

Induction and Modulation of EVs by Cigarette Smoke and Their Relevance in Lung Disease: Recent Advances
Abstract
:1. Introduction
2. Lung Diseases Associated with CS-Induced Release of EVs
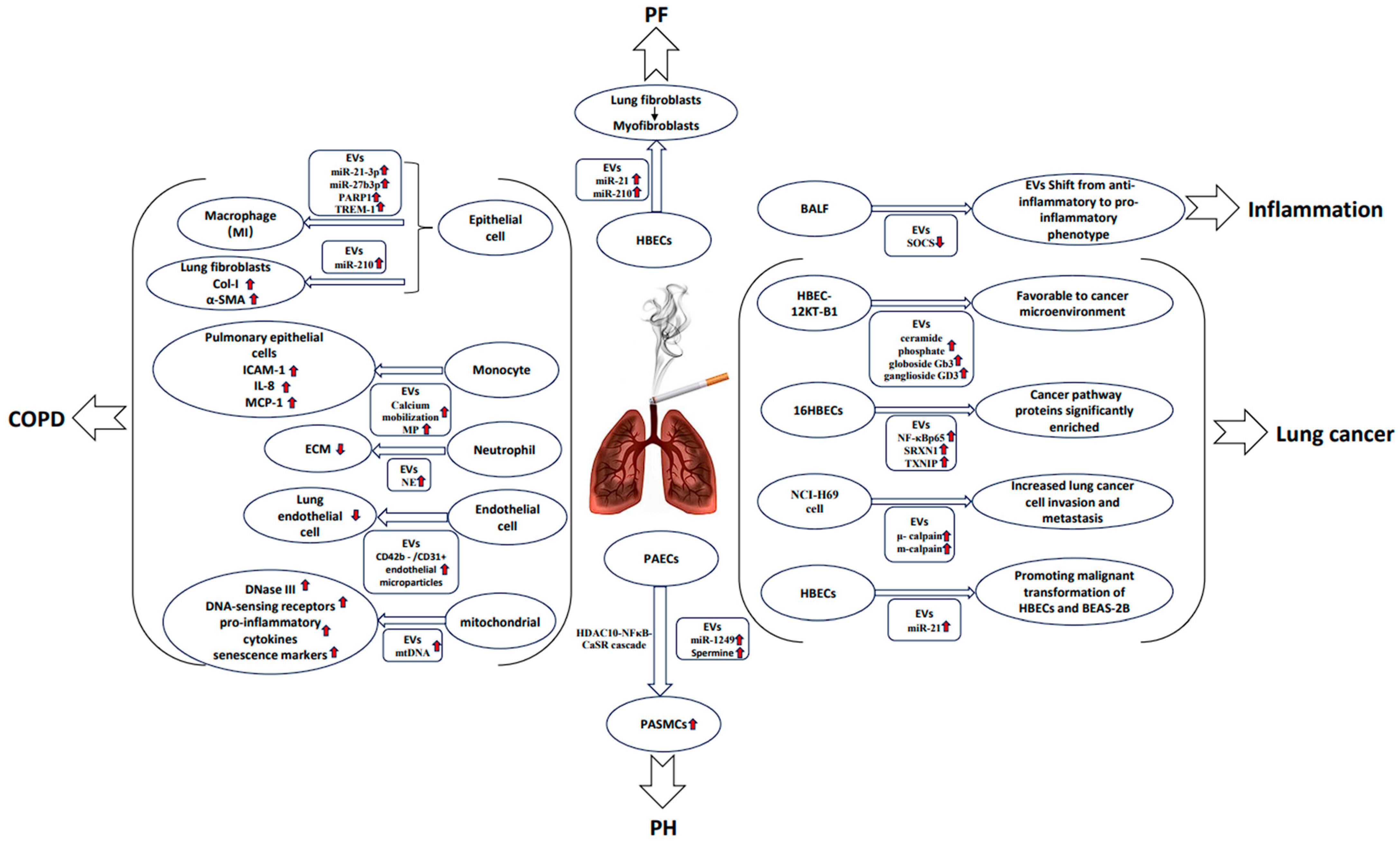
2.1. COPD
2.2. Lung Cancer
2.3. Pulmonary Fibrosis
2.4. Pulmonary Hypertension
3. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mossina, A.; Lukas, C.; Merl-Pham, J.; Uhl, F.E.; Mutze, K.; Schamberger, A.; Staab-Weijnitz, C.; Jia, J.; Yildirim, A.Ö.; Königshoff, M.; et al. Cigarette Smoke Alters the Secretome of Lung Epithelial Cells. Proteomics 2017, 17, 1600243. [Google Scholar] [CrossRef]
- Soleimani, F.; Dobaradaran, S.; De-la-Torre, G.E.; Schmidt, T.C.; Saeedi, R. Content of Toxic Components of Cigarette, Cigarette Smoke vs. Cigarette Butts: A Comprehensive Systematic Review. Sci. Total Environ. 2022, 813, 152667. [Google Scholar] [CrossRef] [PubMed]
- Mackay, J.; Eriksen, M.; Shafey, O. The Tobacco Atlas, 2nd ed.; American Cancer Society: Atlanta, GA, USA, 2006; pp. 1106–1107. [Google Scholar]
- World Health Statistics 2019: Monitoring Health for the SDGs, Sustainable Development Goals. Available online: https://www.who.int/publications-detail-redirect/9789241565707 (accessed on 14 September 2022).
- Singh, S.; Hu, X.; Dixelius, C. Dynamics of Nucleic Acid Mobility. Genetics 2023, 225, iyad132. [Google Scholar] [CrossRef]
- Lacroix, R.; Dignat-George, F. Microparticles: New Protagonists in Pericellular and Intravascular Proteolysis. Semin. Thromb. Hemost. 2013, 39, 033–039. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Extracellular Vesicles Released in Response to Respiratory Exposures: Implications for Chronic Disease. J. Toxicol. Environ. Health B 2018, 21, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and Functional Characterization of Endothelial Microparticles Released by Cigarette Smoke. Sci. Rep. 2016, 6, 31596. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-J.; Liu, Y.; Chen, Y.; Yu, D.; Williams, K.J.; Liu, M.-L. Novel Proteolytic Microvesicles Released from Human Macrophages after Exposure to Tobacco Smoke. Am. J. Pathol. 2013, 182, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Mobarrez, F.; Antoniewicz, L.; Bosson, J.A.; Kuhl, J.; Pisetsky, D.S.; Lundbäck, M. The Effects of Smoking on Levels of Endothelial Progenitor Cells and Microparticles in the Blood of Healthy Volunteers. PLoS ONE 2014, 9, e90314. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, E.; Lee, M.Y. Exosomes as Diagnostic Biomarkers in Cancer. Mol. Cell. Toxicol. 2018, 14, 113–122. [Google Scholar] [CrossRef]
- Fujita, Y.; Kosaka, N.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular Vesicles in Lung Microenvironment and Pathogenesis. Trends Mol. Med. 2015, 21, 533–542. [Google Scholar] [CrossRef]
- Ryu, A.-R.; Kim, D.H.; Kim, E.; Lee, M.Y. The Potential Roles of Extracellular Vesicles in Cigarette Smoke-Associated Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 4692081. [Google Scholar] [CrossRef]
- Celli, B.; Fabbri, L.; Criner, G.; Martinez, F.J.; Mannino, D.; Vogelmeier, C.; Montes De Oca, M.; Papi, A.; Sin, D.D.; Han, M.K.; et al. Definition and Nomenclature of Chronic Obstructive Pulmonary Disease: Time for Its Revision. Am. J. Respir. Crit. Care Med. 2022, 206, 1317–1325. [Google Scholar] [CrossRef]
- Orozco-Levi, M.; Colmenares-Mejia, C.; Ruiz, J.; Valencia-Baron, Y.D.; Ramirez-Sarmiento, A.; Quintero-Lesmes, D.C.; Serrano, N.C. Effect of Antioxidants in the Treatment of COPD Patients: Scoping Review. J. Nutr. Metab. 2021, 2021, 7463391. [Google Scholar] [CrossRef] [PubMed]
- Jehan Peerzada, K. Chronic Obstructive Pulmonary Disease: An Update on Therapeutics and Pathophysiological Understanding. In Chronic Lung Diseases; Springer: Singapore, 2020; pp. 157–180. [Google Scholar] [CrossRef]
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated Role of Cigarette Smoking, Oxidative Stress, and Immune Response in COPD and Corresponding Treatments. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, E.; Zinellu, A.; Fois, A.G.; Pau, M.C.; Scano, V.; Piras, B.; Carru, C.; Pirina, P. Oxidative Stress Biomarkers in Chronic Obstructive Pulmonary Disease Exacerbations: A Systematic Review. Antioxidants 2021, 10, 710. [Google Scholar] [CrossRef] [PubMed]
- Benedikter, B.J.; Volgers, C.; Van Eijck, P.H.; Wouters, E.F.M.; Savelkoul, P.H.M.; Reynaert, N.L.; Haenen, G.R.M.M.; Rohde, G.G.U.; Weseler, A.R.; Stassen, F.R.M. Cigarette Smoke Extract Induced Exosome Release Is Mediated by Depletion of Exofacial Thiols and Can Be Inhibited by Thiol-Antioxidants. Free Radic. Biol. Med. 2017, 108, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Araya, J.; Ito, S.; Kobayashi, K.; Kosaka, N.; Yoshioka, Y.; Kadota, T.; Hara, H.; Kuwano, K.; Ochiya, T. Suppression of Autophagy by Extracellular Vesicles Promotes Myofibroblast Differentiation in COPD Pathogenesis. J. Extracell. Vesicles 2015, 4, 28388. [Google Scholar] [CrossRef]
- Puttur, F.; Gregory, L.G.; Lloyd, C.M. Airway Macrophages as the Guardians of Tissue Repair in the Lung. Immunol. Cell Biol. 2019, 97, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef] [PubMed]
- Finicelli, M.; Digilio, F.A.; Galderisi, U.; Peluso, G. The Emerging Role of Macrophages in Chronic Obstructive Pulmonary Disease: The Potential Impact of Oxidative Stress and Extracellular Vesicle on Macrophage Polarization and Function. Antioxidants 2022, 11, 464. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.E.; Finney-Hayward, T.K.; Quint, J.K.; Thomas, C.M.R.; Tudhope, S.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E. Defective Macrophage Phagocytosis of Bacteria in COPD. Eur. Respir. J. 2010, 35, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking Alters Alveolar Macrophage Recognition and Phagocytic Ability. Am. J. Respir. Cell Mol. Biol. 2007, 37, 748–755. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, H.; Shi, R.; Fan, W.; Zhang, J.; Su, W.; Wang, Y.; Li, P. MiRNAomics Analysis Reveals the Promoting Effects of Cigarette Smoke Extract-Treated Beas-2B-Derived Exosomes on Macrophage Polarization. Biochem. Biophys. Res. Commun. 2021, 572, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, H.; Fan, W.; Zhang, J.; Yao, Y.; Su, W.; Wang, Y.; Li, P. Naringenin Suppresses BEAS-2B-Derived Extracellular Vesicular Cargoes Disorder Caused by Cigarette Smoke Extract Thereby Inhibiting M1 Macrophage Polarization. Front. Immunol. 2022, 13, 930476. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Q.; Yu, Q.; Xiao, J.; Zhao, H. Cigarette Smoke Extract-Treated Airway Epithelial Cells-Derived Exosomes Promote M1 Macrophage Polarization in Chronic Obstructive Pulmonary Disease. Int. Immunopharmacol. 2021, 96, 107700. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, W.; Wang, Z.; Chen, J.; Ding, M.; Han, L. TREM-1 Deficiency Attenuates the Inflammatory Responses in LPS-Induced Murine Endometritis. Microb. Biotechnol. 2019, 12, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; He, X.; Bian, Y.; Guo, Q.; Zheng, K.; Zhao, Y.; Lu, C.; Liu, B.; Xu, X.; Zhang, G.; et al. Triptolide Modulates TREM-1 Signal Pathway to Inhibit the Inflammatory Response in Rheumatoid Arthritis. Int. J. Mol. Sci. 2016, 17, 498. [Google Scholar] [CrossRef]
- Tan, C.; Gurien, S.D.; Royster, W.; Aziz, M.; Wang, P. Extracellular CIRP Induces Inflammation in Alveolar Type II Cells via TREM-1. Front. Cell Dev. Biol. 2020, 8, 579157. [Google Scholar] [CrossRef]
- Xia, H.; Wu, Y.; Zhao, J.; Li, W.; Lu, L.; Ma, H.; Cheng, C.; Sun, J.; Xiang, Q.; Bian, T.; et al. The Aberrant Cross-Talk of Epithelium-Macrophages via METTL3-Regulated Extracellular Vesicle MiR-93 in Smoking-Induced Emphysema. Cell Biol. Toxicol. 2022, 38, 167–183. [Google Scholar] [CrossRef]
- Miao, Y.; Wu, J.; Wu, R.; Wang, E.; Wang, J. Circ_0040929 Serves as Promising Biomarker and Potential Target for Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2022, 17, 2079–2092. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, N.; Oshins, R.; Mehrad, B.; Lascano, J.E.; Qiang, X.; West, J.R.; Holliday, L.S.; Lee, J.; Wiesemann, G.; Eydgahi, S.; et al. Cigarette Smoke Exposed Airway Epithelial Cell-Derived EVs Promote Pro-Inflammatory Macrophage Activation in Alpha-1 Antitrypsin Deficiency. Respir. Res. 2022, 23, 232. [Google Scholar] [CrossRef] [PubMed]
- Cordazzo, C.; Petrini, S.; Neri, T.; Lombardi, S.; Carmazzi, Y.; Pedrinelli, R.; Paggiaro, P.; Celi, A. Rapid Shedding of Proinflammatory Microparticles by Human Mononuclear Cells Exposed to Cigarette Smoke Is Dependent on Ca2+ Mobilization. Inflamm. Res. 2014, 63, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Abdul Roda, M.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell 2019, 176, 113–126.e15. [Google Scholar] [CrossRef]
- Liu, H.; Ding, L.; Zhang, Y.; Ni, S. Circulating Endothelial Microparticles Involved in Lung Function Decline in a Rat Exposed in Cigarette Smoke Maybe from Apoptotic Pulmonary Capillary Endothelial Cells. J. Thorac. Dis. 2014, 6, 649–655. [Google Scholar] [CrossRef]
- Saxena, A.; Walters, M.S.; Shieh, J.-H.; Shen, L.-B.; Gomi, K.; Downey, R.J.; Crystal, R.G.; Moore, M.A.S. Extracellular Vesicles from Human Airway Basal Cells Respond to Cigarette Smoke Extract and Affect Vascular Endothelial Cells. Sci. Rep. 2021, 11, 6104. [Google Scholar] [CrossRef] [PubMed]
- Curradi, G.; Walters, M.S.; Ding, B.-S.; Rafii, S.; Hackett, N.R.; Crystal, R.G. Airway Basal Cell Vascular Endothelial Growth Factor-Mediated Cross-Talk Regulates Endothelial Cell-Dependent Growth Support of Human Airway Basal Cells. Cell. Mol. Life Sci. 2012, 69, 2217–2231. [Google Scholar] [CrossRef]
- Xu, H.; Ling, M.; Xue, J.; Dai, X.; Sun, Q.; Chen, C.; Liu, Y.; Zhou, L.; Liu, J.; Luo, F.; et al. Exosomal MicroRNA-21 Derived from Bronchial Epithelial Cells Is Involved in Aberrant Epithelium-Fibroblast Cross-Talk in COPD Induced by Cigarette Smoking. Theranostics 2018, 8, 5419–5433. [Google Scholar] [CrossRef]
- Giordano, L.; Farnham, A.; Dhandapani, P.K.; Salminen, L.; Bhaskaran, J.; Voswinckel, R.; Rauschkolb, P.; Scheibe, S.; Sommer, N.; Beisswenger, C.; et al. Alternative Oxidase Attenuates Cigarette Smoke–induced Lung Dysfunction and Tissue Damage. Am. J. Respir. Cell Mol. Biol. 2019, 60, 515–522. [Google Scholar] [CrossRef]
- Giordano, L.; Gregory, A.D.; Pérez Verdaguer, M.; Ware, S.A.; Harvey, H.; DeVallance, E.; Brzoska, T.; Sundd, P.; Zhang, Y.; Sciurba, F.C.; et al. Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD. Cells 2022, 11, 369. [Google Scholar] [CrossRef]
- Global Initiative for Chronic Obstructive Lung Disease Incorporated. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. 2018. Available online: https://goldcopd.org/ (accessed on 2 September 2023).
- Feller, D.; Kun, J.; Ruzsics, I.; Rapp, J.; Sarosi, V.; Kvell, K.; Helyes, Z.; Pongracz, J.E. Cigarette Smoke-Induced Pulmonary Inflammation Becomes Systemic by Circulating Extracellular Vesicles Containing Wnt5a and Inflammatory Cytokines. Front. Immunol. 2018, 9, 1724. [Google Scholar] [CrossRef]
- Chiaradia, E.; Sansone, A.; Ferreri, C.; Tancini, B.; Latella, R.; Tognoloni, A.; Gambelunghe, A.; dell’Omo, M.; Urbanelli, L.; Giovagnoli, S.; et al. Phospholipid Fatty Acid Remodeling and Carbonylated Protein Increase in Extracellular Vesicles Released by Airway Epithelial Cells Exposed to Cigarette Smoke Extract. Eur. J. Cell Biol. 2023, 102, 151285. [Google Scholar] [CrossRef]
- Singh, K.P.; Maremanda, K.P.; Li, D.; Rahman, I. Exosomal MicroRNAs Are Novel Circulating Biomarkers in Cigarette, Waterpipe Smokers, E-Cigarette Users and Dual Smokers. BMC Med. Genom. 2020, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, C.; Wang, W.; Wu, D.; Wang, W.; Ye, X.; Yang, Q. Analysis of Serum Exosome MicroRNAs in the Rat Model of Chronic Obstructive Pulmonary Disease. Am. J. Transl. Res. 2023, 15, 138–150. [Google Scholar] [PubMed]
- Sundar, I.K.; Li, D.; Rahman, I. Small RNA-Sequence Analysis of Plasma-Derived Extracellular Vesicle MiRNAs in Smokers and Patients with Chronic Obstructive Pulmonary Disease as Circulating Biomarkers. J. Extracell. Vesicles 2019, 8, 1684816. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Peto, R. Mortality in Relation to Smoking: 20 Years’ Observations on Male British Doctors. Br. Med. J. 1976, 2, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Bourdonnay, E.; Zasłona, Z.; Penke, L.R.K.; Speth, J.M.; Schneider, D.J.; Przybranowski, S.; Swanson, J.A.; Mancuso, P.; Freeman, C.M.; Curtis, J.L.; et al. Transcellular Delivery of Vesicular SOCS Proteins from Macrophages to Epithelial Cells Blunts Inflammatory Signaling. J. Exp. Med. 2015, 212, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Baarsma, H.A.; Skronska-Wasek, W.; Mutze, K.; Ciolek, F.; Wagner, D.E.; John-Schuster, G.; Heinzelmann, K.; Günther, A.; Bracke, K.R.; Dagouassat, M.; et al. Noncanonical WNT-5A Signaling Impairs Endogenous Lung Repair in COPD. J. Exp. Med. 2017, 214, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Malyla, V.; Paudel, K.R.; De Rubis, G.; Hansbro, N.G.; Hansbro, P.M.; Dua, K. Cigarette Smoking Induces Lung Cancer Tumorigenesis via Upregulation of the WNT/β-Catenin Signaling Pathway. Life Sci. 2023, 326, 121787. [Google Scholar] [CrossRef] [PubMed]
- Machala, M.; Slavik, J.; Kovac, O.; Prochazkova, J.; Pencikova, K.; Parenicova, M.; Strakova, N.; Kotoucek, J.; Kulich, P.; Mollerup, S.; et al. Changes in Sphingolipid Profile of Benzo[a]Pyrene-Transformed Human Bronchial Epithelial Cells Are Reflected in the Altered Composition of Sphingolipids in Their Exosomes. Int. J. Mol. Sci. 2021, 22, 9195. [Google Scholar] [CrossRef]
- Wang, W.; Zeng, R.; Liu, M.; Chen, M.; Wei, S.; Li, B.; Yu, S. Exosome Proteomics Study of the Effects of Traditional Cigarettes and Electronic Cigarettes on Human Bronchial Epithelial Cells. Toxicol. In Vitro 2023, 86, 105516. [Google Scholar] [CrossRef]
- Schuller, H.M. Mechanisms of Smoking-Related Lung and Pancreatic Adenocarcinoma Development. Nat. Rev. Cancer 2002, 2, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Glading, A.; Überall, F.; Keyse, S.M.; Lauffenburger, D.A.; Wells, A. Membrane Proximal ERK Signaling Is Required for M-Calpain Activation Downstream of Epidermal Growth Factor Receptor Signaling. J. Biol. Chem. 2001, 276, 23341–23348. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.J.; Deng, X.M. Tobacco-Specific Nitrosamine 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone Induces Phosphorylation of μ- and m-Calpain in Association with Increased Secretion, Cell Migration, and Invasion. J. Biol. Chem. 2004, 279, 53683–53690. [Google Scholar] [CrossRef] [PubMed]
- Momi, N.; Kaur, S.; Rachagani, S.; Ganti, A.K.; Batra, S.K. Smoking and MicroRNA Dysregulation: A Cancerous Combination. Trends Mol. Med. 2014, 20, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Shi, J. Considering Exosomal MiR-21 as a Biomarker for Cancer. J. Clin. Med. 2016, 5, 42. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G. MicroRNA-21 (MiR-21) Represses Tumor Suppressor PTEN and Promotes Growth and Invasion in Non-Small Cell Lung Cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, F.; Wang, B.; Li, H.; Xu, Y.; Liu, X.; Shi, L.; Lu, X.; Xu, W.; Lu, L.; et al. STAT3-Regulated Exosomal MiR-21 Promotes Angiogenesis and Is Involved in Neoplastic Processes of Transformed Human Bronchial Epithelial Cells. Cancer Lett. 2016, 370, 125–135. [Google Scholar] [CrossRef]
- Héliot, A.; Landkocz, Y.; Roy Saint-Georges, F.; Gosset, P.; Billet, S.; Shirali, P.; Courcot, D.; Martin, P.J. Smoker Extracellular Vesicles Influence Status of Human Bronchial Epithelial Cells. Int. J. Hyg. Environ. Health 2017, 220, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Deng, J.; Han, Z.; Cui, Y.; He, R.; Gu, Y.; Zhang, Q. CircRNA_0026344 via Exosomal MiR-21 Regulation of Smad7 Is Involved in Aberrant Cross-Talk of Epithelium-Fibroblasts during Cigarette Smoke-Induced Pulmonary Fibrosis. Toxicol. Lett. 2021, 347, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Zhang, Y.; Wan, R.; Jiang, M.; Xu, Y.; Zhang, Q. MiR-21 Mediates Nickel Nanoparticle-Induced Pulmonary Injury and Fibrosis. Nanotoxicology 2020, 14, 1175–1197. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.; Peinado, V.I.; Ramirez, J.; Melgosa, T.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Characterization of Pulmonary Vascular Remodelling in Smokers and Patients with Mild COPD. Eur. Respir. J. 2002, 19, 632–638. [Google Scholar] [CrossRef]
- Albert Barbera, J. Mechanisms of Development of Chronic Obstructive Pulmonary Disease-Associated Pulmonary Hypertension. Pulm. Circ. 2013, 3, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Tan, R.; Sun, M.; Yuan, L.; Ruiz, M.; Dupuis, J.; Hu, Q.; Zhu, L. MiR-1249 on Endothelial Extracellular Vesicles Mediates Cigarette Smoke–Induced Pulmonary Hypertension by Inhibiting HDAC10 (Histone Deacetylase 10)-NFκB (Nuclear Factor ΚB)-CaSR (Calcium-Sensing Receptor) Cascade. Hypertension 2022, 79, 2721–2732. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xiao, R.; Zhang, X.; Lang, Y.; Liu, F.; Yu, Z.; Zhang, J.; Su, Y.; Lu, Y.; Wang, T.; et al. Spermine on Endothelial Extracellular Vesicles Mediates Smoking-Induced Pulmonary Hypertension Partially through Calcium-Sensing Receptor. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.X.; Geibel, J.P.; Hebert, S.C. Extracellular Polyamines Regulate Fluid Secretion in Rat Colonic Crypts via the Extracellular Calcium-Sensing Receptor. Gastroenterology 2004, 126, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Nolte, A.; Knoepp, F.; Giordano, L.; Pecina, P.; Hüttemann, M.; Grossman, L.I.; Weissmann, N.; Sommer, N. Mitochondrial Oxygen Sensing of Acute Hypoxia in Specialized Cells-Is There a Unifying Mechanism? Biochim. Biophys. Acta (BBA)-Bioenerg. 2022, 1863, 148911. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Disease/Disorder | Sample/Subjects | Effect of Cigarette Smoke | Reference |
|---|---|---|---|
| COPD | Primary human lung microvascular endothelial cells | CS exposure significantly upregulated miRNAs let-7d, -126, -125-5p, and -22 in human cell EMPs and mouse circulating granules. | [8] Serban et al. |
| COPD | Human bronchial lung epithelial cells (BEAS-2B) | CSE augmented the release of the EV subtype exosomes, which could be prevented by scavenging thiolreactive components using NAC or GSH. | [20] Benedikter et al. |
| COPD | Primary human bronchial epithelial cells (HBECs) and lung fibroblasts (LFs) | CS upregulates the HBECS-derived exosome miR-210 to promote myofibroblast differentiation in LF. | [21] Fujita et al. |
| COPD | BEAS-2B | CS increased the levels of miR-21-3p, miR-27b-3p, and PARP1 protein in BEAS-2B-derived EVs, thus promoting M1-type polarization of macrophages. | [27] Chen, Z et al. |
| COPD | Mouse airway epithelial cells and mouse macrophage cell line | CSE-induced MAECS-derived EVs can cause lung injury in mice by upregulating TREM-1 expression and promoting M1 macrophage polarization. | [29] Wang et al. |
| COPD | Macrophages and bronchial epithelial cells | CS induces METTL3-mediated transfer of mature miR-93 from bronchial epithelial cells to macrophages via EVs. | [33] Xia et al. |
| COPD | HBECs, serum from smokers and nonsmokers | The expression of serum exosome circ_0040929 in smokers is upregulated, targeting miR-515-5p and thereby increasing the expression of IGFBP3. | [34] Miao et al. |
| COPD | AATD macrophages and bronchial epithelial cells | CS-induced EVs induced granulocyte-macrophage colony-stimulating factor and IL-8 expression in AATD macrophages. | [35] Khodayari et al. |
| COPD | Human mononuclear cells and lung epithelial cells | CSE-induced MP in human mononuclear cells upregulates pro-inflammatory mediators ICAM-1, IL-8, and MCP-1 in lung epithelial cells. | [37] Genschmer et al. |
| COPD | Wistar rats | CS induced a significant increase in plasma CD42b-/CD31 + EMPs level, resulting in endothelial cell apoptosis and endocortical stress injury. | [38] Liu et al. |
| COPD | Immortalized human airway basal cell line and endothelial cell | CSE-treated BCi-NS1.1-derived EVs promote endothelial cell survival and may in turn promote airway remodeling. | [39] Saxena et al. |
| COPD | Untreated or CSE-treated HBECs and bronchial fibroblast cells | CSE treatment increased the level of HBECS-derived exosome miR-21, which promoted the differentiation of airway fibroblasts into myofibroblasts. | [41] Xu et al. |
| COPD | BEAS-2B | Exposure to CSE decreases mitochondrial membrane potential, increases oxidative stress, dysregulates mitochondrial dynamics, and triggers the release of mitochondrial DNA encapsulated in EVs. | [42] Giordano et al. |
| COPD | Mouse and human model systems | CS-induced upregulation of WNT-5A in serum EVs from COPD affecting M1/M2 macrophage polarization. | [45] Feller et al. |
| COPD | BEAS-2B | CSE treatment increases oxidized protein content and saturated fatty acid/monounsaturated fatty acid ratio. | [46] Chiaradia et al. |
| COPD | Plasma EVs from smoker and nonsmoker | Seven differentially expressed microRNAs in plasma EVs when comparing the smoking and nonsmoking groups. | [47] Singh et al. |
| COPD | Lung tissue of COPD rats induced by CSE | The expression of miR-5-182p and miR-5-185p was significantly downregulated in lung tissue of COPD rats induced by CSE compared with control rats. | [48] Ouyang et al. |
| COPD | BEAS-2B cells and human monocytes (U937) | Eight miRNAs expressed in the CSE-derived exosomes of BEAS-2B cells were significantly increased; a trend of reduced expression of most exosomal miRNAs from CSE-treated U937 cells was found. | [49] Sundar et al. |
| COPD | Human Broncho Alveolar Lavages | CS can alter lung EVs profile that can influence surrounding bronchial epithelial cells. | [64] Héliot et al. |
| Lung cancer | HBECs | CS treatment can induce tumorigenesis of healthy cells by upregulating WNT/β-catenin signaling in vitro and human lung cancer patients. | [54] Malyla et al. |
| Lung cancer | HBECs | CSE induces significant enrichment of DEEPs produced by HBEC in the cancer pathway. | [56] Wang et al. |
| Lung cancer | Human lung cancer cells | NNK significantly enhanced the expression levels of μ- and m-calpain in NCI-H69-derived EVs. | [58] Glading et al. |
| Lung cancer | HBEC-12KT cells and HBEC-12KT-B1 cells | Levels of ceramide phosphate, bulboside Gb3, and ganglioside GD3 were increased in exosomes derived from BaP-transformed HBEC-12KT-B1 cells. | [59] Xu et al. |
| Lung cancer | HBECs | CS promoted miR-21 levels in normal HBECs and angiogenesis of human umbilical vein endothelial cells. | [63] Liu et al. |
| PF | HBECs | CS upregulates miR-21 in HBECS-derived EVs, activates TGF-β1/Smads pathway, and induces fibroblast differentiation, leading to the formation of excessive ECM. | [65] Bai et al. |
| PH | eEVs, PAECs and PASMCs | The exosome miR-1249 produced by CSE-treated PAECs promotes the hyperproliferative and anti-apoptotic state of PASMC, thus promoting the development of PH. | [69] Su et al. |
| PH | Smooth muscle cells and eEVs from cigarette-smoking human | CS exposure increased spermine positive eEVs production and spermine content. | [70] Zhu et al. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, M.; Zou, M.; Yao, Y.; Wu, H.; Su, W.; Wang, Y.; Li, P. Induction and Modulation of EVs by Cigarette Smoke and Their Relevance in Lung Disease: Recent Advances. J. Respir. 2023, 3, 164-177. https://doi.org/10.3390/jor3040016
Zhong M, Zou M, Yao Y, Wu H, Su W, Wang Y, Li P. Induction and Modulation of EVs by Cigarette Smoke and Their Relevance in Lung Disease: Recent Advances. Journal of Respiration. 2023; 3(4):164-177. https://doi.org/10.3390/jor3040016
Chicago/Turabian StyleZhong, Mengli, Muhan Zou, Yue Yao, Hao Wu, Weiwei Su, Yonggang Wang, and Peibo Li. 2023. "Induction and Modulation of EVs by Cigarette Smoke and Their Relevance in Lung Disease: Recent Advances" Journal of Respiration 3, no. 4: 164-177. https://doi.org/10.3390/jor3040016