
Three-Dimensional Airway Spheroids and Organoids for Cystic Fibrosis Research


Abstract
:1. Introduction
1.1. Novel Therapies for CF
1.2. In Vivo and In Vitro Models for CF
1.3. Spheroids and Organoids for CF
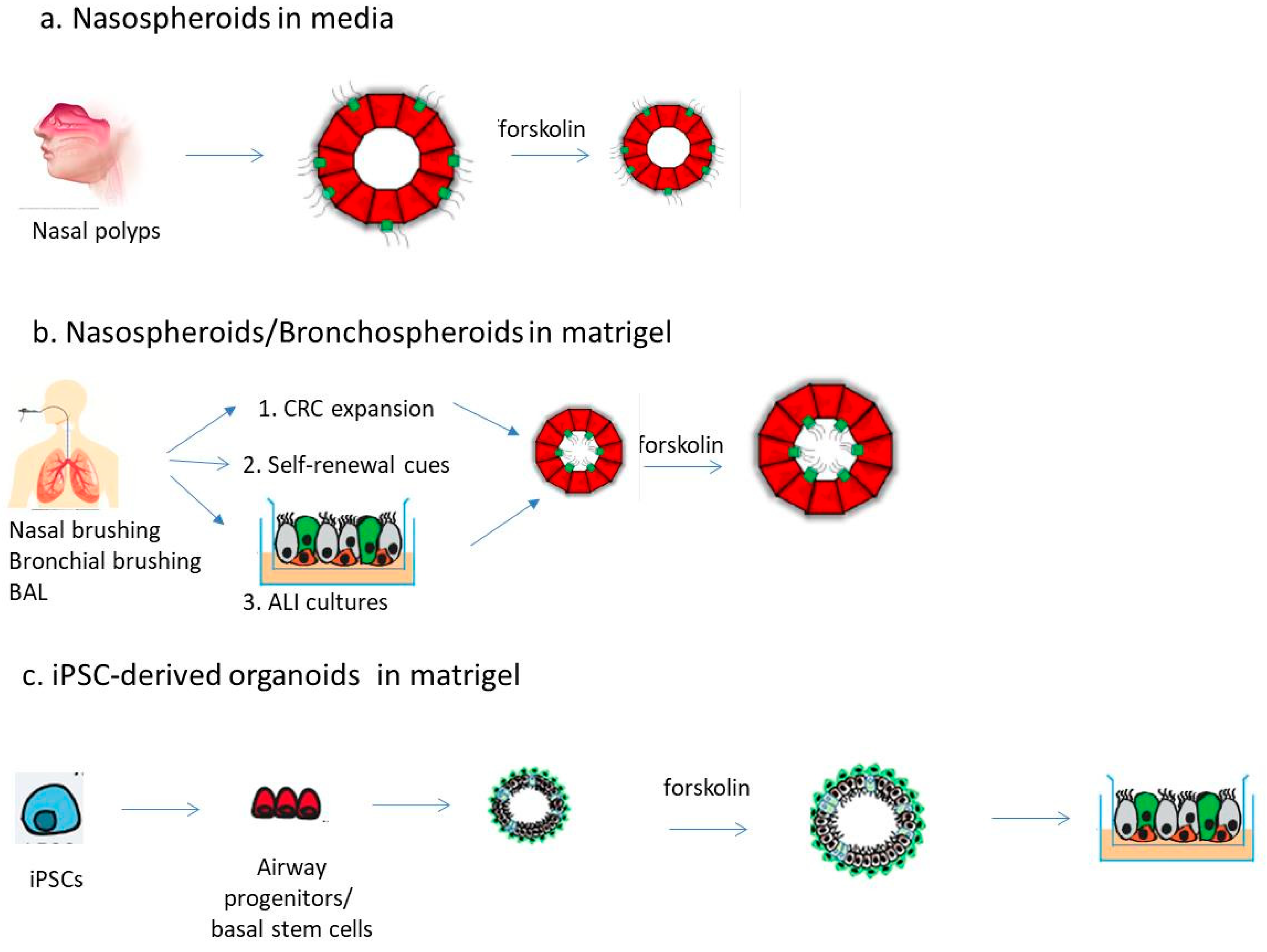
2. Airway Spheroids from Primary Airway Epithelial Cells
2.1. Spheroids from Differentiated Nasal Epithelial Cells
2.2. Spheroids from Airway Stem/Progenitor Cells
3. Airway Organoids from iPSCs
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Cuevas-Ocana, S.; Laselva, O.; Avolio, J.; Nenna, R. The era of CFTR modulators: Improvements made and remaining challenges. Breathe 2020, 16, 200016. [Google Scholar] [CrossRef]
- Dechecchi, M.C.; Tamanini, A.; Cabrini, G. Molecular basis of cystic fibrosis: From bench to bedside. Ann. Transl. Med. 2018, 6, 334. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR modulators: The changing face of cystic fibrosis in the era of precision medicine. Front. Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbry, P.; Cavard, A.; Chanson, M.; Jaffe, A.B.; Plasschaert, L.W. Regeneration of airway epithelial cells to study rare cell states in cystic fibrosis. J. Cyst. Fibros. 2020, 19, S42–S46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, C.A.; Da Tan, C.; Tarran, R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J. Physiol. 2013, 591, 4377–4387. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Bergeron, C.; Cantin, A.M. New therapies to correct the cystic fibrosis basic defect. Int. J. Mol. Sci. 2021, 22, 6193. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W. Lumacaftor—Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor—Ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445—Tezacaftor—Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+tezacaftor+ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Laselva, O.; Ardelean, M.C.; Bear, C.E. Phenotyping rare CFTR mutations reveal functional expression defects restored by TRIKAFTA. J. Pers. Med. 2021, 11, 301. [Google Scholar] [CrossRef]
- Yang, S.J.; Chen, H.M.; Hsieh, C.H.; Hsu, J.T.; Yeh, C.N.; Yeh, T.S.; Hwang, T.L.; Jan, Y.Y.; Chen, M.F. Akt pathway is required for oestrogen-mediated attenuation of lung injury in a rodent model of cerulein-induced acute pancreatitis. Injury 2011, 42, 638–642. [Google Scholar] [CrossRef]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659—tezacaftor—Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor—Tezacaftor—Ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor—Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.T.; Sharifi, N.A.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Bedwell, D.M.; Kaenjak, A.; Benos, D.J.; Bebok, Z.; Bubien, J.K.; Hong, J.; Tousson, A.; Clancy, J.P.; Sorscher, E.J. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat. Med. 1997, 3, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Valley, H.C.; Bukis, K.M.; Bell, A.; Cheng, Y.; Wong, E.; Jordan, N.J.; Allaire, N.E.; Sivachenko, A.; Liang, F.; Bihler, H.; et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J. Cyst. Fibros. 2019, 18, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021, 20, 436–442. [Google Scholar] [CrossRef]
- Aksit, M.A.; Bowling, A.D.; Evans, T.A.; Joynt, A.T.; Osorio, D.; Patel, S.; West, N.; Merlo, C.; Sosnay, P.R.; Cutting, G.R.; et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J. Cyst. Fibros. 2019, 18, 606–613. [Google Scholar] [CrossRef]
- Keenan, M.M.; Huang, L.; Jordan, N.J.; Wong, E.; Cheng, Y.; Valley, H.C.; Mahiou, J.; Liang, F.; Bihler, H.; Mense, M.; et al. Nonsense-mediated RNA decay pathway inhibition restores expression and function of W1282X CFTR. Am. J. Respir. Cell Mol. Biol. 2019, 61, 290–300. [Google Scholar] [CrossRef]
- Laselva, O.; Eckford, P.D.; Bartlett, C.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Functional rescue of c.3846G>A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J. Cyst. Fibros. 2020, 19, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Ouyang, H.; Laselva, O.; Bartlett, C.; Zhou, Z.P.; Duan, C.; Gunawardena, T.; Avolio, J.; Bear, C.E.; Gonska, T.; et al. A helper-dependent adenoviral vector rescues CFTR to wild-type functional levels in cystic fibrosis epithelial cells harbouring class I mutations. Eur. Respir. J. 2020, 56, 2000205. [Google Scholar] [CrossRef] [PubMed]
- Erwood, S.; Laselva, O.; Bily, T.M.I.; Brewer, R.A.; Rutherford, A.H.; Bear, C.E.; Ivakine, E.A. Allele-specific prevention of nonsense-mediated decay in cystic fibrosis using homology-independent genome editing. Mol. Ther. Methods Clin. Dev. 2020, 17, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Mention, K.; Cavusoglu-Doran, K.; Sanz, D.J.; Bacalhau, M.; Lopes-Pacheco, M.; Harrison, P.T.; Farinha, C.M. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282X-CFTR mutation. J. Cyst. Fibros. 2021, 21, S1569–S1993. [Google Scholar]
- Bandara, R.A.; Chen, Z.R.; Hu, J. Potential of helper-dependent adenoviral vectors in CRISPR-cas9-mediated lung gene therapy. Cell Biosci. 2021, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Allan, K.M.; Farrow, N.; Donnelley, M.; Jaffe, A.; Waters, S.A. Treatment of cystic fibrosis: From gene- to cell-based therapies. Front. Pharmacol. 2021, 12, 639475. [Google Scholar] [CrossRef] [PubMed]
- King, N.E.; Suzuki, S.; Barilla, C.; Hawkins, F.J.; Randell, S.H.; Reynolds, S.D.; Stripp, B.R.; Davis, B.R. Correction of airway stem cells: Genome editing approaches for the treatment of cystic fibrosis. Hum. Gene Ther. 2020, 31, 956–972. [Google Scholar] [CrossRef]
- McCarron, A.; Donnelley, M.; Parsons, D. Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res. 2018, 19, 54. [Google Scholar] [CrossRef]
- Mou, H.; Brazauskas, K.; Rajagopal, J. Personalized medicine for cystic fibrosis: Establishing human model systems. Pediatr. Pulmonol. 2015, 50, S14–S23. [Google Scholar] [CrossRef]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. -Cell Physiol. 2010, 298, C866–C874. [Google Scholar] [CrossRef] [Green Version]
- Haggie, P.M.; Phuan, P.W.; Tan, J.A.; Xu, H.; Avramescu, R.G.; Perdomo, D.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Lukacs, G.L.; et al. Correctors and potentiators rescue function of the truncated W1282X-cystic fibrosis transmembrane regulator (CFTR) translation product. J. Biol. Chem. 2017, 292, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, S.; Di Gioia, S.; di Toma, L.; Conese, M. Human cellular models for the investigation of lung inflammation and mucus production in cystic fibrosis. Anal. Cell. Pathol. 2018, 2018, 3839803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, D.E.; Brewington, J.J. Nasal epithelial cell-based models for individualized study in cystic fibrosis. Int. J. Mol. Sci. 2021, 22, 4448. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dousmanis, A.G.; Nairn, A.C.; Gadsby, D.C. Distinct Mg2+-dependent steps rate limit opening and closing of a single CFTR Cl− channel. J. Gen. Physiol. 2002, 119, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Devor, D.C.; Bridges, R.J.; Pilewski, J.M. Pharmacological modulation of ion transport across wild-type and deltaf508 CFTR-expressing human bronchial epithelia. Am. J. Physiol. -Cell Physiol. 2000, 279, C461–C479. [Google Scholar] [CrossRef] [PubMed]
- Randell, S.H.; Fulcher, M.L.; O’Neal, W.; Olsen, J.C. Primary epithelial cell models for cystic fibrosis research. Methods Mol. Biol. 2011, 742, 285–310. [Google Scholar]
- Gkatzis, K.; Taghizadeh, S.; Huh, D.; Stainier, D.Y.R.; Bellusci, S. Use of three-dimensional organoids and lung-on-a-chip methods to study lung development, regeneration and disease. Eur. Respir. J. 2018, 52, 1800876. [Google Scholar] [CrossRef]
- Lu, T.; Cao, Y.; Zhao, P.; Shen, S.; Xi, Y. Organoid: A powerful tool to study lung regeneration and disease. Cell Regen 2021, 10, 21. [Google Scholar] [CrossRef]
- van der Vaart, J.; Clevers, H. Airway organoids as models of human disease. J. Intern. Med. 2021, 289, 604–613. [Google Scholar] [CrossRef]
- Nikolic, M.Z.; Rawlins, E.L. Lung organoids and their use to study cell-cell interaction. Curr. Pathobiol. Rep. 2017, 5, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.; Houwen, R.H.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- van Mourik, P.; Beekman, J.M.; van der Ent, C.K. Intestinal organoids to model cystic fibrosis. Eur. Respir. J. 2019, 54, 1802379. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Gogorza Gondra, R.A.; Kruisselbrink, E.; Vonk, A.M.; Janssens, H.M.; de Winter-de Groot, K.M.; van der Ent, C.K.; Beekman, J.M. Optimal correction of distinct CFTR folding mutants in rectal cystic fibrosis organoids. Eur. Respir. J. 2016, 48, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Furstova, E.; Vonk, A.M.; Ferrante, M.; Verfaillie, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekman, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef]
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of organoid swelling and in vivo biomarkers of CFTR function to determine effects of lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for the F508del mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592. [Google Scholar] [CrossRef]
- Zomer-van Ommen, D.D.; de Poel, E.; Kruisselbrink, E.; Oppelaar, H.; Vonk, A.M.; Janssens, H.M.; van der Ent, C.K.; Hagemeijer, M.C.; Beekman, J.M. Comparison of ex vivo and in vitro intestinal cystic fibrosis models to measure CFTR-dependent ion channel activity. J. Cyst. Fibros. 2018, 17, 316–324. [Google Scholar] [CrossRef]
- Xia, S.; Bozóky, Z.; Laselva, O.; Di Paola, M.; Ahmadi, S.; Jiang, J.X.; Pitstick, A.; Jiang, C.; Rotin, D.; Mayhew, C.N.; et al. High-throughput functional analysis of CFTR and other apically localized channels in iPSC derived intestinal organoids. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gotoh, S.; Ito, I.; Nagasaki, T.; Yamamoto, Y.; Konishi, S.; Korogi, Y.; Matsumoto, H.; Muro, S.; Hirai, T.; Funato, M.; et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep. 2014, 3, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Castillon, N.; Hinnrasky, J.; Zahm, J.M.; Kaplan, H.; Bonnet, N.; Corlieu, P.; Klossek, J.M.; Taouil, K.; Avril-Delplanque, A.; Peault, B.; et al. Polarized expression of cystic fibrosis transmembrane conductance regulator and associated epithelial proteins during the regeneration of human airway surface epithelium in three-dimensional culture. Lab. Investig. 2002, 82, 989–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimbellot, J.S.; Leach, J.M.; Chaudhry, I.G.; Quinney, N.L.; Boyles, S.E.; Chua, M.; Aban, I.; Jaspers, I.; Gentzsch, M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2, e95734. [Google Scholar] [CrossRef] [Green Version]
- Hild, M.; Jaffe, A.B. Production of 3-D airway organoids from primary human airway basal cells and their use in high-throughput screening. Curr. Protoc. Stem Cell Biol. 2016, 37, IE.9.1–IE.9.15. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Bottinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.S.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi(R) and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Jiang, J.; Ahmadi, S.; Lew, A.; Laselva, O.; Xia, S.; Bartlett, C.; Ip, W.; Wellhauser, L.; Ouyang, H.; et al. ORKAMBI-mediated rescue of mucociliary clearance in cystic fibrosis primary respiratory cultures is enhanced by arginine uptake, arginase inhibition, and promotion of nitric oxide signaling to the cystic fibrosis transmembrane conductance regulator channel. Mol. Pharmacol. 2019, 96, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.S.; Irony-Tur Sinai, M.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C.; et al. Antisense oligonucleotide-based drug development for cystic fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021, 20, 865–875. [Google Scholar] [CrossRef]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J., 3rd; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3, e99385. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A.; Sousa, L.; Barreto, C.; Amaral, M.D. Changes in transcriptome of native nasal epithelium expressing F508del-CFTR and intersecting data from comparable studies. Respir. Res. 2013, 14, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might brushed nasal cells be a surrogate for CFTR modulator clinical response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized medicine based on nasal epithelial cells: Comparative studies with rectal biopsies and intestinal organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef]
- Martinovich, K.M.; Iosifidis, T.; Buckley, A.G.; Looi, K.; Ling, K.M.; Sutanto, E.N.; Kicic-Starcevich, E.; Garratt, L.W.; Shaw, N.C.; Montgomery, S.; et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci. Rep. 2017, 7, 17971. [Google Scholar] [CrossRef]
- Chapman, S.; Liu, X.; Meyers, C.; Schlegel, R.; McBride, A.A. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J. Clin. Investig. 2010, 120, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Bukowy-Bieryllo, Z. Long-term differentiating primary human airway epithelial cell cultures: How far are we? Cell Commun. Signal. 2021, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD signaling inhibition enables long-term expansion of diverse epithelial basal cells. Cell Stem Cell 2016, 19, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Awatade, N.T.; Wong, S.L.; Capraro, A.; Pandzic, E.; Slapetova, I.; Zhong, L.; Turgutoglu, N.; Fawcett, L.K.; Whan, R.M.; Jaffe, A.; et al. Significant functional differences in differentiated conditionally reprogrammed (CRC)- and feeder-free dual SMAD inhibited-expanded human nasal epithelial cells. J. Cyst. Fibros. 2021, 20, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.S.; Procida, K.; Larsen, P.L.; Holstein-Rathlou, N.H.; Frederiksen, O. Water permeability in human airway epithelium. Pflugers. Arch. 2005, 451, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Bridges, M.A.; Walker, D.C.; Harris, R.A.; Wilson, B.R.; Davidson, A.G. Cultured human nasal epithelial multicellular spheroids: Polar cyst-like model tissues. Biochem. Cell Biol. 1991, 69, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Marthin, J.K.; Stevens, E.M.; Larsen, L.A.; Christensen, S.T.; Nielsen, K.G. Patient-specific three-dimensional explant spheroids derived from human nasal airway epithelium: A simple methodological approach for ex vivo studies of primary ciliary dyskinesia. Cilia 2017, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugebauer, P.; Endepols, H.; Mickenhagen, A.; Walger, M. Ciliogenesis in submersion and suspension cultures of human nasal epithelial cells. Eur. Arch. Otorhinolaryngol. 2003, 260, 325–330. [Google Scholar] [CrossRef]
- Gamarra, F.; Bergner, A.; Stauss, E.; Stocker, I.; Grundler, S.; Huber, R.M. Rotation frequency of human bronchial and nasal epithelial spheroids as an indicator of mucociliary function. Respiration 2006, 73, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J., 3rd; Ostmann, A.J.; Strecker, L.M.; Szczesniak, R.D.; Clancy, J.P. Detection of CFTR function and modulation in primary human nasal cell spheroids. J. Cyst. Fibros. 2018, 17, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human nasal epithelial organoids for therapeutic development in cystic fibrosis. Genes 2020, 11, 603. [Google Scholar] [CrossRef]
- Awatade, N.T.; Wong, S.L.; Hewson, C.K.; Fawcett, L.K.; Kicic, A.; Jaffe, A.; Waters, S.A. Human primary epithelial cell models: Promising tools in the era of cystic fibrosis personalized medicine. Front. Pharmacol. 2018, 9, 1429. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, P.S.; Frederiksen, O.; Holstein-Rathlou, N.H.; Larsen, P.L.; Qvortrup, K. Ion transport in epithelial spheroids derived from human airway cells. Am. J. Physiol. -Lung Cell. Mol. Physiol. 1999, 276, L75–L80. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.S.; Holstein-Rathlou, N.H.; Larsen, P.L.; Qvortrup, K.; Frederiksen, O. Fluid absorption related to ion transport in human airway epithelial spheroids. Am. J. Physiol. -Lung Cell. Mol. Physiol. 1999, 277, L1096–L1103. [Google Scholar] [CrossRef]
- Pedersen, P.S.; Braunstein, T.H.; Jorgensen, A.; Larsen, P.L.; Holstein-Rathlou, N.H.; Frederiksen, O. Stimulation of aquaporin-5 and transepithelial water permeability in human airway epithelium by hyperosmotic stress. Pflügers Arch. -Eur. J. Physiol. 2007, 453, 777–785. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.; Brewington, J.J.; Harkness, B.; Clancy, J.P.; Trapnell, B.C. Personalised CFTR pharmacotherapeutic response testing and therapy of cystic fibrosis. Eur. Respir. J. 2018, 51, 1702457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatngalim, G.D.; Rodenburg, L.W.; Aalbers, B.L.; Raeven, H.H.M.; Aarts, E.M.; Silva, I.A.L.; Nijenhuis, W.; Vrendenbarg, S.; Kruisselbrink, E.; Brunsveld, J.E.; et al. CFTR modulator response measurements in subjects with cystic fibrosis using 2D differentiated nasal epithelia converted into spheroids. bioRxiv 2021. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Chung, M.I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L. Lung organoids: Current uses and future promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Q.; Choi, K.M.; Sicard, D.; Tschumperlin, D.J. Human airway organoid engineering as a step toward lung regeneration and disease modeling. Biomaterials 2017, 113, 118–132. [Google Scholar] [CrossRef] [Green Version]
- Danahay, H.; Pessotti, A.D.; Coote, J.; Montgomery, B.E.; Xia, D.; Wilson, A.; Yang, H.; Wang, Z.; Bevan, L.; Thomas, C.; et al. Notch2 is required for inflammatory cytokine-driven goblet cell metaplasia in the lung. Cell Rep. 2015, 10, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [Green Version]
- Sprott, R.F.; Ritzmann, F.; Langer, F.; Yao, Y.; Herr, C.; Kohl, Y.; Tschernig, T.; Bals, R.; Beisswenger, C. Flagellin shifts 3D bronchospheres towards mucus hyperproduction. Respir. Res. 2020, 21, 222. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Vonk, A.M.; Silva, I.A.; Botelho, H.M.; Bor, R.V.; van Mourik, P.; Heida-Michel, S.; Aguilera, B.; Mullenders, J.; Boj, S.F.; et al. High reproducibility of forskolin-induced swelling of intestinal organoids across three academic laboratories. Pediatric. Pulmonol. 2019, 54, S326. [Google Scholar]
- Wilkinson, D.C.; Alva-Ornelas, J.A.; Sucre, J.M.; Vijayaraj, P.; Durra, A.; Richardson, W.; Jonas, S.J.; Paul, M.K.; Karumbayaram, S.; Dunn, B.; et al. Development of a three-dimensional bioengineering technology to generate lung tissue for personalized disease modeling. Stem Cells Transl. Med. 2017, 6, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Green, M.D.; Chen, A.; Nostro, M.C.; d’Souza, S.L.; Schaniel, C.; Lemischka, I.R.; Gouon-Evans, V.; Keller, G.; Snoeck, H.W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.X.; Islam, M.N.; O’Neill, J.; Hu, Z.; Yang, Y.G.; Chen, Y.W.; Mumau, M.; Green, M.D.; Vunjak-Novakovic, G.; Bhattacharya, J.; et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, H.; Zhao, R.; Sherwood, R.; Ahfeldt, T.; Lapey, A.; Wain, J.; Sicilian, L.; Izvolsky, K.; Musunuru, K. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell 2012, 10, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.P.; Bear, C.E.; Chin, S.; Pasceri, P.; Thompson, T.O.; Huan, L.J.; Ratjen, F.; Ellis, J.; Rossant, J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 2012, 30, 876–882. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.P.; Chin, S.; Xia, S.; Garner, J.; Bear, C.E.; Rossant, J. Efficient generation of functional CFTR-expressing airway epithelial cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 363–381. [Google Scholar] [CrossRef]
- Firth, A.L.; Dargitz, C.T.; Qualls, S.J.; Menon, T.; Wright, R.; Singer, O.; Gage, F.H.; Khanna, A.; Verma, I.M. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E1723–E1730. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [Green Version]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell 2017, 20, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019, 14, 518–540. [Google Scholar] [CrossRef]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Berical, A.; Lee, R.E.; Lu, J.; Beermann, M.L.; LeSeur, J.A.; Mithal, A.; Thomas, D.; Ranallo, N.; Peasley, M.; Stuffer, A.; et al. A multimodal iPSC platform for cystic fibrosis drug testing. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jiang, J.X.; Wellhauser, L.; Laselva, O.; Utkina, I.; Bozoky, Z.; Gunawardena, T.; Ngan, Z.; Xia, S.; Eckford, P.D.W.; Ratjen, F.; et al. A new platform for high-throughput therapy testing on iPSC-derived, immature airway from Cystic Fibrosis Patients. bioRxiv 2021. [Google Scholar]
- Ngan, S.Y.; Quach, H.; Dierolf, J.; Laselva, O.; Lee, J.-A.; Huang, E.; Mangos, M.; Xia, S.; Wong, A.P. Modeling lung cell development using human pluripotent stem cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hawkins, F.J.; Suzuki, S.; Beermann, M.L.; Barilla, C.; Wang, R.; Villacorta-Martin, C.; Berical, A.; Jean, J.C.; Le Suer, J.; Matte, T.; et al. Derivation of airway basal stem cells from human pluripotent stem cells. Cell Stem Cell 2021, 28, 79–95. [Google Scholar] [CrossRef]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019, 26, 1701–1708. [Google Scholar] [CrossRef] [Green Version]
- Konishi, S.; Gotoh, S.; Tateishi, K.; Yamamoto, Y.; Korogi, Y.; Nagasaki, T.; Matsumoto, H.; Muro, S.; Hirai, T.; Ito, I.; et al. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 2016, 6, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.; Chu, H.; Poon, V.K.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Study | Spheroid Model | Morphology and Antigen Expression | Function | Pharmacological Treatment |
|---|---|---|---|---|
| Pedersen et al., 1999 [92] | CF (n = 9) and non-CF (n = 17) subjects. Free-floating spheroids from nasal polyp-epithelial sheets. | “Lumen-out” configuration. The apical ciliated membrane facing the bath, and the basolateral cell membrane pointing toward a fluid-filled lumen. | Transepithelial PD measurements compatible with the presence of an amiloride-sensitive Na+ absorption and ATP-sensitive Cl- channel in the apical membrane. CF spheroids PD was not changed by the increase in cAMP. | None. |
| Pedersen et al., 1999 [93] | CF (n = 7) and non-CF (n = 15) subjects. Free-floating spheroids from nasal polyp-epithelial sheets. | “Lumen-out” configuration. The apical ciliated membrane facing the bath, and the basolateral cell membrane pointing toward a fluid-filled lumen. | Fluid absorption rates were equal in non-CF and CF spheroids. Amiloride inhibited fluid absorption to a lower residual level in non-CF than in CF spheroids. | None. |
| Pedersen et al., 2007 [94] | CF (n = 4) and non-CF (n = 5) subjects. | “Lumen-out” configuration. The apical ciliated membrane facing the bath, and the basolateral cell membrane pointing toward a fluid-filled lumen. | Hyperosmotic treatment caused an increase in epithelial water permeability without changing fluid absorption rates. | None. |
| Guimbellot et al., 2017 [68] | CF (n = 3) and non-CF (n = 9) subjects. CF genotypes: F508del/F508del; F508del/I618T; G551D/F508del | “Lumen-out” configuration. The apical ciliated membrane facing the bath, and the basolateral cell membrane pointing toward a fluid-filled lumen. CFTR expression at the level of apical region. | Shrinking of non-CF spheroids upon the increase in cAMP levels. CF spheroids showed diminished volume reduction following CFTR activation. | Lumacaftor–Ivacaftor treatment partially restored cross-sectional area reduction of CF nanospheroids. |
| Brewington et al., 2018 [89] | CF (n = 19) and non-CF (n = 6) subjects. CF genotypes: F508del/F508del (n = 9); others with at least one non-F508del mutation | “Lumen-in” configuration. Cilia on the luminal surface. Positive for E-cadherin, luminal F actin and alpha tubulin, and the mucin MUC5AC. | Non-CF spheroids swelled upon CFTR stimulation. NEC spheroids from F508del homozygotes shrank following CFTR stimulation. | F508del homozygous spheroids swelled when pre-treated with Ivacaftor and Lumacaftor, or incubated at 27 °C. |
| McCarthy et al., 2018 [95] | Nasal curettage. One CF patient heterozygous for Ser1159Pro and F508del. | “Lumen-in” configuration. | Nasospheroids did not swell in the FIS assay. | Nasospheroids swelled in response to Lumacaftor–Ivacaftor. Following ex vivo studies, the patients commenced in vivo therapy. |
| Sachs et al., 2019 [70] | Lung biospies and BAL specimens (“bronchospheroids”). Non-CF and CF subjects. CF genotypes: F508el/F508del (n = 3), F508del/G542X (n = 1), R334W/R334W (n = 1). | “Lumen-in” configuration. Cilia on the luminal surface. Positive for basal marker keratin-5 (KRT5), club cell marker secretoglobin family 1A member 1 (SCGB1A1), cilia marker acetylated a-tubulin, or secretory cell marker mucin 5AC (MUC5AC). | In non-CF spheroids, spheroids swelled upon forskolin and Eact stimulation. | Forskolin-induced swelling was reduced in CF compared to wild-type spheroids, and correlated with the severity of the tested CFTR genotypes. It could be augmented with Lumacaftor and Ivacaftor. Eact induced a swelling similar to that induced by forskolin. |
| Liu et al., 2020 [90] | Nasal brushing. CF (n = 36) and non-CF (n = 12) subjects. CF genotypes: F508del/F508del (n = 5); others with at least one minimal or residual function mutation | “Lumen-in” configuration. Cilia on the luminal surface. Positive for MUC5AC and MUC5B, ZO-1, CFTR, and FOXI1. | The baseline luminal ratio and the FIS assay distinguish between non-CF and CF spheroids and also between CF with different genotypes. | None. |
| Amatngalim et al., 2021 [96] | Nasal brushing. CF (n = 22) and CF (n = 22) subjects (F508del/F508del). | 3D spheroids with a “lumen-in configuration” derived from 2D differentiated ALI-NEC cultures. β-tubulin IV+ cilia and MUC5AC+ secretory cells inside of the spheroid and p63+ and KRT5+ basal cells. | FIS measured in non-CF spheroids was significantly higher compared to CF spheroids, while Eact induced a more significant swelling in CF spheroids compared to non-CF ones. | No response of CF organoids to Ivacaftor–Lumacaftor while a detectable swelling was obtained when cells were grown at ALI in the presence of neuregulin and IL-1β. Under these conditions, a high increase in FIS was obtained with Ivacaftor–Tezacaftor–Elexacaftor treatment. |
| Study | Organoid Model | Morphology and Antigen Expression | Function | Pharmacological Treatment |
|---|---|---|---|---|
| McCauley et al., [111] | Proximalized airway organoids. One non-CF subject and two CF patients homozygous for F508del. | “Lumen-in” configuration. | Little, if any, swelling was observed in either CF lines after exposure to forskolin as compared to the wild-type line. | The gene-corrected F508del/WT organoids significantly swelled in response to forskolin treatment. |
| Berical et al., [115] | Proximalized airway organoids. Three non-CF subjects and five CF patients (1 homozygous for W1282X, three homozygous for F508del, one homozygous for G551D). | “Lumen-in” configuration. CFTR expression at similar levels to primary HBE cultures | A small but statistically significant basal swelling in G551D organoids, but no detectable basal FIS in F508del and W1282X organoids. | FIS increased in G551D organoids after treatment with Ivacaftor. Treatment of F508del organoids with the first-generation correctors (Lumacaftor, Tezacaftor) had a small effect on FIS, while a robust increase was obtained with the triple combination Ivacaftor–Tezacaftor–Elexacaftor. In W1282X organoids, combinatorial treatment with G418, SMG1i, and Ivacaftor–Tezacaftor–Elexacaftor led to a significant increase in FIS. |
| Ngan et al., 2021 [117] | Fetal lung-derived organoids. Non-CF hiPSC lines. | “Lumen-in” configuration. By immunofluorescence: expression of basal cell marker KRT5, ciliated cell marker FOXJ1, luminal epithelial cell marker KRT8, secretory cell marker MUC16. By qPCR: expression of ΔNP63 and KRT14, acetylated a-tubulin, cytokeratin-8/18 (KRT8/18), and secretoglobulin 1A1 (SCGB1A1) and mucin 5ac (MUC5AC). CFTR co-expressed at the lumen side with ZO-1. | The FLiPR analysis on spheroids 2–3 days after seeding on collagen-coated plates found an increase in fluorescence activity indicative of CFTR function upon forskolin-induction that was inhibited with CFTR inhibitor-172. Significant swelling was found after 24 h of forskolin stimulation. | None. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laselva, O.; Conese, M. Three-Dimensional Airway Spheroids and Organoids for Cystic Fibrosis Research. J. Respir. 2021, 1, 229-247. https://doi.org/10.3390/jor1040022
Laselva O, Conese M. Three-Dimensional Airway Spheroids and Organoids for Cystic Fibrosis Research. Journal of Respiration. 2021; 1(4):229-247. https://doi.org/10.3390/jor1040022
Chicago/Turabian StyleLaselva, Onofrio, and Massimo Conese. 2021. "Three-Dimensional Airway Spheroids and Organoids for Cystic Fibrosis Research" Journal of Respiration 1, no. 4: 229-247. https://doi.org/10.3390/jor1040022