
Translocation of Adenosine A2B Receptor to Mitochondria Influences Cytochrome P450 2E1 Activity after Acetaminophen Overdose

 , , and
, , and 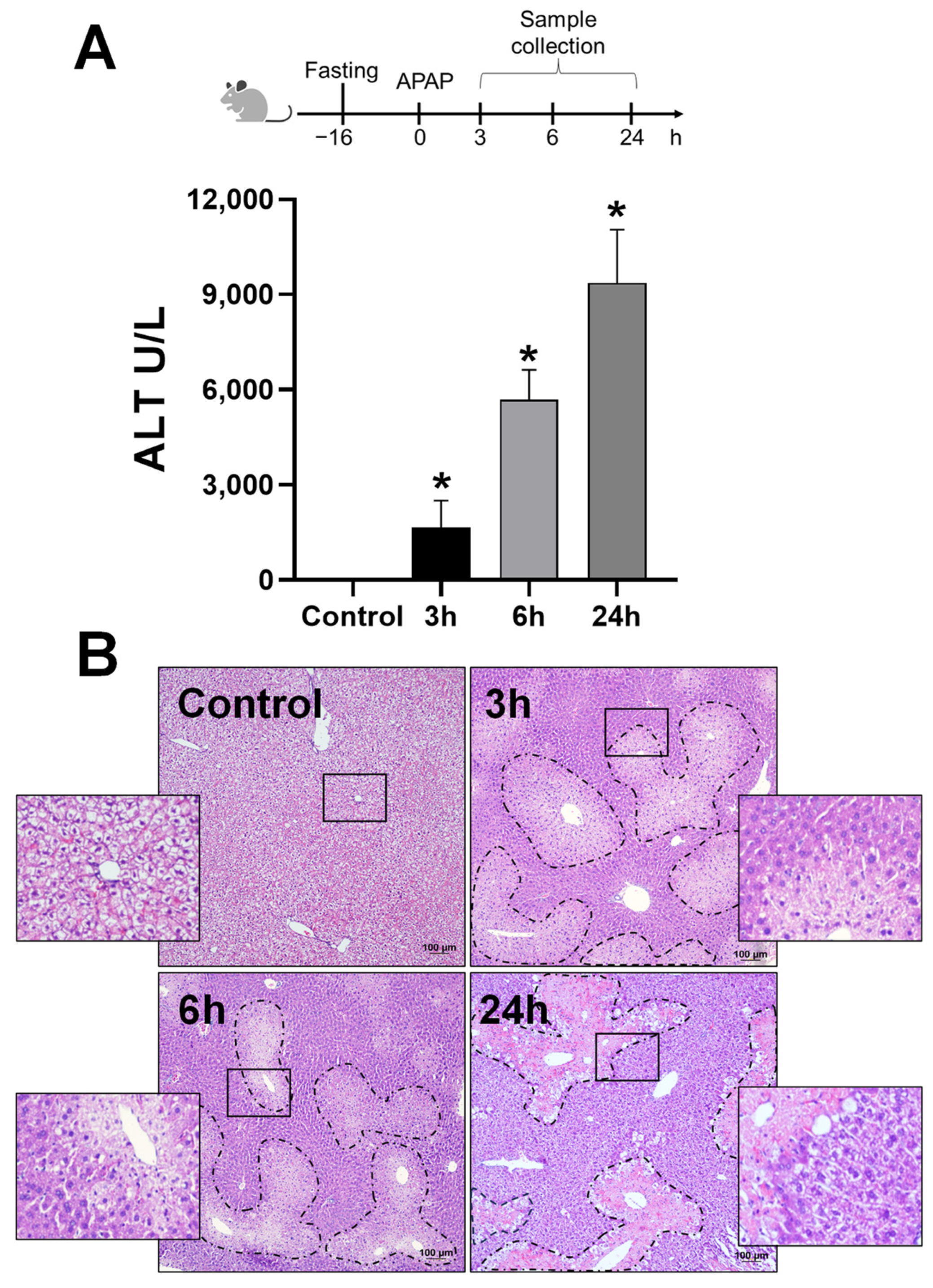{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Biochemical Measurements, Histology, and Cyp2E1 Activity Assay
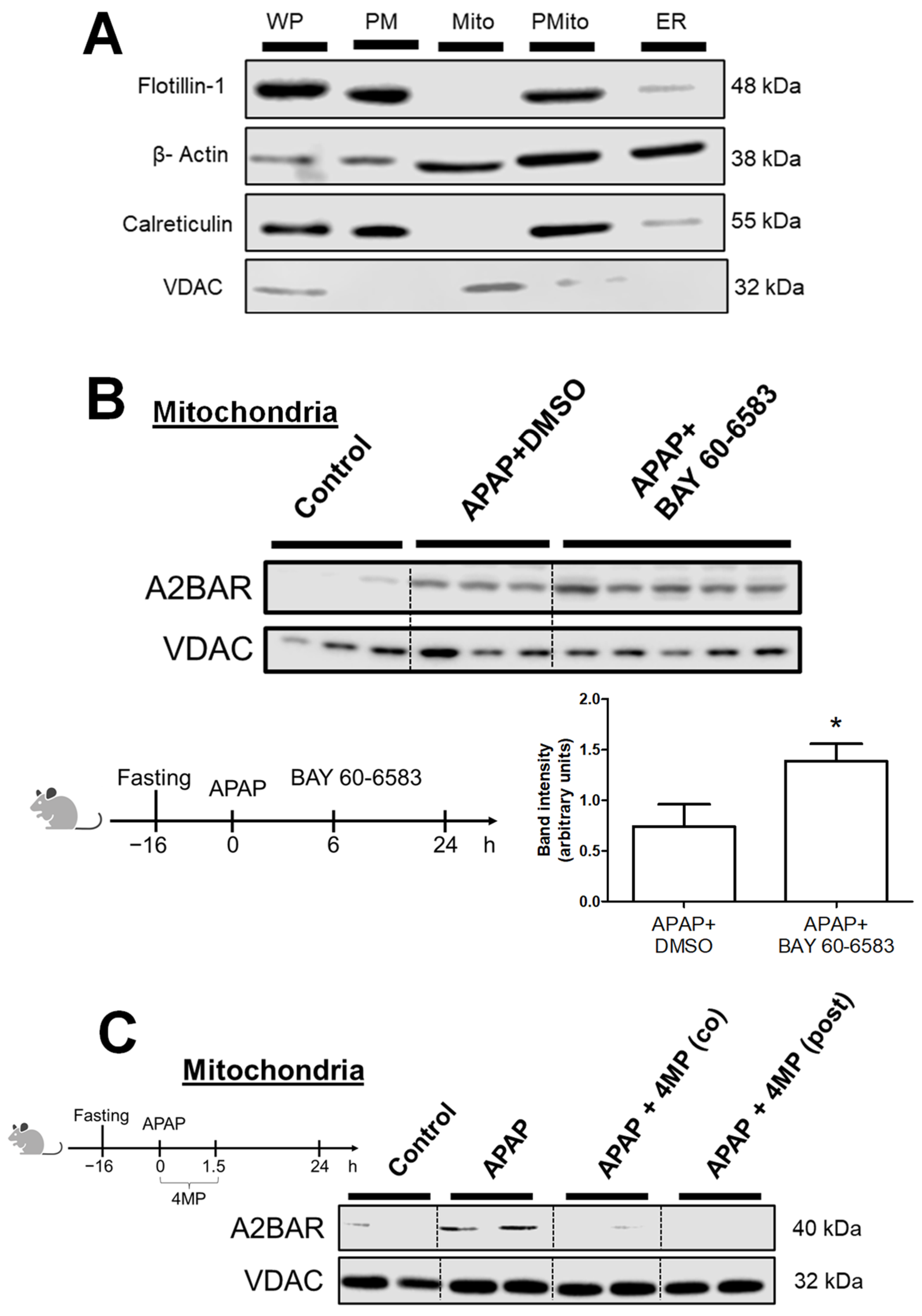
2.3. Mitochondria and Plasma Membrane Separation
2.4. Proteinase K Digestion and Western Blotting
2.5. Molecular Modeling
2.6. Statistics
3. Results
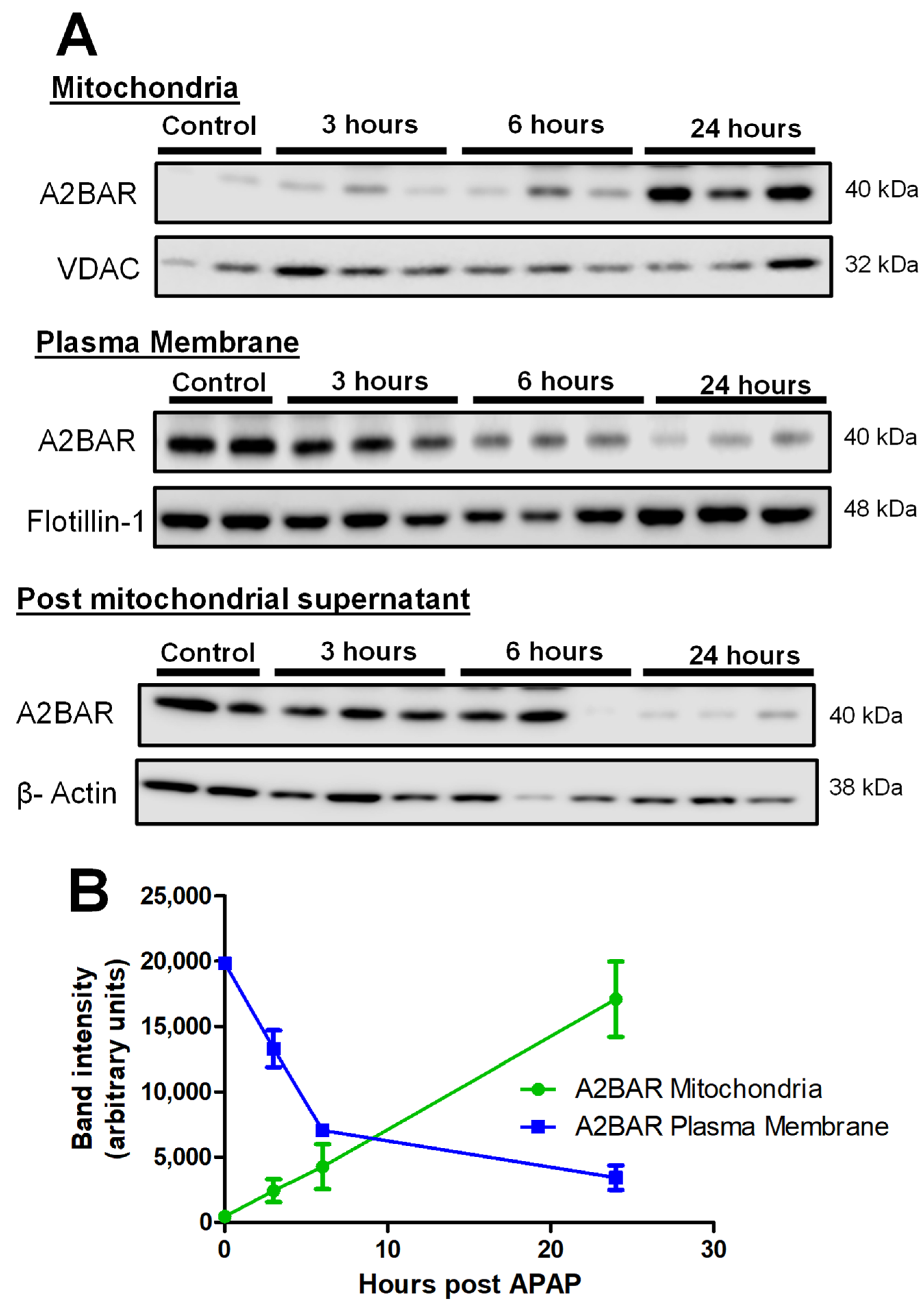
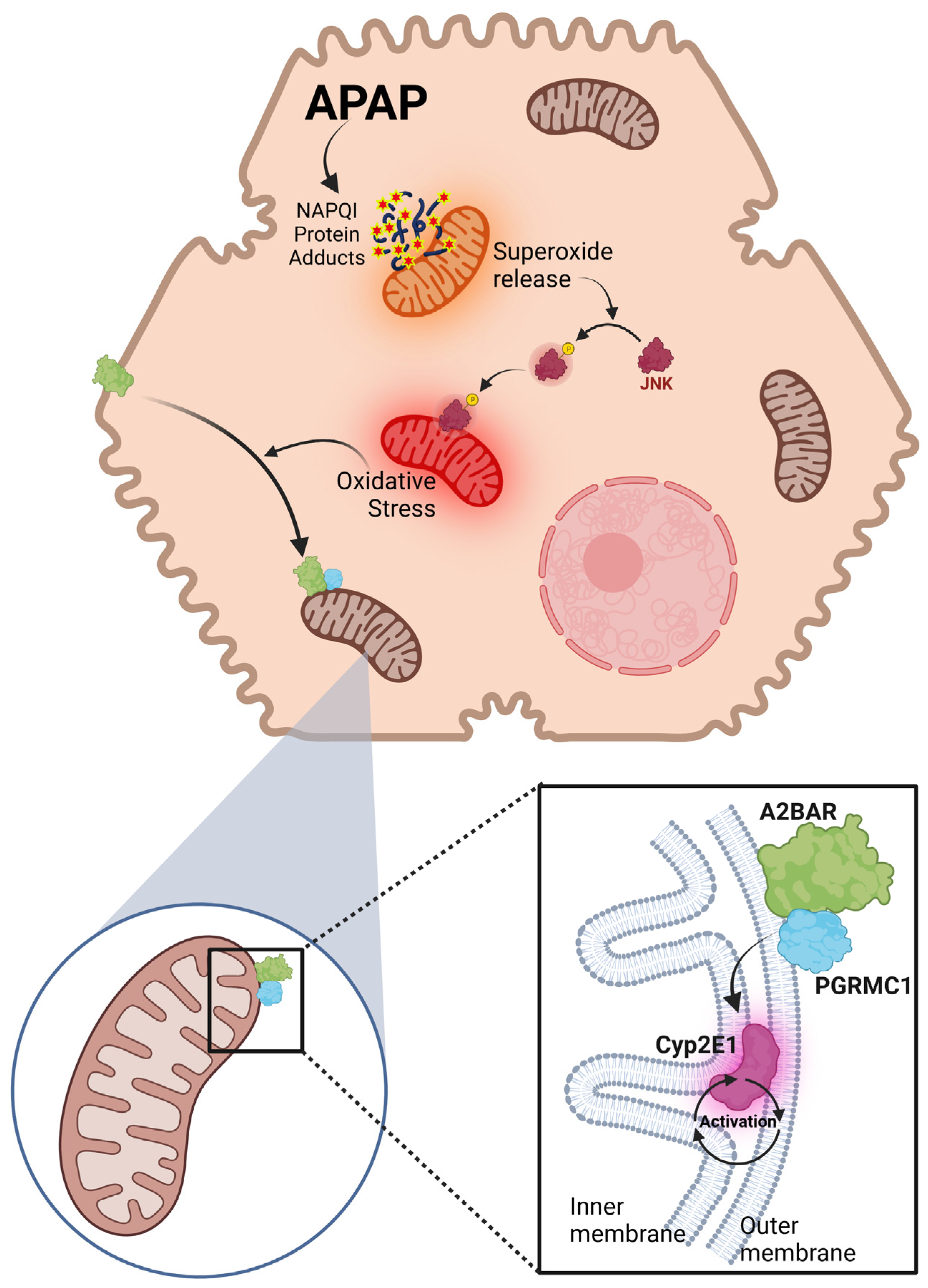
3.1. APAP Overdose Induces Mitochondrial Translocation of A2BAR
3.2. Generation of Reactive Metabolite and JNK Activation Is Required for A2BAR Mitochondrial Translocation
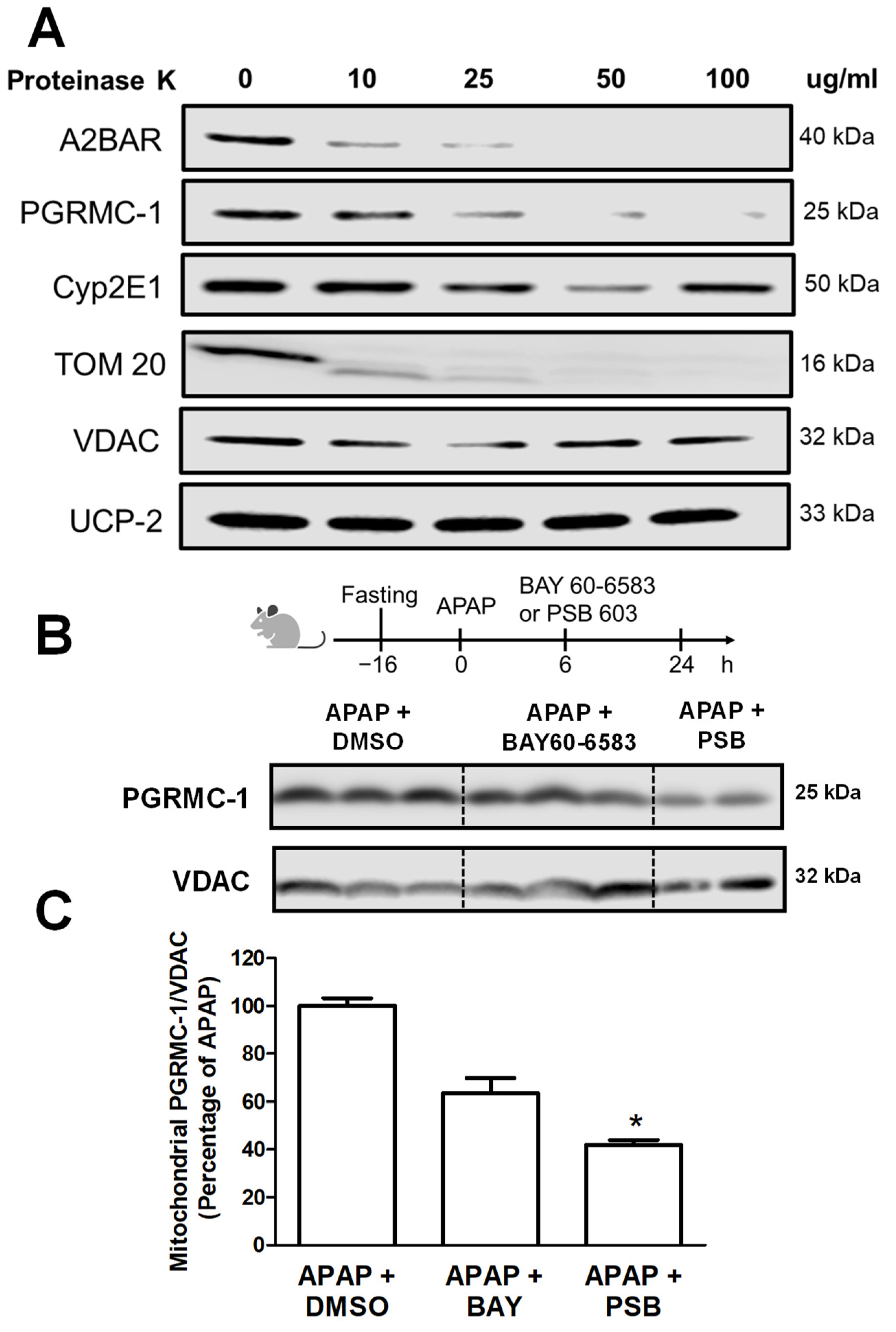
3.3. A2BAR Is Translocated to the Outer Mitochondrial Membrane and Has a Similar Localization to That of PGRMC1
3.4. A2BAR Possibly Binds to PGRMC1 on the Outer Mitochondria Membrane
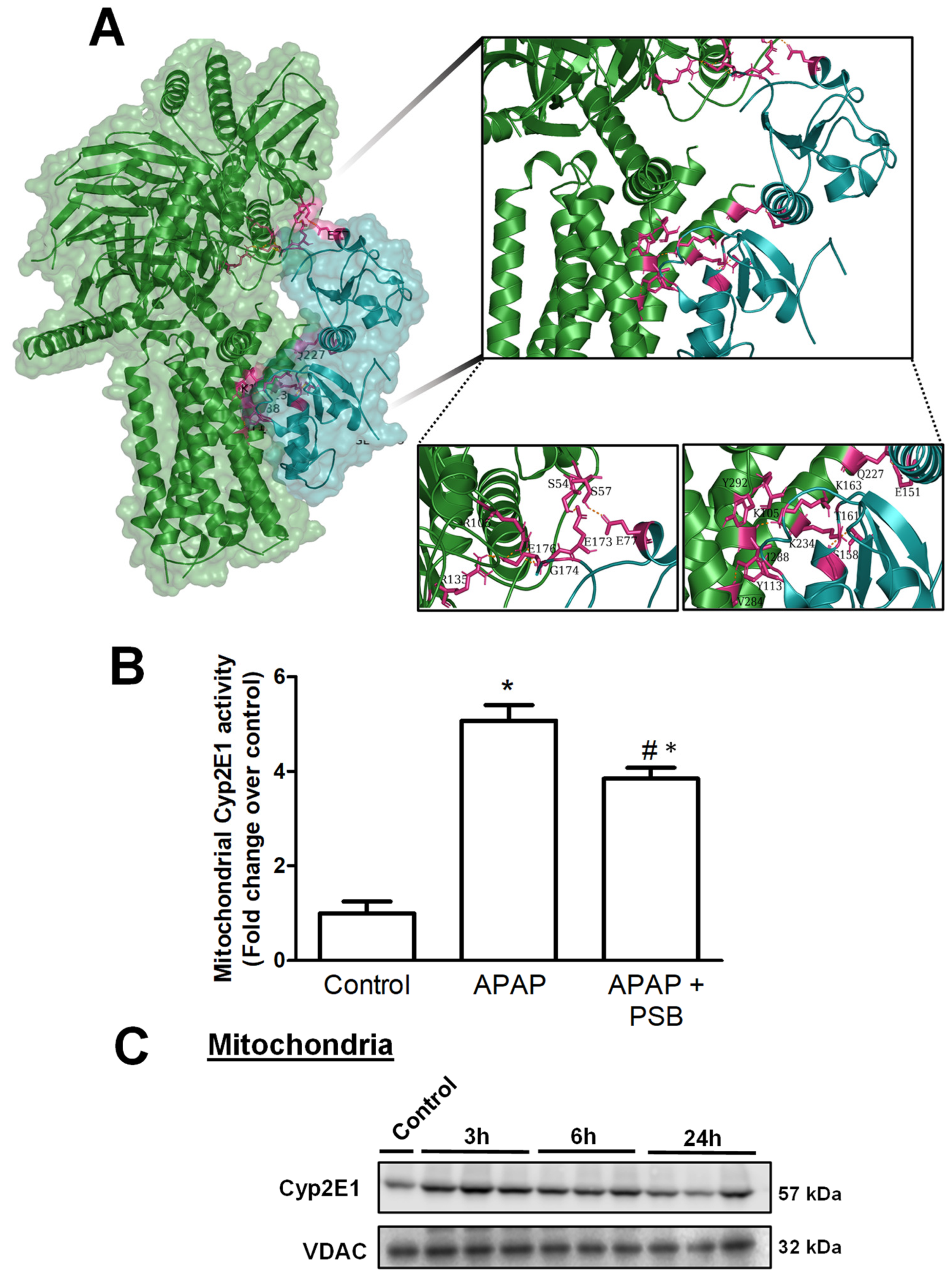
3.5. A2BAR–PGRMC1 Binding Influences Mitochondrial Cyp2E1 Activity after APAP Overdose
4. Discussion
4.1. A2BAR Localization and Trafficking in Hepatocytes
4.2. Mitochondrial Oxidant Stress and A2BAR Trafficking
4.3. A2BAR Binding to PCRMC1 on Mitochondria
4.4. A2BAR–PGRMC1 Binding Modulates CYP2E1 Activity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pasquini, S.; Contri, C.; Borea, P.A.; Vincenzi, F.; Varani, K. Adenosine and Inflammation: Here, There and Everywhere. Int. J. Mol. Sci. 2021, 22, 7685. [Google Scholar] [CrossRef] [PubMed]
- Beukers, M.W.; den Dulk, H.; van Tilburg, E.W.; Brouwer, J.; Ijzerman, A.P. Why are A(2B) receptors low-affinity adenosine receptors? Mutation of Asn273 to Tyr increases affinity of human A(2B) receptor for 2-(1-Hexynyl)adenosine. Mol. Pharmacol. 2000, 58, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Aherne, C.M.; Kewley, E.M.; Eltzschig, H.K. The resurgence of A2B adenosine receptor signaling. Biochim. Biophys. Acta 2011, 1808, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Sitaraman, S.V.; Wang, L.; Wong, M.; Bruewer, M.; Hobert, M.; Yun, C.H.; Merlin, D.; Madara, J.L. The adenosine 2b receptor is recruited to the plasma membrane and associates with E3KARP and Ezrin upon agonist stimulation. J. Biol. Chem. 2002, 277, 33188–33195. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine receptors: Expression, function and regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kolachala, V.; Walia, B.; Balasubramanian, S.; Hall, R.A.; Merlin, D.; Sitaraman, S.V. Agonist-induced polarized trafficking and surface expression of the adenosine 2b receptor in intestinal epithelial cells: Role of SNARE proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1100–G1107. [Google Scholar] [CrossRef] [PubMed]
- Grube, K.; Rudebusch, J.; Xu, Z.; Bockenholt, T.; Methner, C.; Muller, T.; Cuello, F.; Zimmermann, K.; Yang, X.; Felix, S.B.; et al. Evidence for an intracellular localization of the adenosine A2B receptor in rat cardiomyocytes. Basic Res. Cardiol. 2011, 106, 385–396. [Google Scholar] [CrossRef]
- Gnad, T.; Navarro, G.; Lahesmaa, M.; Reverte-Salisa, L.; Copperi, F.; Cordomi, A.; Naumann, J.; Hochhauser, A.; Haufs-Brusberg, S.; Wenzel, D.; et al. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity. Cell Metab. 2020, 32, 56–70.e7. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Mitochondria in Acetaminophen-Induced Liver Injury and Recovery: A Concise Review. Livers 2023, 3, 219–231. [Google Scholar] [CrossRef]
- Lee, S.S.; Buters, J.T.; Pineau, T.; Fernandez-Salguero, P.; Gonzalez, F.J. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J. Biol. Chem. 1996, 271, 12063–12067. [Google Scholar] [CrossRef]
- Cheung, C.; Yu, A.M.; Ward, J.M.; Krausz, K.W.; Akiyama, T.E.; Feigenbaum, L.; Gonzalez, F.J. The cyp2e1-humanized transgenic mouse: Role of cyp2e1 in acetaminophen hepatotoxicity. Drug Metab. Dispos. 2005, 33, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.A.; Anandatheerthavarada, H.K.; Fang, J.K.; Cudic, M.; Otvos, L.; Avadhani, N.G. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N terminus and requires mitochondrial specific electron transfer proteins for activity. J. Biol. Chem. 2001, 276, 24680–24689. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.R.; Mukhopadhyay, D.; Myers, S.L.; Mosher, E.P.; Brookheart, R.T.; Kammers, K.; Sehgal, A.; Selen, E.S.; Wolfgang, M.J.; Bumpus, N.N.; et al. Progesterone receptor membrane component 1 (PGRMC1) binds and stabilizes cytochromes P450 through a heme-independent mechanism. J. Biol. Chem. 2021, 297, 101316. [Google Scholar] [CrossRef] [PubMed]
- Piel, R.B., 3rd; Shiferaw, M.T.; Vashisht, A.A.; Marcero, J.R.; Praissman, J.L.; Phillips, J.D.; Wohlschlegel, J.A.; Medlock, A.E. A Novel Role for Progesterone Receptor Membrane Component 1 (PGRMC1): A Partner and Regulator of Ferrochelatase. Biochemistry 2016, 55, 5204–5217. [Google Scholar] [CrossRef]
- Duan, L.; Woolbright, B.L.; Jaeschke, H.; Ramachandran, A. Late Protective Effect of Netrin-1 in the Murine Acetaminophen Hepatotoxicity Model. Toxicol. Sci. 2020, 175, 168–181. [Google Scholar] [CrossRef]
- Duan, L.; Sanchez-Guerrero, G.; Jaeschke, H.; Ramachandran, A. Activation of the adenosine A2B receptor even beyond the therapeutic window of N-acetylcysteine accelerates liver recovery after an acetaminophen overdose. Food Chem. Toxicol. 2022, 163, 112911. [Google Scholar] [CrossRef]
- Akakpo, J.Y.; Ramachandran, A.; Duan, L.; Schaich, M.A.; Jaeschke, M.W.; Freudenthal, B.D.; Ding, W.X.; Rumack, B.H.; Jaeschke, H. Delayed Treatment with 4-Methylpyrazole Protects against Acetaminophen Hepatotoxicity in Mice by Inhibition of c-Jun n-Terminal Kinase. Toxicol. Sci. 2019, 170, 57–68. [Google Scholar] [CrossRef]
- Molck, C.; Ryall, J.; Failla, L.M.; Coates, J.L.; Pascussi, J.M.; Heath, J.K.; Stewart, G.; Hollande, F. The A(2b) adenosine receptor antagonist PSB-603 promotes oxidative phosphorylation and ROS production in colorectal cancer cells via adenosine receptor-independent mechanism. Cancer Lett. 2016, 383, 135–143. [Google Scholar] [CrossRef]
- Kotanska, M.; Szafarz, M.; Mika, K.; Dziubina, A.; Bednarski, M.; Muller, C.E.; Sapa, J.; Kiec-Kononowicz, K. PSB 603—A known selective adenosine A2B receptor antagonist—Has anti-inflammatory activity in mice. Biomed. Pharmacother. 2021, 135, 111164. [Google Scholar] [CrossRef]
- Akakpo, J.Y.; Ramachandran, A.; Kandel, S.E.; Ni, H.M.; Kumer, S.C.; Rumack, B.H.; Jaeschke, H. 4-Methylpyrazole protects against acetaminophen hepatotoxicity in mice and in primary human hepatocytes. Hum. Exp. Toxicol. 2018, 37, 1310–1322. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A. PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. Bio Protoc. 2017, 7, e2124. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xin, W.; Yang, X.M.; Kuno, A.; Rich, T.C.; Cohen, M.V.; Downey, J.M. A2B adenosine receptors inhibit superoxide production from mitochondrial complex I in rabbit cardiomyocytes via a mechanism sensitive to Pertussis toxin. Br. J. Pharmacol. 2011, 163, 995–1006. [Google Scholar] [CrossRef]
- Ramachandran, A.; Umbaugh, D.S.; Jaeschke, H. Mitochondrial Dynamics in Drug-Induced Liver Injury. Livers 2021, 1, 102–115. [Google Scholar] [CrossRef]
- Umbaugh, D.S.; Nguyen, N.T.; Jaeschke, H.; Ramachandran, A. Mitochondrial Membrane Potential Drives Early Change in Mitochondrial Morphology after Acetaminophen Exposure. Toxicol. Sci. 2021, 180, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dey, S.; Matsunami, H. Calreticulin: Roles in cell-surface protein expression. Membranes 2014, 4, 630–641. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen hepatotoxicity: A mitochondrial perspective. Adv. Pharmacol. 2019, 85, 195–219. [Google Scholar]
- Nguyen, N.T.; Du, K.; Akakpo, J.Y.; Umbaugh, D.S.; Jaeschke, H.; Ramachandran, A. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol. Lett. 2021, 338, 21–31. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef]
- Walther, D.M.; Rapaport, D. Biogenesis of mitochondrial outer membrane proteins. Biochim. Biophys. Acta 2009, 1793, 42–51. [Google Scholar] [CrossRef]
- Sepuri, N.B.; Yadav, S.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mitochondrial targeting of intact CYP2B1 and CYP2E1 and N-terminal truncated CYP1A1 proteins in Saccharomyces cerevisiae--role of protein kinase A in the mitochondrial targeting of CYP2E1. FEBS J. 2007, 274, 4615–4630. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Nakane, T.; Koike, I.; Yamamoto, T.; Sugiura, Y.; Harada, E.; Sugase, K.; Shimamura, T.; Ohmura, M.; Muraoka, K.; et al. Haem-dependent dimerization of PGRMC1/Sigma-2 receptor facilitates cancer proliferation and chemoresistance. Nat. Commun. 2016, 7, 11030. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xu, Y.; Guo, S.; He, X.; Sun, J.; Li, X.; Li, C.; Yin, W.; Cheng, X.; Jiang, H.; et al. Structures of adenosine receptor A(2B)R bound to endogenous and synthetic agonists. Cell Discov. 2022, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, P.R.; Meneely, K.M.; Scott, E.E. Structures of human cytochrome P-450 2E1. Insights into the binding of inhibitors and both small molecular weight and fatty acid substrates. J. Biol. Chem. 2008, 283, 33698–33707. [Google Scholar] [CrossRef]
- Knockaert, L.; Descatoire, V.; Vadrot, N.; Fromenty, B.; Robin, M.A. Mitochondrial CYP2E1 is sufficient to mediate oxidative stress and cytotoxicity induced by ethanol and acetaminophen. Toxicol. In Vitro 2011, 25, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Buters, J.T.; Schiller, C.D.; Chou, R.C. A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem. Pharmacol. 1993, 46, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K.; Crespi, C.L.; Waxman, D.J. Determination of CYP2B6 component of 7-ethoxy-4-trifluoromethylcoumarin O-deethylation activity in human liver microsomes. Methods Mol. Biol. 2006, 320, 97–102. [Google Scholar] [PubMed]
- Vecchio, E.A.; White, P.J.; May, L.T. The adenosine A(2B) G protein-coupled receptor: Recent advances and therapeutic implications. Pharmacol. Ther. 2019, 198, 20–33. [Google Scholar] [CrossRef]
- Mundell, S.; Kelly, E. Adenosine receptor desensitization and trafficking. Biochim. Biophys. Acta 2011, 1808, 1319–1328. [Google Scholar] [CrossRef]
- Rosenberger, P.; Schwab, J.M.; Mirakaj, V.; Masekowsky, E.; Mager, A.; Morote-Garcia, J.C.; Unertl, K.; Eltzschig, H.K. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 2009, 10, 195–202. [Google Scholar] [CrossRef]
- Knowles, H.J. The Adenosine A(2B) Receptor Drives Osteoclast-Mediated Bone Resorption in Hypoxic Microenvironments. Cells 2019, 8, 624. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Sitkovsky, M.V. Adenosine A2A receptor is involved in cell surface expression of A2B receptor. J. Biol. Chem. 2010, 285, 39271–39288. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.K.; Field, M.C.; Tippins, J.R. Regulation of thromboxane receptor signaling at multiple levels by oxidative stress-induced stabilization, relocation and enhanced responsiveness. PLoS ONE 2010, 5, e12798. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Alonso, V.; Esbrit, P.; Friedman, P.A. Oxidation inhibits PTH receptor signaling and trafficking. Biochem. Biophys. Res. Commun. 2017, 482, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Hu, R.K.; Xia, G.C.; Yan, S.H.; Ren, Q.L.; Zhao, J.; Wang, F.H.; Huang, C.C.; Yao, Q.; Tan, Y.; et al. Tyrosine nitrations impaired intracellular trafficking of FSHR to the cell surface and FSH-induced Akt-FoxO3a signaling in human granulosa cells. Aging 2019, 11, 3094–3116. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat. Res. 2012, 751, 77–81. [Google Scholar] [CrossRef] [PubMed]
- St. Hilaire, C.; Koupenova, M.; Carroll, S.H.; Smith, B.D.; Ravid, K. TNF-alpha upregulates the A2B adenosine receptor gene: The role of NAD(P)H oxidase 4. Biochem. Biophys. Res. Commun. 2008, 375, 292–296. [Google Scholar] [CrossRef]
- Herring, S.E.; Mao, S.; Bhalla, M.; Tchalla, E.Y.I.; Kramer, J.M.; Bou Ghanem, E.N. Mitochondrial ROS production by neutrophils is required for host antimicrobial function against Streptococcus pneumoniae and is controlled by A2B adenosine receptor signaling. PLoS Pathog. 2022, 18, e1010700. [Google Scholar] [CrossRef]
- Jaeschke, H.; Duan, L.; Nguyen, N.; Ramachandran, A. Mitochondrial Damage and Biogenesis in Acetaminophen-induced Liver Injury. Liver Res. 2019, 3, 150–156. [Google Scholar] [CrossRef]
- Du, K.; McGill, M.R.; Xie, Y.; Bajt, M.L.; Jaeschke, H. Resveratrol prevents protein nitration and release of endonucleases from mitochondria during acetaminophen hepatotoxicity. Food Chem. Toxicol. 2015, 81, 62–70. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.A. Choose your partners for the next dance: Implied PGRMC1 roles in membrane trafficking and mitochondrial modulation. Fertil. Steril. 2020, 113, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Powell, D.W.; Bard, M.; Eckstein, J.; Barbuch, R.; Link, A.J.; Espenshade, P.J. Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metab. 2007, 5, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Debose-Boyd, R.A. A helping hand for cytochrome p450 enzymes. Cell Metab. 2007, 5, 81–83. [Google Scholar] [CrossRef]
- Cahill, M.A. Progesterone receptor membrane component 1: An integrative review. J. Steroid Biochem. Mol. Biol. 2007, 105, 16–36. [Google Scholar] [CrossRef]
- Ranganathan, P.; Mohamed, R.; Jayakumar, C.; Ramesh, G. Guidance cue netrin-1 and the regulation of inflammation in acute and chronic kidney disease. Mediat. Inflamm. 2014, 2014, 525891. [Google Scholar] [CrossRef]
- Lee, S.R.; Heo, J.H.; Jo, S.L.; Kim, G.; Kim, S.J.; Yoo, H.J.; Lee, K.P.; Kwun, H.J.; Shin, H.J.; Baek, I.J.; et al. Progesterone receptor membrane component 1 reduces cardiac steatosis and lipotoxicity via activation of fatty acid oxidation and mitochondrial respiration. Sci. Rep. 2021, 11, 8781. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Guerrero, G.; Umbaugh, D.S.; Ramachandran, A.A.; Artigues, A.; Jaeschke, H.; Ramachandran, A. Translocation of Adenosine A2B Receptor to Mitochondria Influences Cytochrome P450 2E1 Activity after Acetaminophen Overdose. Livers 2024, 4, 15-30. https://doi.org/10.3390/livers4010002
Sanchez-Guerrero G, Umbaugh DS, Ramachandran AA, Artigues A, Jaeschke H, Ramachandran A. Translocation of Adenosine A2B Receptor to Mitochondria Influences Cytochrome P450 2E1 Activity after Acetaminophen Overdose. Livers. 2024; 4(1):15-30. https://doi.org/10.3390/livers4010002
Chicago/Turabian StyleSanchez-Guerrero, Giselle, David S. Umbaugh, Abhay A. Ramachandran, Antonio Artigues, Hartmut Jaeschke, and Anup Ramachandran. 2024. "Translocation of Adenosine A2B Receptor to Mitochondria Influences Cytochrome P450 2E1 Activity after Acetaminophen Overdose" Livers 4, no. 1: 15-30. https://doi.org/10.3390/livers4010002