
Role of Pyroptosis in Acetaminophen-Induced Hepatotoxicity


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pyroptotic Cell Death Signaling
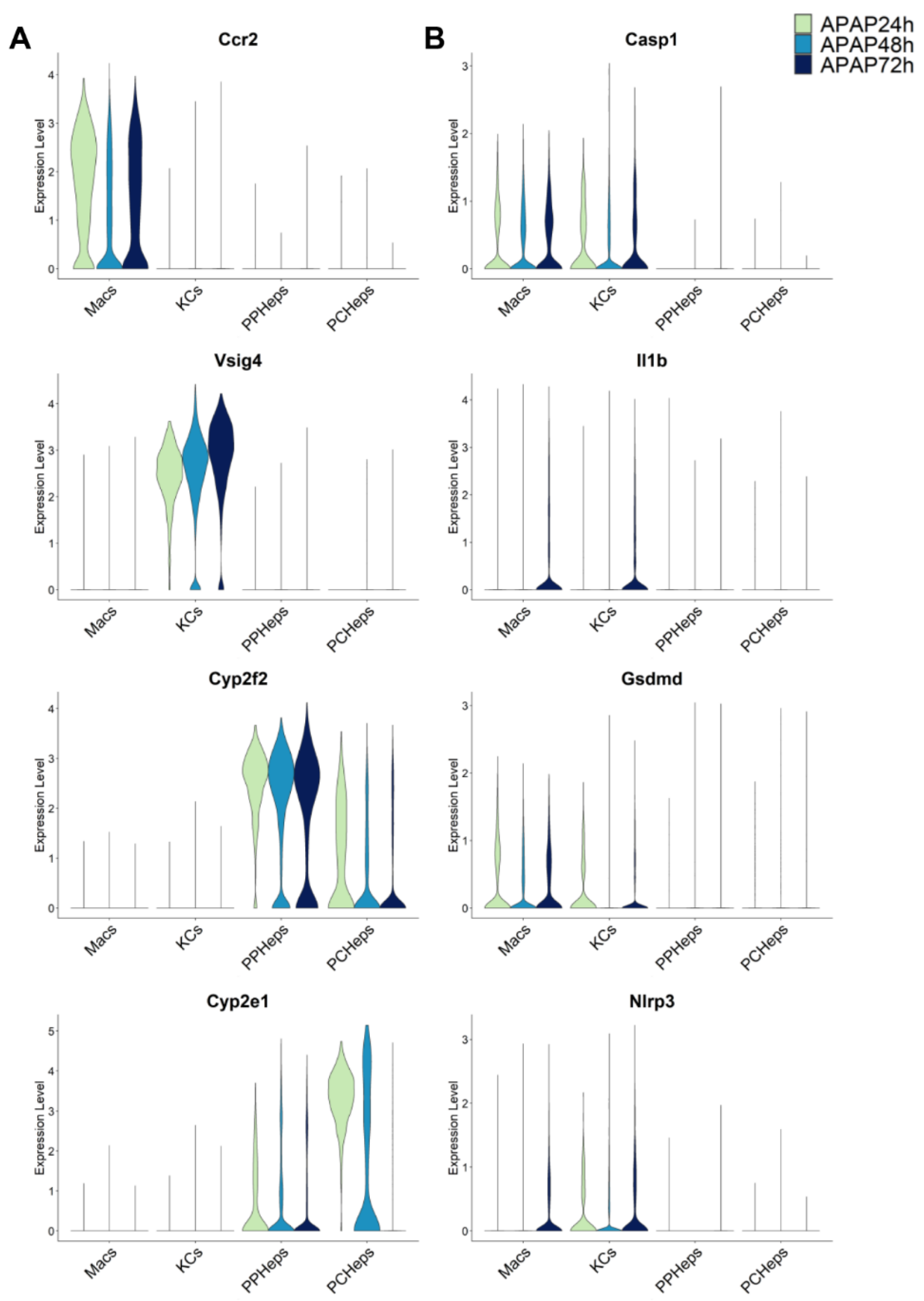
3. Evidence for Pyroptosis in APAP Toxicity
4. Evidence against Pyroptosis in Acetaminophen-Induced Liver Injury
5. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jaeschke, H. Acetaminophen: Dose-Dependent Drug Hepatotoxicity and Acute Liver Failure in Patients. Dig. Dis. 2015, 33, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Wendon, J. Acute liver failure. N. Engl. J. Med. 2013, 369, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Stravitz, R.T.; Lee, W.M. Acute liver failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185–194. [Google Scholar]
- Jollow, D.J.; Mitchell, J.R.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [Google Scholar]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Rumack, B.H.; Bateman, D.N. Acetaminophen and acetylcysteine dose and duration: Past, present and future. Clin. Toxicol. 2012, 50, 91–98. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Du, K.; Akakpo, J.Y.; Umbaugh, D.S.; Jaeschke, H.; Ramachandran, A. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol. Lett. 2021, 338, 21–31. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, C.; Lemasters, J.J.; Jaeschke, H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2010, 246, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, K.; Kim, J.S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Akakpo, J.Y.; Ramachandran, A.; Kandel, S.E.; Ni, H.M.; Kumer, S.C.; Rumack, B.H.; Jaeschke, H. 4-Methylpyrazole protects against acetaminophen hepatotoxicity in mice and in primary human hepatocytes. Hum. Exp. Toxicol. 2018, 37, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.M.; Padilla-Jones, A.; Fisher, E.S.; Akakpo, J.Y.; Jaeschke, H.; Rumack, B.H.; Gerkin, R.D.; Curry, S.C. The Effect of 4-Methylpyrazole on Oxidative Metabolism of Acetaminophen in Human Volunteers. J. Med. Toxicol. 2020, 16, 169–176. [Google Scholar] [CrossRef]
- Akakpo, J.Y.; Ramachandran, A.; Duan, L.; Schaich, M.A.; Jaeschke, M.W.; Freudenthal, B.D.; Ding, W.X.; Rumack, B.H.; Jaeschke, H. Delayed Treatment With 4-Methylpyrazole Protects Against Acetaminophen Hepatotoxicity in Mice by Inhibition of c-Jun n-Terminal Kinase. Toxicol. Sci. 2019, 170, 57–68. [Google Scholar] [CrossRef]
- Akakpo, J.Y.; Ramachandran, A.; Curry, S.C.; Rumack, B.H.; Jaeschke, H. Comparing N-acetylcysteine and 4-methylpyrazole as antidotes for acetaminophen overdose. Arch. Toxicol. 2022, 96, 453–465. [Google Scholar] [CrossRef]
- Kaiser, S.K.; Dart, R.C. The roles of antidotes in emergency situations. Emerg. Med. Clin. N. Am. 2022, 40, 381–394. [Google Scholar] [CrossRef]
- Farber, J.L. The biochemical pathology of toxic cell death. Monogr. Pathol. 1985, 26, 19–31. [Google Scholar]
- Poli, G.; Albano, E.; Dianzani, M.U. The role of lipid peroxidation in liver damage. Chem. Phys. Lipids 1987, 45, 117–142. [Google Scholar] [CrossRef]
- Jaeschke, H.; Bajt, M.L. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 2006, 89, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, A.; Jaeschke, H. Acetaminophen hepatotoxicity. Semin. Liver Dis. 2019, 39, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H.; Ramachandran, A.; Chao, X.; Ding, W.X. Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch. Toxicol. 2019, 93, 3491–3502. [Google Scholar] [CrossRef]
- Iorga, A.; Dara, L. Cell death in drug-induced liver injury. Adv. Pharmacol. 2019, 85, 31–74. [Google Scholar]
- Ray, S.D.; Mumaw, V.R.; Raje, R.R.; Fariss, M.W. Protection of acetaminophen-induced hepatocellular apoptosis and necrosis by cholesteryl hemisuccinate pretreatment. J. Pharmacol. Exp. Ther. 1996, 279, 1470–1483. [Google Scholar]
- El-Hassan, H.; Anwar, K.; Macanas-Pirard, P.; Crabtree, M.; Chow, S.C.; Johnson, V.L.; Lee, P.C.; Hinton, R.H.; Price, S.C.; Kass, G.E. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: Roles of cytochrome c, Bax, Bid, and caspases. Toxicol. Appl. Pharmacol. 2003, 191, 118–129. [Google Scholar] [CrossRef]
- Gujral, J.S.; Knight, T.R.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Mode of cell death after acetaminophen overdose in mice: Apoptosis or oncotic necrosis? Toxicol. Sci. 2002, 67, 322–328. [Google Scholar] [CrossRef]
- Jaeschke, H.; Duan, L.; Akakpo, J.Y.; Farhood, A.; Ramachandran, A. The role of apoptosis in acetaminophen hepatotoxicity. Food Chem. Toxicol. 2018, 118, 709–718. [Google Scholar] [CrossRef]
- Zhang, Y.F.; He, W.; Zhang, C.; Liu, X.J.; Lu, Y.; Wang, H.; Zhang, Z.H.; Chen, X.; Xu, D.X. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol. Lett. 2014, 225, 445–453. [Google Scholar] [CrossRef]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Barilla, R.; et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015, 6, e1759. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, T.R.; Fariss, M.W.; Farhood, A.; Jaeschke, H. Role of lipid peroxidation as a mechanism of liver injury after acetaminophen overdose in mice. Toxicol. Sci. 2003, 76, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, K.; Kim, J.S.; Uchiyama, A.; Jaeschke, H.; Lemasters, J.J. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol. Sci. 2010, 117, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kholmukhamedov, A.; Lindsey, C.C.; Beeson, C.C.; Jaeschke, H.; Lemasters, J.J. Translocation of iron from lysosomes to mitochondria during acetaminophen-induced hepatocellular injury: Protection by starch-desferal and minocycline. Free Radic. Biol. Med. 2016, 97, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Adelusi, O.B.; Ramachandran, A.; Lemasters, J.J.; Jaeschke, H. The role of iron in lipid peroxidation and protein nitration during acetaminophen-induced liver injury in mice. Toxicol. Appl. Pharmacol. 2022, 445, 116043. [Google Scholar] [CrossRef]
- Jaeschke, H.; Adelusi, O.B.; Ramachandran, A. Ferroptosis and Acetaminophen Hepatotoxicity: Are We Going Down Another Rabbit Hole? Gene Expr. 2021, 20, 169–178. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Wang, Z.; Sun, R.; Zou, B.; Li, R.; Liu, D.; Lin, M.; Zhou, J.; Ning, S.; et al. Peroxiredoxin 3 Inhibits Acetaminophen-Induced Liver Pyroptosis Through the Regulation of Mitochondrial ROS. Front. Immunol. 2021, 12, 652782. [Google Scholar] [CrossRef]
- Liu, T.; Yang, L.; Gao, H.; Zhuo, Y.; Tu, Z.; Wang, Y.; Xun, J.; Zhang, Q.; Zhang, L.; Wang, X. 3,4-dihydroxyphenylethyl alcohol glycoside reduces acetaminophen-induced acute liver failure in mice by inhibiting hepatocyte ferroptosis and pyroptosis. PeerJ 2022, 10, e13082. [Google Scholar] [CrossRef] [PubMed]
- Rousta, A.M.; Mirahmadi, S.M.; Shahmohammadi, A.; Mehrabi, Z.; Fallah, S.; Baluchnejadmojarad, T.; Roghani, M. Therapeutic Potential of Isorhamnetin following Acetaminophen-Induced Hepatotoxicity through Targeting NLRP3/NF-κB/Nrf2. Drug Res. 2022, 72, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [Green Version]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Torre-Minguela, C.; Barberà-Cremades, M.; Gómez, A.I.; Martín-Sánchez, F.; Pelegrín, P. Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci. Rep. 2016, 6, 22586. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Rühl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, M.; Ferrer-Sueta, G.; Radi, R. Kinetic studies on peroxynitrite reduction by peroxiredoxins. Methods Enzymol. 2008, 441, 173–196. [Google Scholar]
- Trujillo, M.; Ferrer-Sueta, G.; Thomson, L.; Flohé, L.; Radi, R. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell. Biochem. 2007, 44, 83–113. [Google Scholar]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, T.R.; Ho, Y.S.; Farhood, A.; Jaeschke, H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: Protection by glutathione. J. Pharmacol. Exp. Ther. 2002, 303, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, K.; Kumagai, K.; Ito, K.; Arakawa, S.; Ando, Y.; Oda, S.; Yamoto, T.; Manabe, S. Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. Toxicol. Pathol. 2009, 37, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Lebofsky, M.; Weinman, S.A.; Jaeschke, H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2011, 251, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.; Farhood, A.; Jaeschke, H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch. Toxicol. 2017, 91, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Farhood, A.; Jaeschke, H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol. Appl. Pharmacol. 2010, 247, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, H.; Zhu, J.; Nan, N.; Lin, Y.; Zhuang, X.; Li, L.; Zhang, Y.; Huang, P. Inhibition of TWEAK/Tnfrsf12a axis protects against acute liver failure by suppressing RIPK1-dependent apoptosis. Cell Death Discov. 2022, 8, 328. [Google Scholar] [CrossRef]
- Yang, C.; Sun, P.; Deng, M.; Loughran, P.; Li, W.; Yi, Z.; Li, S.; Zhang, X.; Fan, J.; Billiar, T.R.; et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis. 2019, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Concha, N.O.; Abdel-Meguid, S.S. Controlling apoptosis by inhibition of caspases. Curr. Med. Chem. 2002, 9, 713–726. [Google Scholar] [CrossRef]
- Jaeschke, H.; Fisher, M.; Lawson, J.A.; Simmons, C.A.; Farhood, A.; Jones, D.A. Activation of caspase 3 (CPP32)-like proteases is essential for TNF-alpha-induced hepatic parenchymal cell apoptosis and neutrophil-mediated necrosis in a murine endotoxin shock model. J. Immunol. 1998, 160, 3480–3486. [Google Scholar] [PubMed]
- Jaeschke, H.; Farhood, A.; Cai, S.X.; Tseng, B.Y.; Bajt, M.L. Protection against TNF-induced liver parenchymal cell apoptosis during endotoxemia by a novel caspase inhibitor in mice. Toxicol. Appl. Pharmacol. 2000, 169, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Bajt, M.L.; Vonderfecht, S.L.; Jaeschke, H. Differential protection with inhibitors of caspase-8 and caspase-3 in murine models of tumor necrosis factor and Fas receptor-mediated hepatocellular apoptosis. Toxicol. Appl. Pharmacol. 2001, 175, 243–252. [Google Scholar] [CrossRef]
- Rodriguez, I.; Matsuura, K.; Ody, C.; Nagata, S.; Vassalli, P. Systemic injection of a tripeptide inhibits the intracellular activation of CPP32-like proteases in vivo and fully protects mice against Fas-mediated fulminant liver destruction and death. J. Exp. Med. 1996, 184, 2067–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Künstle, G.; Leist, M.; Uhlig, S.; Revesz, L.; Feifel, R.; MacKenzie, A.; Wendel, A. ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-alpha. Immunol. Lett. 1997, 55, 5–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajt, M.L.; Lawson, J.A.; Vonderfecht, S.L.; Gujral, J.S.; Jaeschke, H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: Evidence for a postmitochondrial processing of caspase-8. Toxicol. Sci. 2000, 58, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, J.A.; Fisher, M.A.; Simmons, C.A.; Farhood, A.; Jaeschke, H. Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol. Appl. Pharmacol. 1999, 156, 179–186. [Google Scholar] [CrossRef]
- Jaeschke, H.; Cover, C.; Bajt, M.L. Role of caspases in acetaminophen-induced liver injury. Life Sci. 2006, 78, 1670–1676. [Google Scholar] [CrossRef]
- Hu, B.; Colletti, L.M. CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. Hepatology 2010, 52, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Williams, C.D.; Farhood, A. No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatology 2011, 53, 718–719. [Google Scholar] [CrossRef] [Green Version]
- Kelava, T.; Cavar, I.; Culo, F. Influence of small doses of various drug vehicles on acetaminophen-induced liver injury. Can. J. Physiol. Pharmacol. 2010, 88, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, J.; Du, J.; Zhuo, Z.; Yang, S.; Zhang, W.; Wang, W.; Zhang, S.; Iwakura, Y.; Meng, G.; et al. Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell. Mol. Immunol. 2018, 15, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Nguyen, N.T.; McGill, M.R.; Sharpe, M.R.; Curry, S.C.; Jaeschke, H. Generation of pro-and anti-inflammatory mediators after acetaminophen overdose in surviving and non-surviving patients. Toxicol. Lett. 2022, 367, 59–66. [Google Scholar] [CrossRef]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, P.E.; Oliveira, A.G.; Pereira, R.V.; David, B.A.; Gomides, L.F.; Saraiva, A.M.; Pires, D.A.; Novaes, J.T.; Patricio, D.O.; Cisalpino, D.; et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2015, 61, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Umbaugh, D.S.; Ramachandran, A.; Jaeschke, H. Spatial Reconstruction of the Early Hepatic Transcriptomic Landscape After an Acetaminophen Overdose Using Single-Cell RNA-Sequencing. Toxicol. Sci. 2021, 182, 327–345. [Google Scholar] [CrossRef]
- Walesky, C.M.; Kolb, K.E.; Winston, C.L.; Henderson, J.; Kruft, B.; Fleming, I.; Ko, S.; Monga, S.P.; Mueller, F.; Apte, U.; et al. Functional compensation precedes recovery of tissue mass following acute liver injury. Nat. Commun. 2020, 11, 5785. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Veg, T.; Manco, R.; Dan, S.; Papinutti, D.; Lifshitz, A.; Kolodziejczyk, A.A.; Bahar Halpern, K.; Elinav, E.; Itzkovitz, S. The spatiotemporal program of zonal liver regeneration following acute injury. Cell Stem Cell 2022, 29, 973–989.e10. [Google Scholar] [CrossRef]
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.T.; Floros, K.V.; Elkholi, R.; Corrigan, K.A.; Kushnareva, Y.; Wieder, S.Y.; Lindtner, C.; Serasinghe, M.N.; Asciolla, J.J.; Buettner, C.; et al. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol. Cell 2015, 57, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajt, M.L.; Farhood, A.; Lemasters, J.J.; Jaeschke, H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 2008, 324, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A. Acetaminophen-induced apoptosis: Facts versus fiction. J. Clin. Transl. Res. 2020, 6, 36–47. [Google Scholar] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaeschke, H.; Umbaugh, D.S.; Ramachandran, A. Role of Pyroptosis in Acetaminophen-Induced Hepatotoxicity. Livers 2022, 2, 425-435. https://doi.org/10.3390/livers2040032
Jaeschke H, Umbaugh DS, Ramachandran A. Role of Pyroptosis in Acetaminophen-Induced Hepatotoxicity. Livers. 2022; 2(4):425-435. https://doi.org/10.3390/livers2040032
Chicago/Turabian StyleJaeschke, Hartmut, David S. Umbaugh, and Anup Ramachandran. 2022. "Role of Pyroptosis in Acetaminophen-Induced Hepatotoxicity" Livers 2, no. 4: 425-435. https://doi.org/10.3390/livers2040032