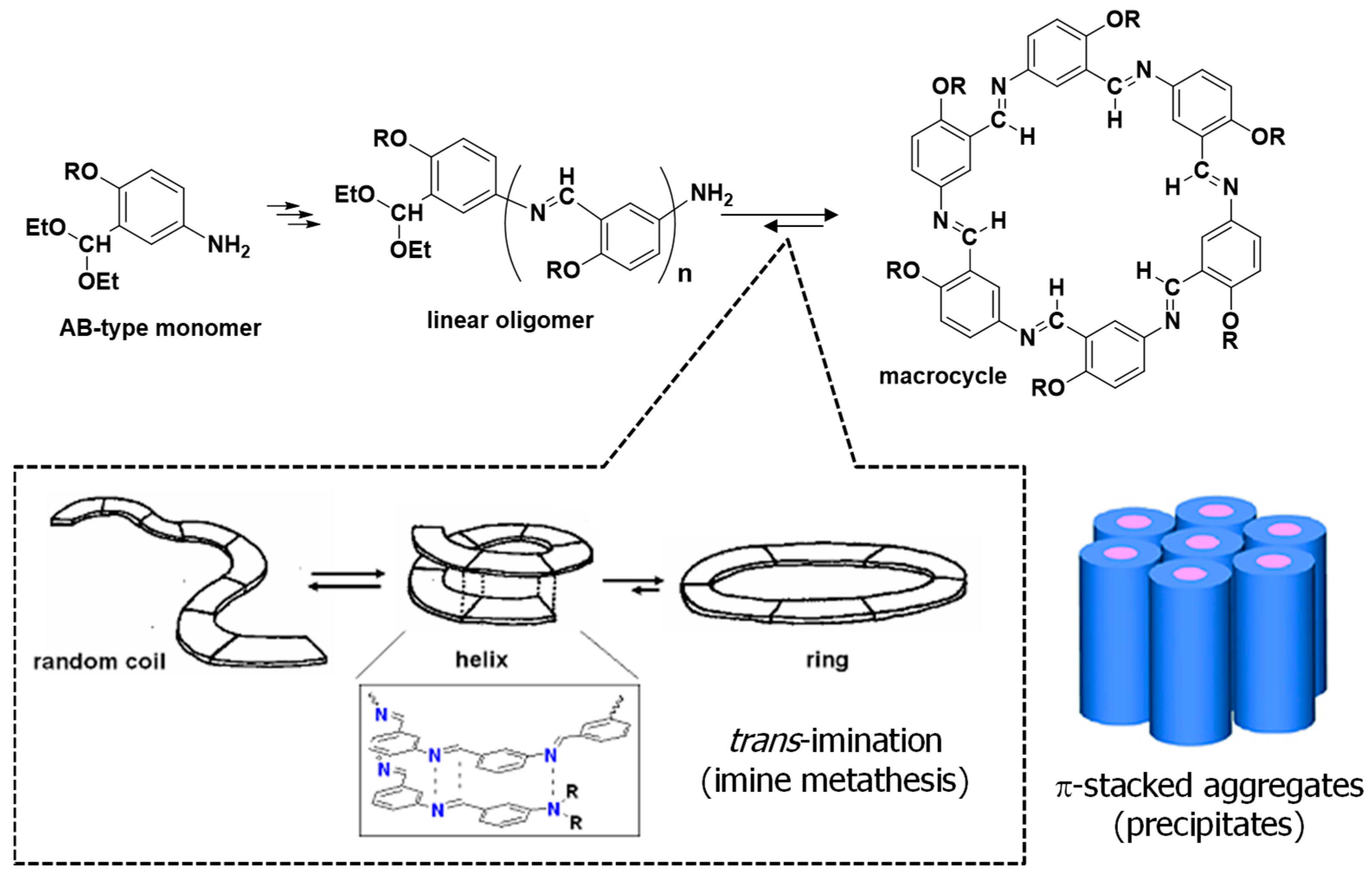
2.3. Synthesis of Hexakis(2-alkoxy-1,5-phenyleneimine) Macrocycles
2.3.1. Hexakis(2-octyloxy-1,5-phenyleneimine) Macrocycle (OcO–Cm6)
2.3.2. Hexakis(2-((S)-(–)-3,7-dimethyloctyloxy)-1,5-phenyleneimine) Macrocycle ((–)BCO–Cm6)
2.3.3. Hexakis(2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy-1,5-phenyleneimine) Macrocycle (TEGO–Cm6)
3. Results
3.1. Preparation of Monomers
3.2. Synthesis of Hexakis(2-alkoxy-1,5-phenyleneimine) Macrocycles
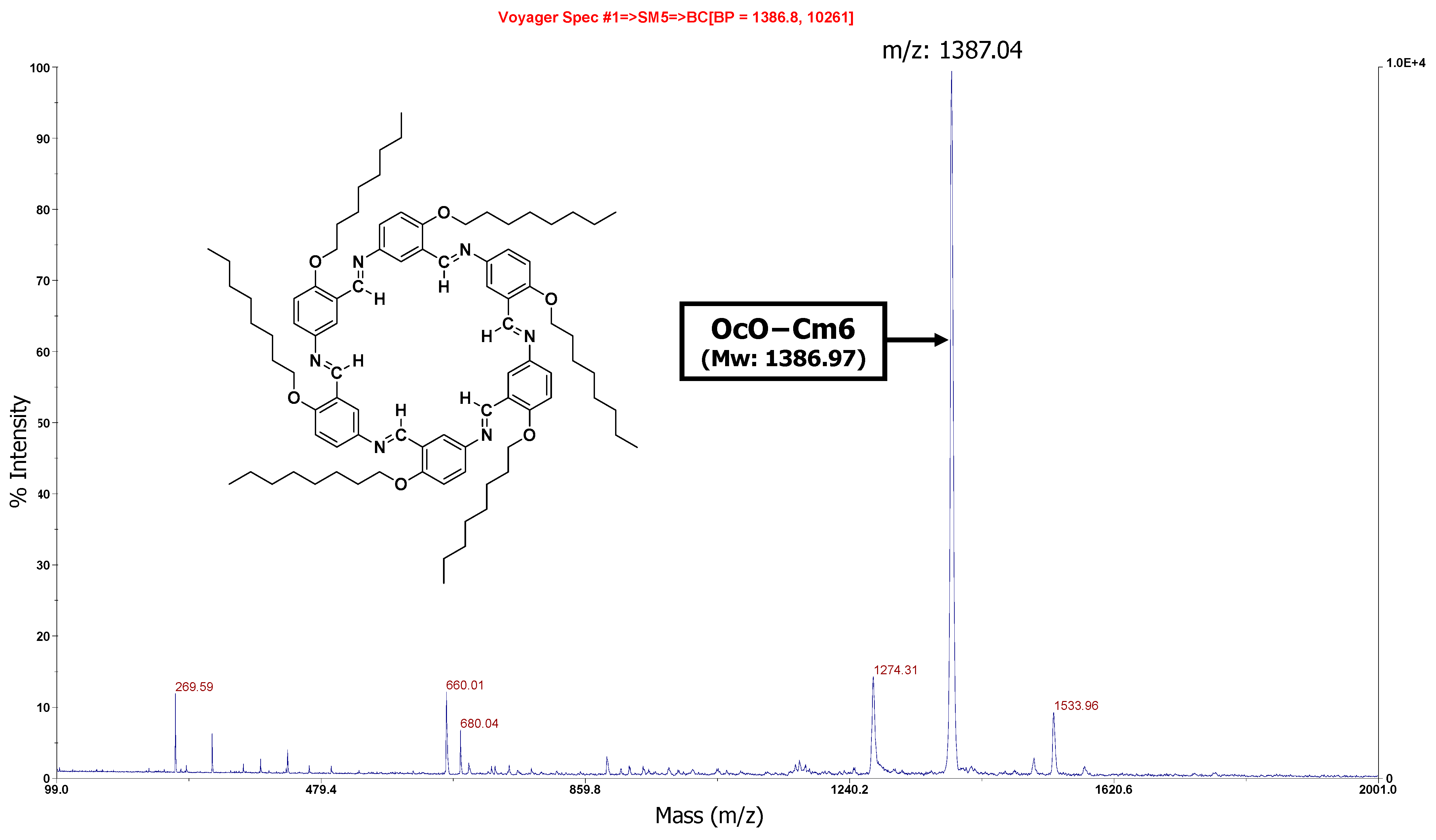
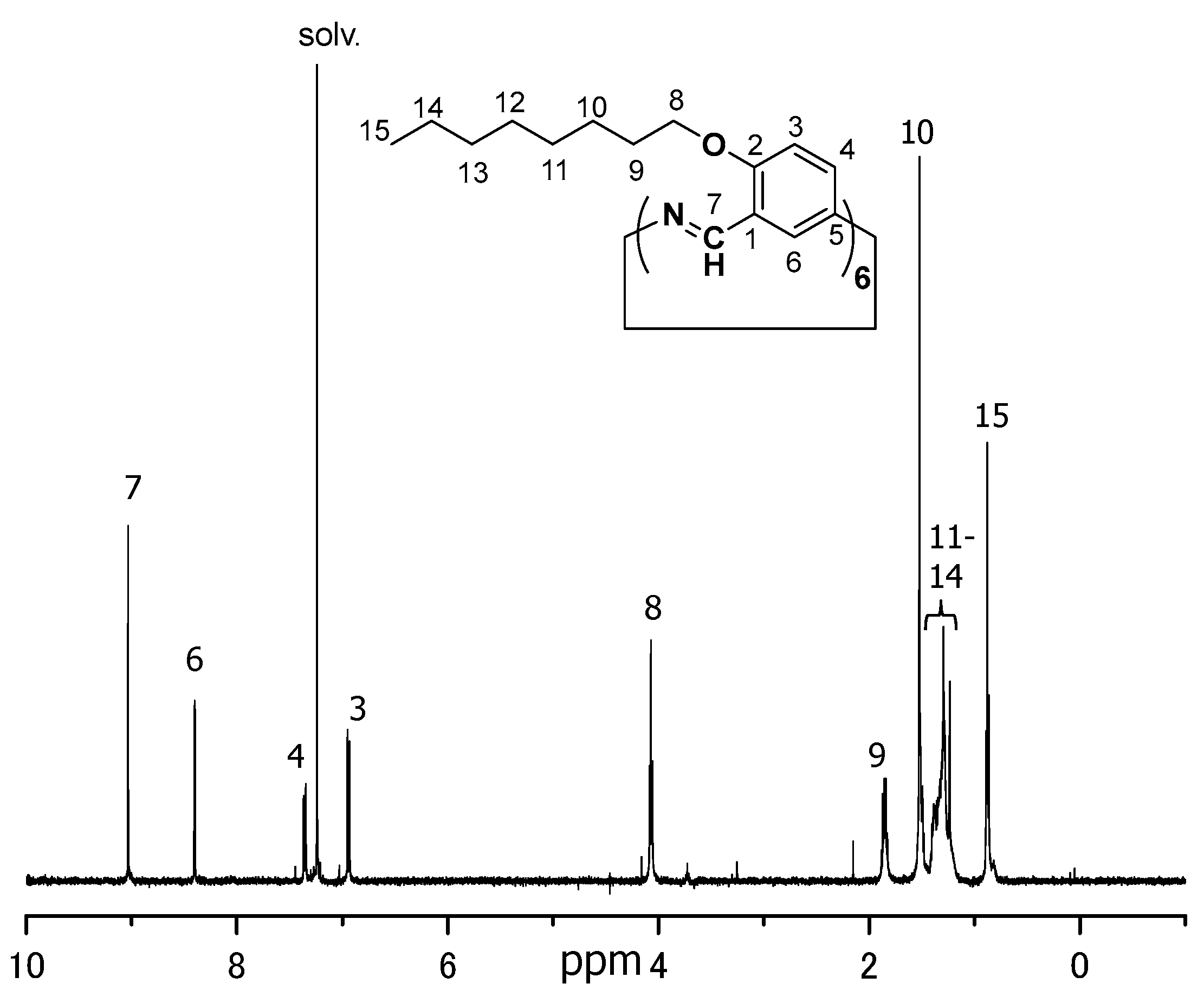
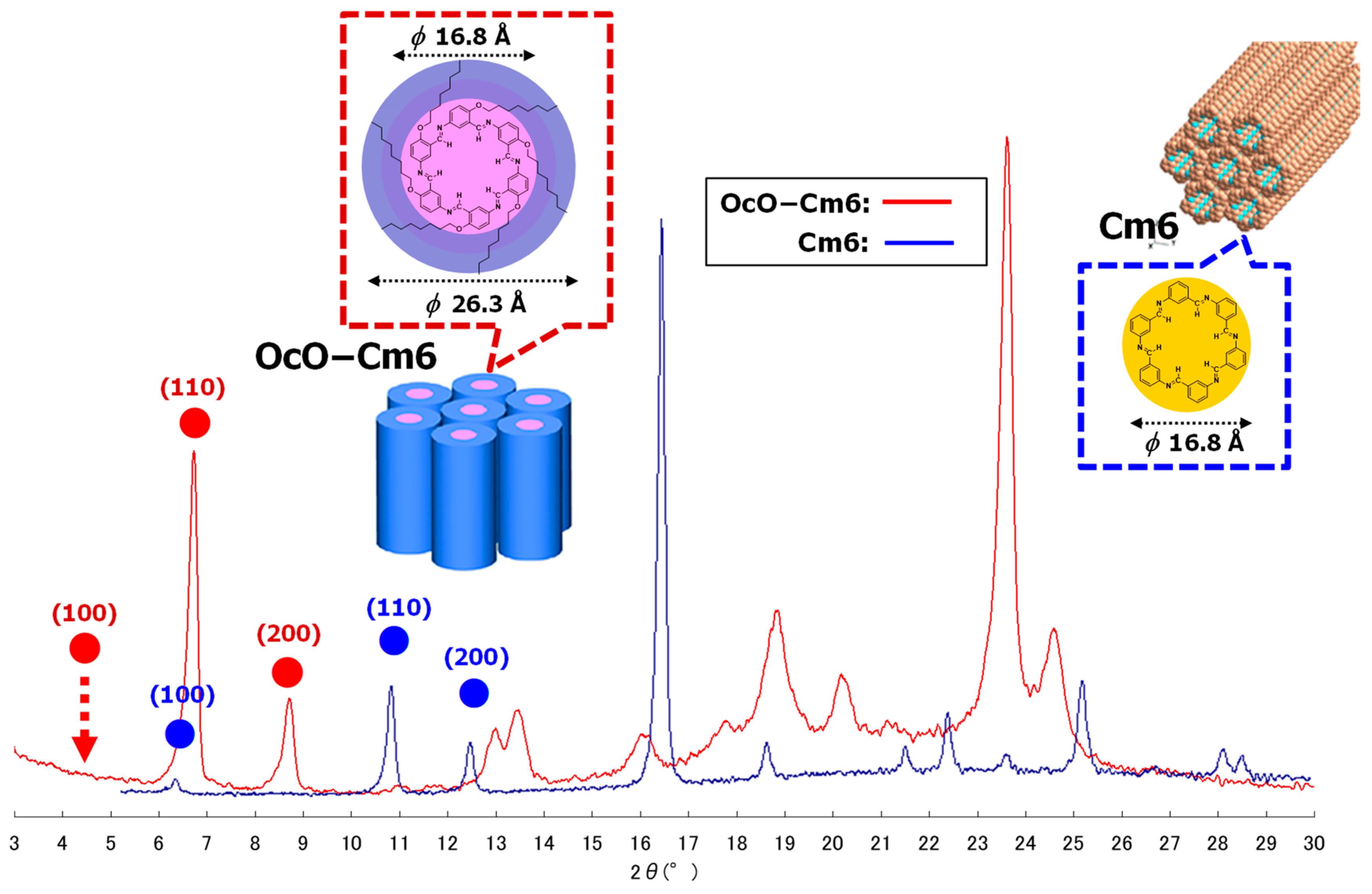
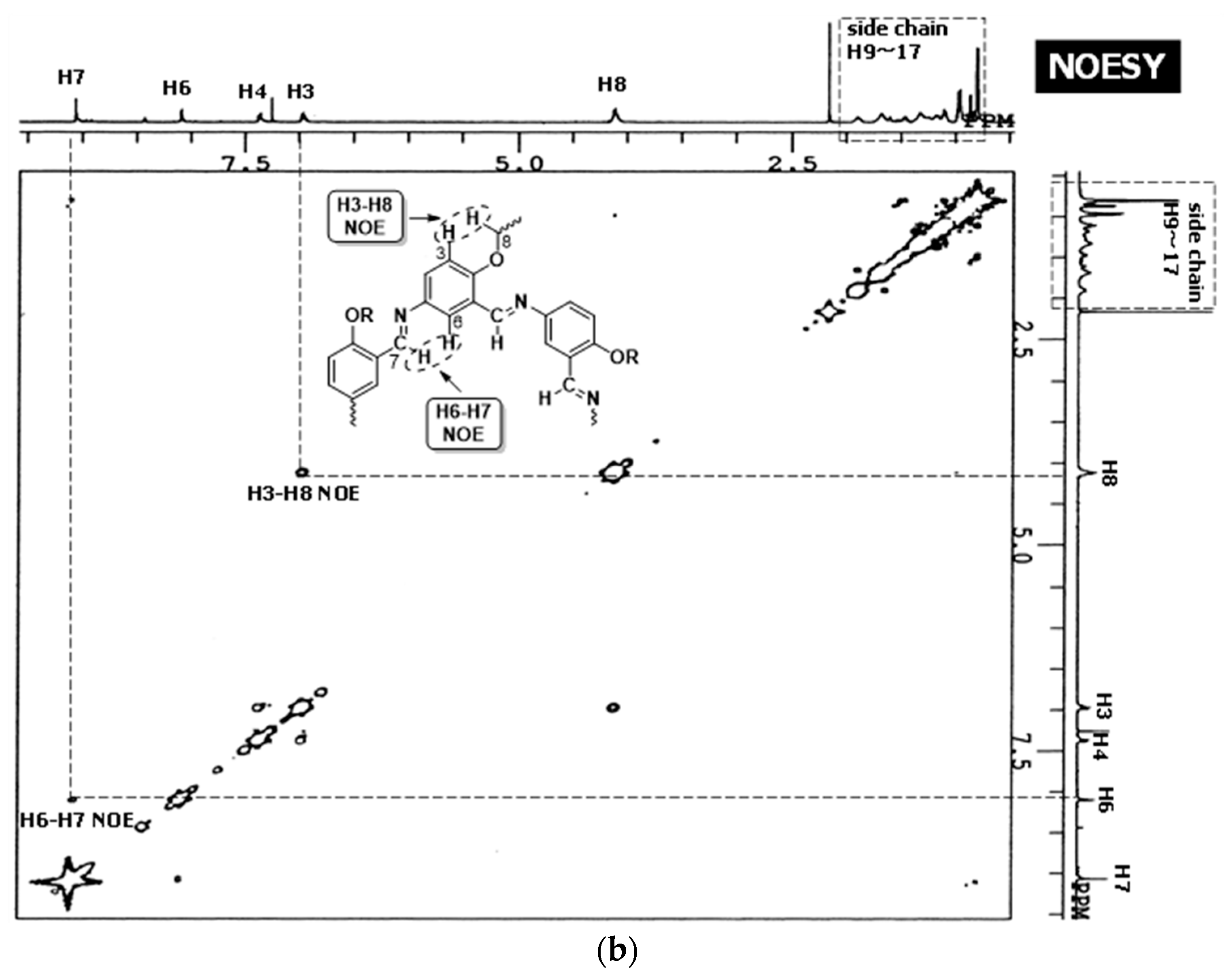
3.2.1. Hexakis(2-octyloxy-1,5-phenyleneimine) Macrocycle (OcO–Cm6)
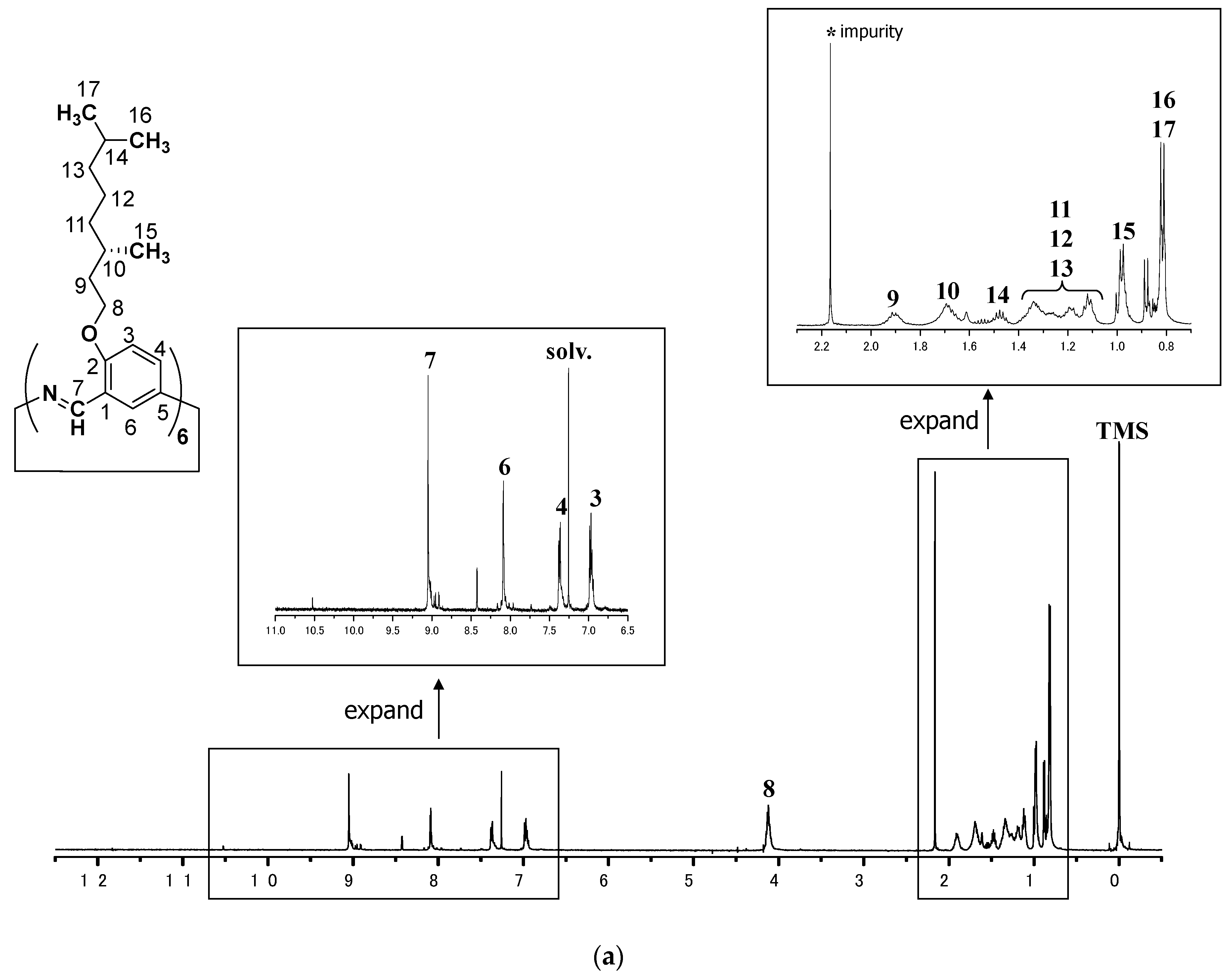
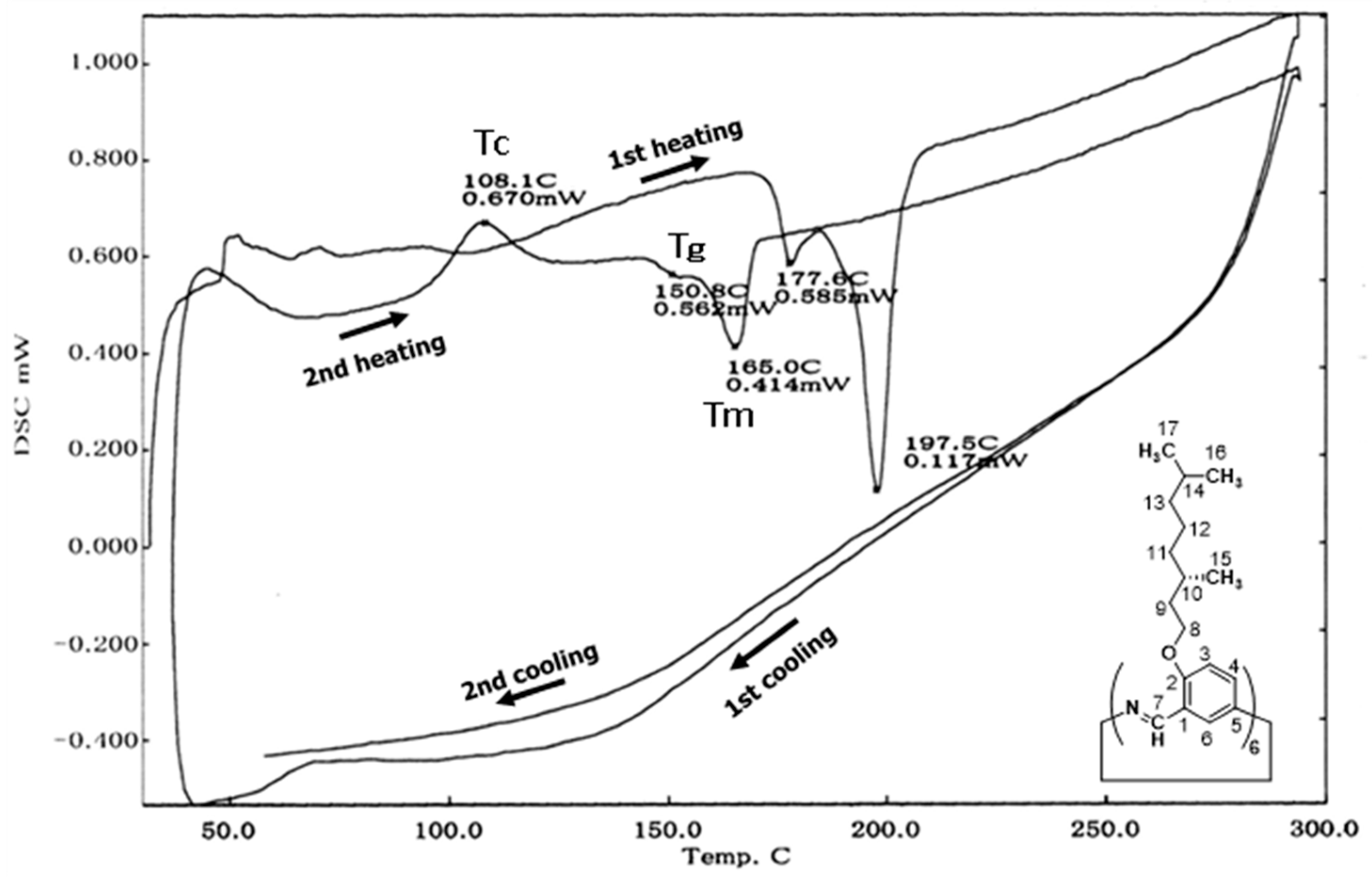
3.2.2. Hexakis(2-((S)-(–)-3,7-dimethyloctyloxy)-1,5-phenyleneimine) Macrocycle ((–)BCO–Cm6)
3.2.3. Hexakis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy-1,5-phenyleneimine) Macrocycle (TEGO–Cm6)
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Moore, J.S. Shape-persistent macrocycles: Structures and synthetic approaches from arylene and ethynylene building blocks. Angew. Chem. Int. Ed. 2006, 45, 4416–4439. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhao, M.; Li, S.; Xu, Y.; Loh, T. Macrolide synthesis through intramolecular oxidative cross-coupling of alkenes. Angew. Chem. 2018, 130, 564–568. [Google Scholar] [CrossRef]
- Kotha, S.; Chavan, S.A.; Shaikh, M. Diversity-oriented approach to macrocyclic cyclophane derivatives by Suzuki–Miyaura cross-coupling and olefin metathesis as key steps. J. Org. Chem. 2012, 77, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Moore, J.S. Arylene ethynylene macrocycles prepared by precipitation-driven alkyne metathesis. J. Am. Chem. Soc. 2004, 126, 12796. [Google Scholar] [CrossRef]
- Kotha, S.; Meshram, M.; Dommaraju, Y. Design and synthesis of polycycles, heterocycles, and macrocycles via strategic utilization of ring-closing metathesis. Chem. Rec. 2018, 18, 1613–1632. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lei, Z.; Jin, Y.; Zhang, W. By-design molecular architectures via alkyne metathesis. Chem. Sci. 2021, 12, 9591–9606. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, A.; Huanga, Y.; Zhang, W. Shape-persistent arylenevinylene macrocycles (AVMs) prepared viaacyclic dienem-etathesis macrocyclization (ADMAC). Chem. Commun. 2010, 46, 8258–8260. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, M.J. Conjugated shape-persistent macrocycles via Schiff-base condensation: New motifs forsupramolecular chem-istry. Pure Appl. Chem. 2006, 78, 873–888. [Google Scholar] [CrossRef]
- Lisowski, J. Imine- and amine-type macrocycles derived from chiral diamines and aromatic dianhydrides. Molecules 2022, 27, 4097. [Google Scholar] [CrossRef]
- He, Z.; Ye, G.; Jiang, W. Imine macrocycle with a deep cavity: Guest-selected formation of syn/anti configuration and guest-controlled reconfiguration. Chem. A Eur. J. 2015, 21, 3005–3012. [Google Scholar] [CrossRef]
- Zhao, D.; Moore, J.S. Synthesis and self-association of an imine-containing m-phenylene ethynylene macrocycle. J. Org. Chem. 2002, 67, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Esteve, F.; Antheaume, C.; Lehn, J.-M. Dynamic covalent self-assembly and self-sorting processes in the formation of imine-based macrocycles and macrobicyclic cages. Chem. Sci. 2023, 14, 6631–6642. [Google Scholar] [CrossRef] [PubMed]
- Chinnaraja, E.; Arunachalam, R.; Pillai, R.S.; Peuronen, A.; Rissanen, K.; Subramanian, P.S. One-pot synthesis of [2+2]-helicate-like macrocycle and 2+4-μ4-oxo tetranuclear open frame complexes: Chiroptical properties and asymmetric oxidative coupling of 2-naphthols. Appl. Organomet. Chem. 2020, 34, e5666. [Google Scholar] [CrossRef]
- Chinnaraja, E.; Arunachalam, R.; Samanta, J.; Natarajan, R.; Subramanian, P.S. Desymmetrization of meso diols using enantiopure zinc (II) dimers: Synthesis and chiroptical properties. Appl. Organomet. Chem. 2019, 33, e4827. [Google Scholar] [CrossRef]
- Janczak, J.; Prochowicz, D.; Lewiński, J.; Fairen-Jimenez, D.; Bereta, T.; Lisowski, J. Trinuclear cage-like Zn(II) macrocyclic com-plexes: Enantiomeric recognition and gas adsorption properties. Chem. Eur. J. 2016, 22, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Sarnicka, A.; Starynowicz, P.; Lisowski, J. Controlling the macrocycle size by the stoichiometry of the applied template ion. Chem. Commun. 2012, 48, 2237–2239. [Google Scholar] [CrossRef]
- Byun, J.C.; Lee, N.H.; Mun, D.H.; Park, K.M. Synthesis and characterization of dinuclear copper(II) complexes, [Cu2([20]-DCHDC) (La)2] (La = N3−, NCS− or S2O32−) with tetraazadiphenol macrocyclic ligand having cyclohexane rings. Inorg. Chem. Commun. 2010, 13, 1156–1159. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Y.-G.; Zhou, Y.; Boxer, L.M.; Woolley, F.R.; Zingaro, R.A. Artificial Zinc(II) complexes regulate cell cycle and apop-tosis-related genes in tumor cell lines. ChemBioChem 2007, 8, 332–340. [Google Scholar] [CrossRef]
- Gao, J.; Woolley, F.R.; Zingaro, R.A. Catalytic asymmetric cyclopropanation at a chiral platform. Org. Biomol. Chem. 2005, 3, 2126–2128. [Google Scholar] [CrossRef]
- Gao, J.; Reibenspies, J.H.; Martell, A.E. Structurally defined catalysts for enantioselective oxidative coupling reactions. Angew. Chem. 2003, 115, 6190–6194. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, Q.; Taynton, P.; Zhang, W. Dynamic covalent chemistry approaches toward macrocycles, molecular cages, and polymers. Acc. Chem. Res. 2014, 47, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Negulescu, I.; Zhang, D. Dynamic covalent polymer networks based on degenerative imine bond exchange: Tuning the malleability and self-healing properties by solvent. Macromolecules 2016, 17, 6277–6284. [Google Scholar] [CrossRef]
- Korich, A.L.; Hughes, T.S. Arylene imine macrocycles of C3h and C3 symmetry from reductive imination of nitroformylarenes. Org. Lett. 2008, 10, 5405–5408. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.S.; Korich, A.L. Efficient synthesis of an ortho-phenylene-para-phenylene-imine macrocycle. In Proceedings of the 230th ACS National Meeting, Washington, DC, USA, 28 August–1 September 2005. Abstracts of Papers. [Google Scholar]
- Moriya, Y.; Yamanaka, M.; Mori, K. Synthesis of C3-symmetric macrocyclic triimines from monomers having Boc−protected amine and formyl group. Chem. Lett. 2022, 51, 217–220. [Google Scholar] [CrossRef]
- Matsumoto, T. Highly efficient one-pot synthesis of hexakis(m-phenyleneimine) macrocyle Cm6 and the thermostimulated self-healing property through dynamic covalent chemistry. Polymers 2023, 15, 3542. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.H.; Jones, T.V.; Seyler, H.; Peters, J.O.; Kim, T.H.; Chang, J.Y.; Tew, G.N. Liquid crystalline order from ortho-phenylene ethynylene macrocycles. J. Am. Chem. Soc. 2006, 128, 9264–9265. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Kato, M.; Soumiya, S.; Nakaya, M.; Onoe, J.; Tanaka, K. Columnar liquid crystals from a giant macrocycle mesogen. Angew. Chem. Int. Ed. 2017, 57, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Granata, G.; Petralia, S.; Forte, G.; Conoci, S.; Consoli, G.M.L. Injectable supramolecular nanohydrogel from a micellar self-assembling calix[4]arene derivative and curcumin for a sustained drug release. Mater. Sci. Eng. C 2020, 111, 110842. [Google Scholar] [CrossRef]
- Coh, C.Y.; Mocerino, M.; Ogden, M.I. Macrocyclic gelators. Supramol. Chem. 2013, 25, 555–566. [Google Scholar] [CrossRef]
- Williamson, A.W. Theory of etherification. J. Chem. Soc. 1852, 4, 106–112. [Google Scholar]
- Zhao, D.; Moore, J.S. Folding-driven reversible polymerization of oligo(m-phenylene ethynylene) imines: Solvent and starter sequence studies. Macromolecules 2003, 36, 2712–2720. [Google Scholar] [CrossRef]
- Jin, W.; Fukushima, T.; Niki, M.; Aida, T. Self-assembled graphitic nanotubes with one-handed helical arrays of a chiral am-phiphilic molecular graphene. Proc. Natl. Acad. Sci. USA 2005, 102, 10801–10806. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2θ (deg) | d Value (Å) | 2θ (deg) | d Value (Å) |
|---|---|---|---|
| 4.35 * | 20.29 | 17.81 | 4.98 |
| 6.73 | 13.12 | 18.84 | 4.71 |
| 8.71 | 10.14 | 20.17 | 4.40 |
| 12.99 | 6.81 | 21.13 | 4.20 |
| 13.47 | 6.57 | 23.62 | 3.76 |
| 16.03 | 5.52 | 24.59 | 3.62 |
| Entry | Concentration (mol/L) | Monomer (g) | THF (mL) | H2O (mL) | Gel Time 2 (day) |
|---|---|---|---|---|---|
| 1 | 0.93 | 1.00 | 2.0 | 1.0 | 20 |
| 2 | 0.46 | 5.01 | 20 | 10 | 20 |
| 3 | 0.23 | 2.51 | 20 | 10 | 24 |
| 4 | 0.09 | 1.00 | 20 | 10 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, T. Simple One–Pot Synthesis of Hexakis(2-alkoxy-1,5-phenyleneimine) Macrocycles by Precipitation–Driven Cyclization. Macromol 2024, 4, 1-22. https://doi.org/10.3390/macromol4010001
Matsumoto T. Simple One–Pot Synthesis of Hexakis(2-alkoxy-1,5-phenyleneimine) Macrocycles by Precipitation–Driven Cyclization. Macromol. 2024; 4(1):1-22. https://doi.org/10.3390/macromol4010001
Chicago/Turabian StyleMatsumoto, Toshihiko. 2024. "Simple One–Pot Synthesis of Hexakis(2-alkoxy-1,5-phenyleneimine) Macrocycles by Precipitation–Driven Cyclization" Macromol 4, no. 1: 1-22. https://doi.org/10.3390/macromol4010001