
Effect of Dissolution Time on the Development of All-Cellulose Composites Using the NaOH/Urea Solvent System
 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of All-Cellulose Composites
2.3. Morphological and Structural Characterization of All-Cellulose Composites
2.3.1. Determination of Porosity and Size Reduction
2.3.2. Scanning Electron Microscopy (SEM) Analysis
2.3.3. Brunauer-Emmett-Teller (BET) Surface Area Determination
2.3.4. X-ray Diffraction (XRD) Analysis
2.4. Optical Characterization of All-Cellulose Composites
2.5. Water Vapor Sorption of All-Cellulose Composites
2.6. Mechanical Properties of All-Cellulose Composites
2.7. Statistical Analysis
3. Results
3.1. Morphology
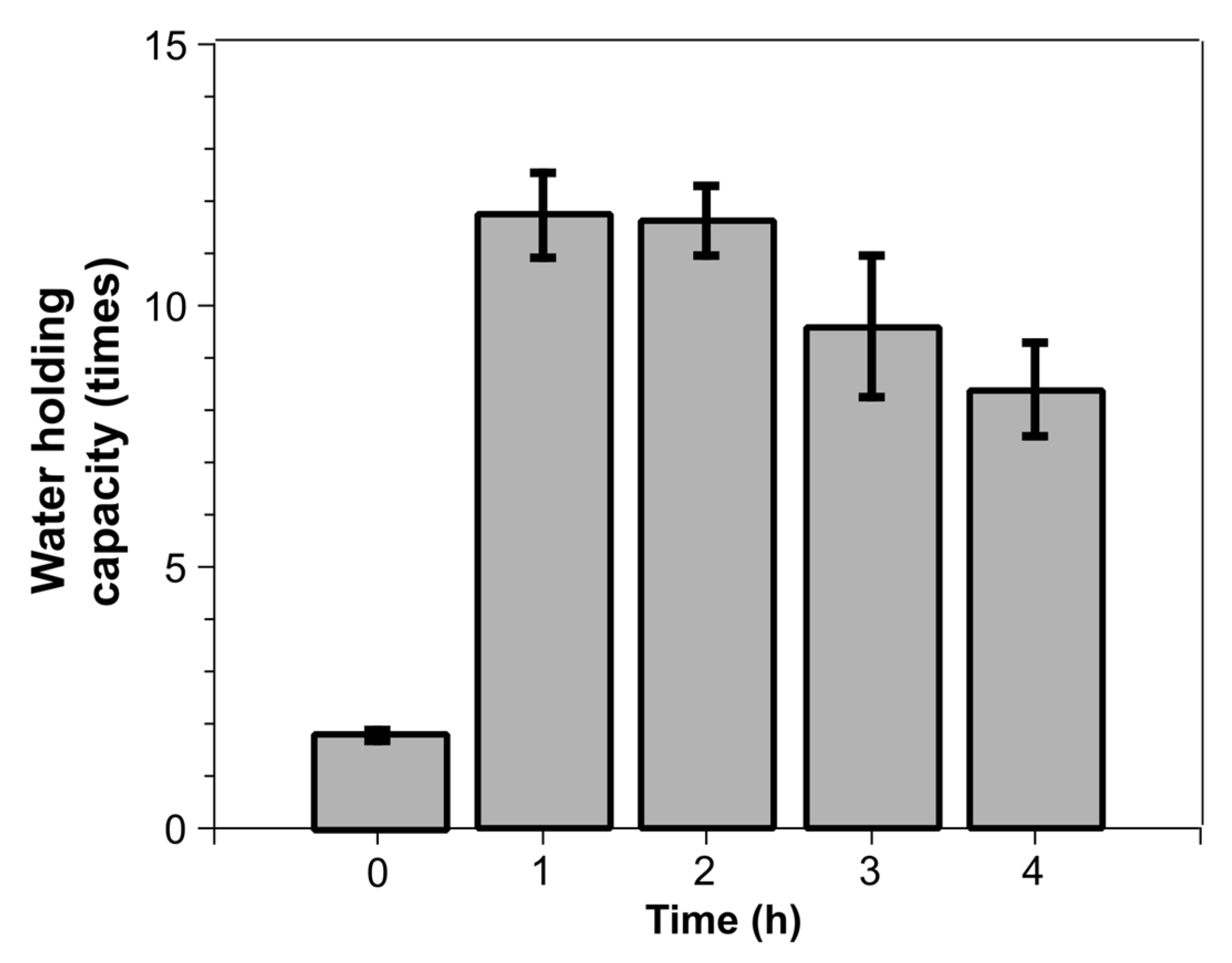
3.2. Water Holding Capacity
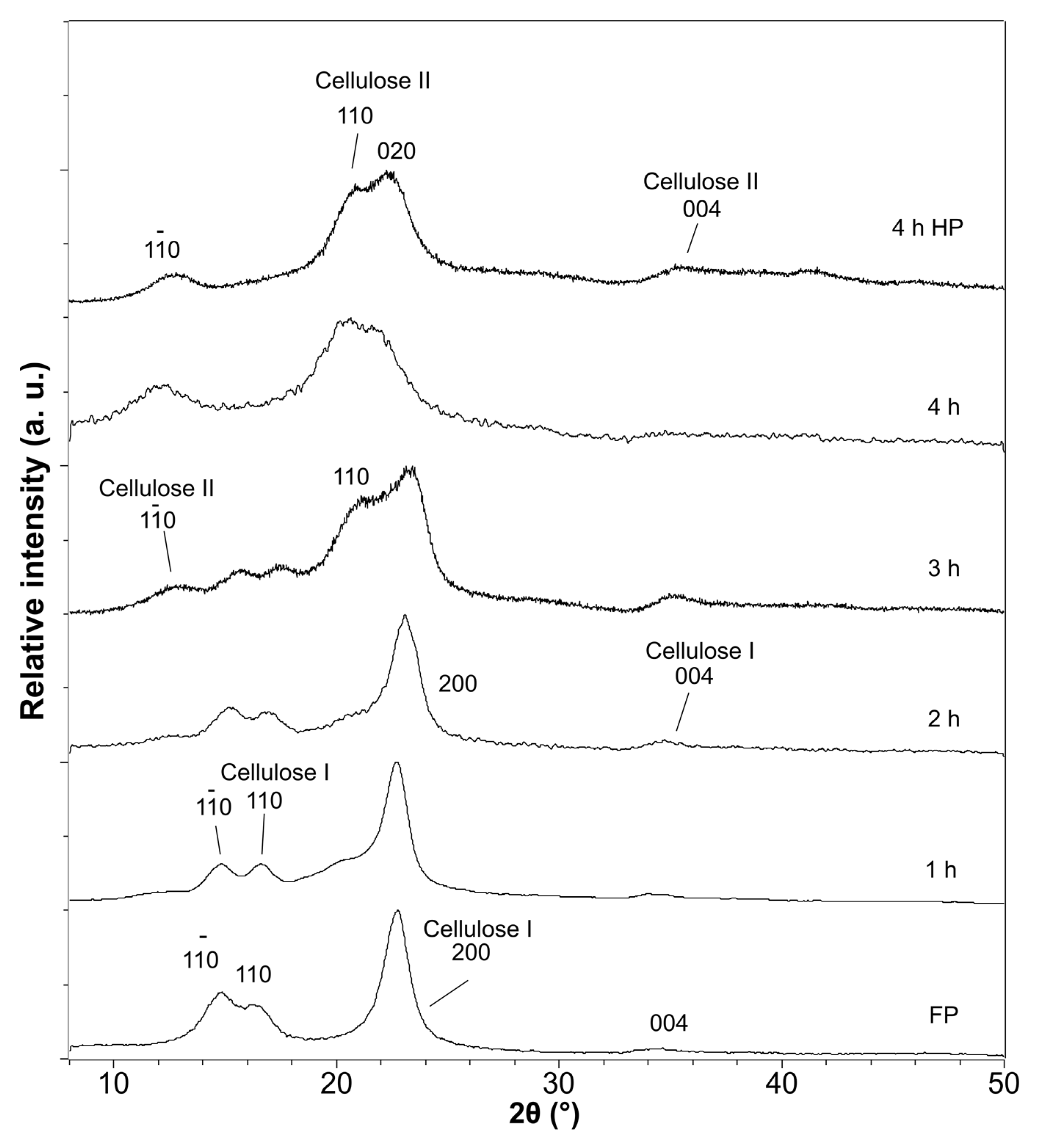
3.3. Crystalline Structure
3.4. Opacity
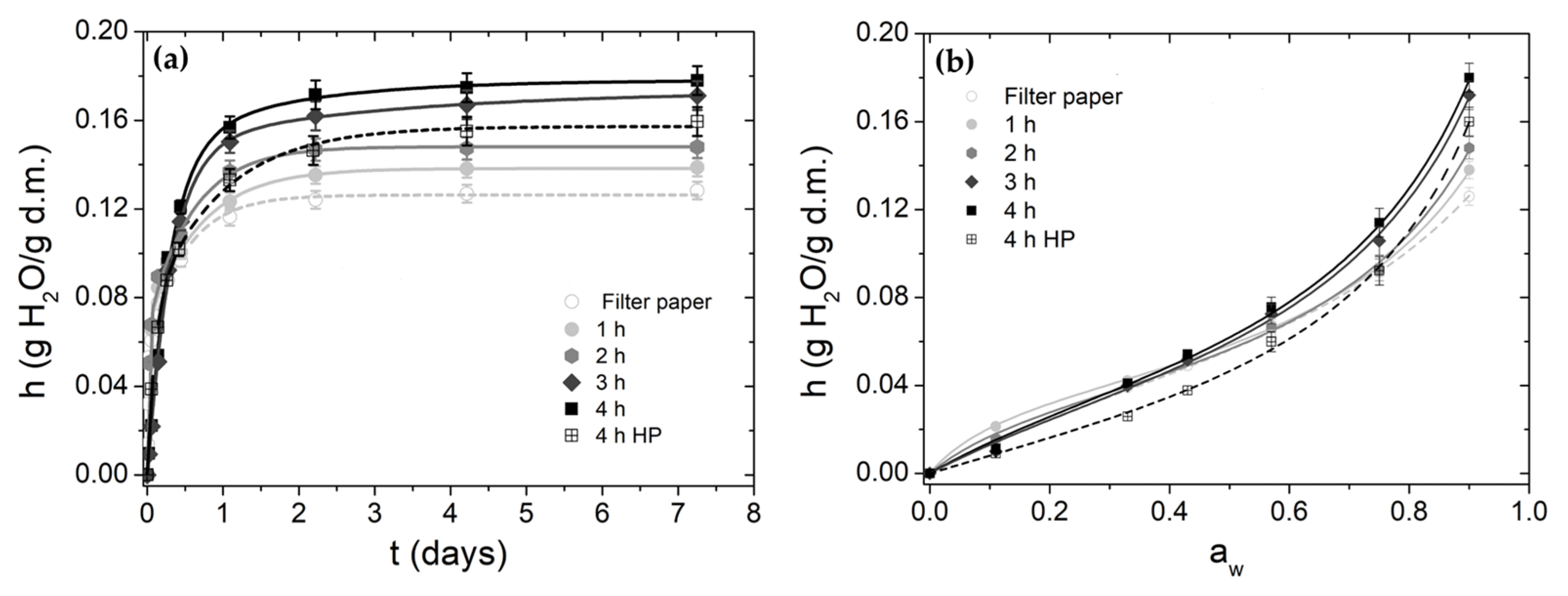
3.5. Water Vapor Sorption
3.6. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mukhopadhyay, S.; Adak, B. Single-Polymer Composites; Chapman and Hall/CRC: Milton, UK, 2018; ISBN 9781351272230. [Google Scholar]
- Capiati, N.J.; Porter, R.S. The Concept of One Polymer Composites Modelled with High Density Polyethylene. J. Mater. Sci. 1975, 10, 1671–1677. [Google Scholar] [CrossRef]
- Matabola, K.P.; De Vries, A.R.; Moolman, F.S.; Luyt, A.S. Single Polymer Composites: A Review. J. Mater. Sci. 2009, 44, 6213–6222. [Google Scholar] [CrossRef]
- Heinze, T.; Koschella, A. Solvents Applied in the Field of Cellulose Chemistry: A Mini Review. Polímeros 2005, 15, 84–90. [Google Scholar] [CrossRef]
- Qi, H.; Yang, Q.; Zhang, L.; Liebert, T.; Heinze, T. The Dissolution of Cellulose in NaOH-Based Aqueous System by Two-Step Process. Cellulose 2011, 18, 237–245. [Google Scholar] [CrossRef]
- Xiong, B.; Zhao, P.; Hu, K.; Zhang, L.; Cheng, G. Dissolution of Cellulose in Aqueous NaOH/Urea Solution: Role of Urea. Cellulose 2014, 21, 1183–1192. [Google Scholar] [CrossRef]
- Budtova, T.; Navard, P. Cellulose in NaOH—Water Based Solvents: A Review. Cellulose 2017, 23, 6–7. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Fang, Y.; Xiang, J.; Ma, Y.; Lu, A.; Kang, H.; Huang, Y.; Guo, H.; Liu, R.; Zhang, L. Intermolecular Interactions and 3D Structure in Cellulose-NaOH-Urea Aqueous System. J. Phys. Chem. B 2014, 118, 10250–10257. [Google Scholar] [CrossRef]
- Li, F.; You, X.; Li, Q.; Qin, D.; Wang, M.; Yuan, S.; Chen, X.; Bi, S. Homogeneous Deacetylation and Degradation of Chitin in NaOH/Urea Dissolution System. Int. J. Biol. Macromol. 2021, 189, 391–397. [Google Scholar] [CrossRef]
- Piltonen, P.; Hildebrandt, N.C.; Westerlind, B.; Valkama, J.P.; Tervahartiala, T.; Illikainen, M. Green and Efficient Method for Preparing All-Cellulose Composites with NaOH/Urea Solvent. Compos. Sci. Technol. 2016, 135, 153–158. [Google Scholar] [CrossRef]
- Labidi, K.; Korhonen, O.; Zrida, M.; Hamzaoui, A.H.; Budtova, T. All-Cellulose Composites from Alfa and Wood Fibers. Ind. Crops Prod. 2019, 127, 135–141. [Google Scholar] [CrossRef]
- Wei, Q.Y.; Lin, H.; Yang, B.; Li, L.; Zhang, L.Q.; Huang, H.D.; Zhong, G.J.; Xu, L.; Li, Z.M. Structure and Properties of All-Cellulose Composites Prepared by Controlling the Dissolution Temperature of a NaOH/Urea Solvent. Ind. Eng. Chem. Res. 2020, 59, 10428–10435. [Google Scholar] [CrossRef]
- Duchemin, B.; Le Corre, D.; Leray, N.; Dufresne, A.; Staiger, M.P. All-Cellulose Composites Based on Microfibrillated Cellulose and Filter Paper via a NaOH-Urea Solvent System. Cellulose 2016, 23, 593–609. [Google Scholar] [CrossRef]
- Nishino, T.; Arimoto, N. All-Cellulose Composite Prepared by Selective Dissolving of Fiber Surface. Biomacromolecules 2007, 8, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- ISO 5351:2010; Pulps—Determination of Limiting Viscosity Number in Cupri-Ethylenediamine (CED) Solution. ISO: Geneva, Switzerland, 2010.
- Kes, M.; Christensen, B.E. A Re-Investigation of the Mark-Houwink-Sakurada Parameters for Cellulose in Cuen: A Study Based on Size-Exclusion Chromatography Combined with Multi-Angle Light Scattering and Viscometry. J. Chromatogr. A 2013, 1281, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Cottet, C.; Salvay, A.G.; Peltzer, M.A.; Fernández-García, M. Incorporation of Poly(Itaconic Acid) with Quaternized Thiazole Groups on Gelatin-Based Films for Antimicrobial-Active Food Packaging. Polymers 2021, 13, 200. [Google Scholar] [CrossRef]
- Alcalá, M.; González, I.; Boufi, S.; Vilaseca, F.; Mutjé, P. All-Cellulose Composites from Unbleached Hardwood Kraft Pulp Reinforced with Nanofibrillated Cellulose. Cellulose 2013, 20, 2909–2921. [Google Scholar] [CrossRef]
- Mwaikambo, L.Y.; Ansell, M.P. The Determination of Porosity and Cellulose Content of Plant Fibers by Density Methods. J. Mater. Sci. Lett. 2001, 20, 2095–2096. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Yam, K.L.; Papadakis, S.E. A Simple Digital Imaging Method for Measuring and Analyzing Color of Food Surfaces. J. Food Eng. 2004, 61, 137–142. [Google Scholar] [CrossRef]
- Riquelme, N.; Díaz-Calderón, P.; Enrione, J.; Matiacevich, S. Effect of Physical State of Gelatin-Plasticizer Based Films on to the Occurrence of Maillard Reactions. Food Chem. 2015, 175, 478–484. [Google Scholar] [CrossRef]
- Delgado, J.F.; Sceni, P.; Peltzer, M.A.; Salvay, A.G.; de la Osa, O.; Wagner, J.R. Development of Innovative Biodegradable Films Based on Biomass of Saccharomyces Cerevisiae. Innov. Food Sci. Emerg. Technol. 2016, 36, 83–91. [Google Scholar] [CrossRef]
- Roos, Y.H.; Drusch, S. Phase Transitions in Foods, 2nd ed.; Academic Press: Cambridge, MA, USA, 2015; 367p. [Google Scholar] [CrossRef]
- Spiess, W.E.L.; Wolf, W.R. Results of the COST 90 Project on Water Activity. Phys. Prop. Foods 1983, 2013, 65–91. [Google Scholar]
- Bedane, A.H.; Eić, M.; Farmahini-Farahani, M.; Xiao, H. Water Vapor Transport Properties of Regenerated Cellulose and Nanofibrillated Cellulose Films. J. Memb. Sci. 2015, 493, 46–57. [Google Scholar] [CrossRef]
- Stępień, A.; Witczak, M.; Witczak, T. Moisture Sorption Characteristics of Food Powders Containing Freeze Dried Avocado, Maltodextrin and Inulin. Int. J. Biol. Macromol. 2020, 149, 256–261. [Google Scholar] [CrossRef]
- ASTM D1708; Standard Test Method for Tensile Properties of Plastics by Use of Microtensile Specimens. ASTM International: West Conshohocken, PA, USA, 2018.
- Adak, B.; Mukhopadhyay, S. All-Cellulose Composite Laminates with Low Moisture and Water Sensitivity. Polymer 2018, 141, 79–85. [Google Scholar] [CrossRef]
- Khakalo, A.; Tanaka, A.; Korpela, A.; Hauru, L.K.J.; Orelma, H. All-Wood Composite Material by Partial Fiber Surface Dissolution with an Ionic Liquid. ACS Sustain. Chem. Eng. 2019, 7, 3195–3202. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Yuan, Z.; Fan, Q.; Dai, X.; Zhao, Y.; Wang, Z.; Qin, M. Characterisation of Cellulose Films Regenerated from Acetone/Water Coagulants. Carbohydr. Polym. 2014, 102, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Kabir, S.M.F.; Sikdar, P.P.; Haque, B.; Bhuiyan, M.A.R.; Ali, A.; Islam, M.N. Cellulose-Based Hydrogel Materials: Chemistry, Properties and Their Prospective Applications. Prog. Biomater. 2018, 7, 153–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, A.D. Idealized Powder Diffraction Patterns for Cellulose Polymorphs. Cellulose 2014, 21, 885–896. [Google Scholar] [CrossRef]
- Nam, S.; French, A.D.; Condon, B.D.; Concha, M. Segal Crystallinity Index Revisited by the Simulation of X-Ray Diffraction Patterns of Cotton Cellulose Iβ and Cellulose II. Carbohydr. Polym. 2016, 135, 1–9. [Google Scholar] [CrossRef]
- Johnson Ford, E.N.; Mendon, S.K.; Thames, S.F.; Rawlins, J.W. X-ray Diffraction of Cotton Treated with Neutralized Vegetable Oil-Based Macromolecular Crosslinkers. J. Eng. Fiber. Fabr. 2010, 5, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhang, L. Rapid Dissolution of Cellulose in LiOH/Urea and NaOH/Urea Aqueous Solutions. Macromol. Biosci. 2005, 5, 539–548. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, C.; Feng, X.; Wu, M.; Tang, Y.; Li, B. Effect of Regeneration Solvent on the Characteristics of Regenerated Cellulose from Lithium Bromide Trihydrate Molten Salt. Cellulose 2020, 27, 9243–9256. [Google Scholar] [CrossRef]
- Li, R.; Wang, S.; Lu, A.; Zhang, L. Dissolution of Cellulose from Different Sources in an NaOH/Urea Aqueous System at Low Temperature. Cellulose 2015, 22, 339–349. [Google Scholar] [CrossRef]
- Nomura, S.; Kugo, Y.; Erata, T. 13C NMR and XRD Studies on the Enhancement of Cellulose II Crystallinity with Low Concentration NaOH Post-Treatments. Cellulose 2020, 27, 3553–3563. [Google Scholar] [CrossRef]
- Rana, A.K.; Frollini, E.; Thakur, V.K. Cellulose Nanocrystals: Pretreatments, Preparation Strategies, and Surface Functionalization. Int. J. Biol. Macromol. 2021, 182, 1554–1581. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.N.; Schliesser, J.; Mittal, A.; Decker, S.R.; Santos, A.F.L.O.M.; Freitas, V.L.S.; Urbas, A.; Lang, B.E.; Heiss, C.; Ribeiro Da Silva, M.D.M.C.; et al. A Thermodynamic Investigation of the Cellulose Allomorphs: Cellulose(Am), Cellulose Iβ(Cr), Cellulose II(Cr), and Cellulose III(Cr). J. Chem. Thermodyn. 2015, 81, 184–226. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chen, G.; Liu, Y.; Liu, Y.; Li, B.; Fang, Z. Transparent and Hazy All-Cellulose Composite Films with Superior Mechanical Properties. ACS Sustain. Chem. Eng. 2018, 6, 6974–6980. [Google Scholar] [CrossRef]
- Retegi, A.; Algar, I.; Martin, L.; Altuna, F.; Stefani, P.; Zuluaga, R.; Gañán, P.; Mondragon, I. Sustainable Optically Transparent Composites Based on Epoxidized Soy-Bean Oil (ESO) Matrix and High Contents of Bacterial Cellulose (BC). Cellulose 2012, 19, 103–109. [Google Scholar] [CrossRef]
- Chen, F.; Bouvard, J.L.; Sawada, D.; Pradille, C.; Hummel, M.; Sixta, H.; Budtova, T. Exploring Digital Image Correlation Technique for the Analysis of the Tensile Properties of All-Cellulose Composites. Cellulose 2021, 28, 4165–4178. [Google Scholar] [CrossRef]
- Pedreschi, F.; León, J.; Mery, D.; Moyano, P. Development of a Computer Vision System to Measure the Color of Potato Chips. Food Res. Int. 2006, 39, 1092–1098. [Google Scholar] [CrossRef]
- Mazeau, K. The Hygroscopic Power of Amorphous Cellulose: A Modeling Study. Carbohydr. Polym. 2015, 117, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.F.; Bovi, J.; Foresti, M.L.; Bernal, C. All-Cellulose Composites Derived from Natural Plant Fibers. In Sustainable Natural Fiber Composites; Khan, A., Manikandan, A., Ramesh, M., Khan, I., Asiri, A.M.A., Eds.; Materials Research Forum LLC: Millersville, PA, USA, 2022. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Porosity (%) | Size Reduction (%) |
|---|---|---|
| FP | 65 ± 1 | --- |
| 1 h | 63 ± 4 | 7 ± 5 |
| 2 h | 46 ± 5 | 53 ± 10 |
| 3 h | 28 ± 1 | 74 ± 6 |
| 4 h | 3 ± 6 | 79 ± 4 |
| 4 h HP | 4 ± 3 | 79 ± 5 |
| Sample | Opacity (Photographs) * | Opacity (Colorimeter) |
|---|---|---|
| FP | 0.94 ± 0.01 | 0.95 ± 0.01 |
| 1 h | 0.95 ± 0.01 | 0.96 ± 0.01 |
| 2 h | 0.94 ± 0.03 | 0.94 ± 0.03 |
| 3 h | 0.78 ± 0.07 | 0.86 ± 0.08 |
| 4 h | 0.71 ± 0.04 | 0.82 ± 0.04 |
| 4 h HP | 0.76 ± 0.06 | 0.74 ± 0.03 |
| Sample | Hydration Kinetics Parameters | Water Sorption Isotherms Parameters | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| h∞ (g/g) | τ1 (days) | Δh1/h∞ | τ2 (days) | Δh2/h∞ | τ (days) | R2 | N (g/g) | c | k | R2 | |
| FP | 0.126 ± 0.007 | 0.010 ± 0.002 | 0.44 ± 0.03 | 0.50 ± 0.05 | 0.56 ± 0.04 | 0.28 ± 0.02 | 0.996 | 0.052 ± 0.006 | 4.1 ± 0.8 | 0.70 ± 0.03 | 0.998 |
| 1 h | 0.138 ± 0.007 | 0.018 ± 0.003 | 0.49 ± 0.03 | 0.68 ± 0.08 | 0.51 ± 0.03 | 0.36 ± 0.04 | 0.996 | 0.040 ± 0.002 | 10.3 ± 1.2 | 0.80 ± 0.01 | 0.999 |
| 2 h | 0.148 ± 0.007 | 0.019 ± 0.003 | 0.51 ± 0.03 | 0.75 ± 0.07 | 0.49 ± 0.03 | 0.38 ± 0.04 | 0.996 | 0.040 ± 0.003 | 7.0 ± 1.3 | 0.82 ± 0.02 | 0.997 |
| 3 h | 0.175 ± 0.013 | 0.31 ± 0.03 | 0.83 ± 0.05 | 1.87 ± 0.31 | 0.17 ± 0.02 | 0.57 ± 0.07 | 0.998 | 0.048 ± 0.006 | 3.8 ± 1.0 | 0.82 ± 0.02 | 0.997 |
| 4 h | 0.180 ± 0.014 | 0.33 ± 0.05 | 0.85 ± 0.06 | 2.03 ± 0.20 | 0.15 ± 0.02 | 0.59 ± 0.07 | 0.998 | 0.049 ± 0.005 | 3.8 ± 1.2 | 0.83 ± 0.02 | 0.997 |
| 4 h HP | 0.162 ± 0.012 | 0.10 ± 0.01 | 0.47 ± 0.04 | 0.93 ± 0.08 | 0.53 ± 0.004 | 0.54 ± 0.08 | 0.998 | 0.043 ± 0.005 | 2.2 ± 0.6 | 0.85 ± 0.02 | 0.998 |
| Sample | Young’s Modulus (GPa) | Tensile Strength (MPa) | Strain at Break (mm/mm) |
|---|---|---|---|
| FP | 1.30 ± 0.13 a | 17.8 ± 2.1 a | 0.07 ± 0.01 a |
| 4 h | 0.53 ± 0.07 b | 14.5 ± 2.7 a | 0.09 ± 0.04 a |
| 4 h HP | 2.14 ± 0.33 c | 46.9 ± 5.4 b | 0.06 ± 0.02 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado, J.F.; Salvay, A.G.; Arroyo, S.; Bernal, C.R.; Foresti, M.L. Effect of Dissolution Time on the Development of All-Cellulose Composites Using the NaOH/Urea Solvent System. Polysaccharides 2023, 4, 65-77. https://doi.org/10.3390/polysaccharides4010005
Delgado JF, Salvay AG, Arroyo S, Bernal CR, Foresti ML. Effect of Dissolution Time on the Development of All-Cellulose Composites Using the NaOH/Urea Solvent System. Polysaccharides. 2023; 4(1):65-77. https://doi.org/10.3390/polysaccharides4010005
Chicago/Turabian StyleDelgado, Juan Francisco, Andrés Gerardo Salvay, Silvana Arroyo, Celina Raquel Bernal, and María Laura Foresti. 2023. "Effect of Dissolution Time on the Development of All-Cellulose Composites Using the NaOH/Urea Solvent System" Polysaccharides 4, no. 1: 65-77. https://doi.org/10.3390/polysaccharides4010005