
The Requirements of Managing Phase I Clinical Trials Risks: The British and Italian Case Studies

 , ,
, ,  ,
,  , , and
, , and 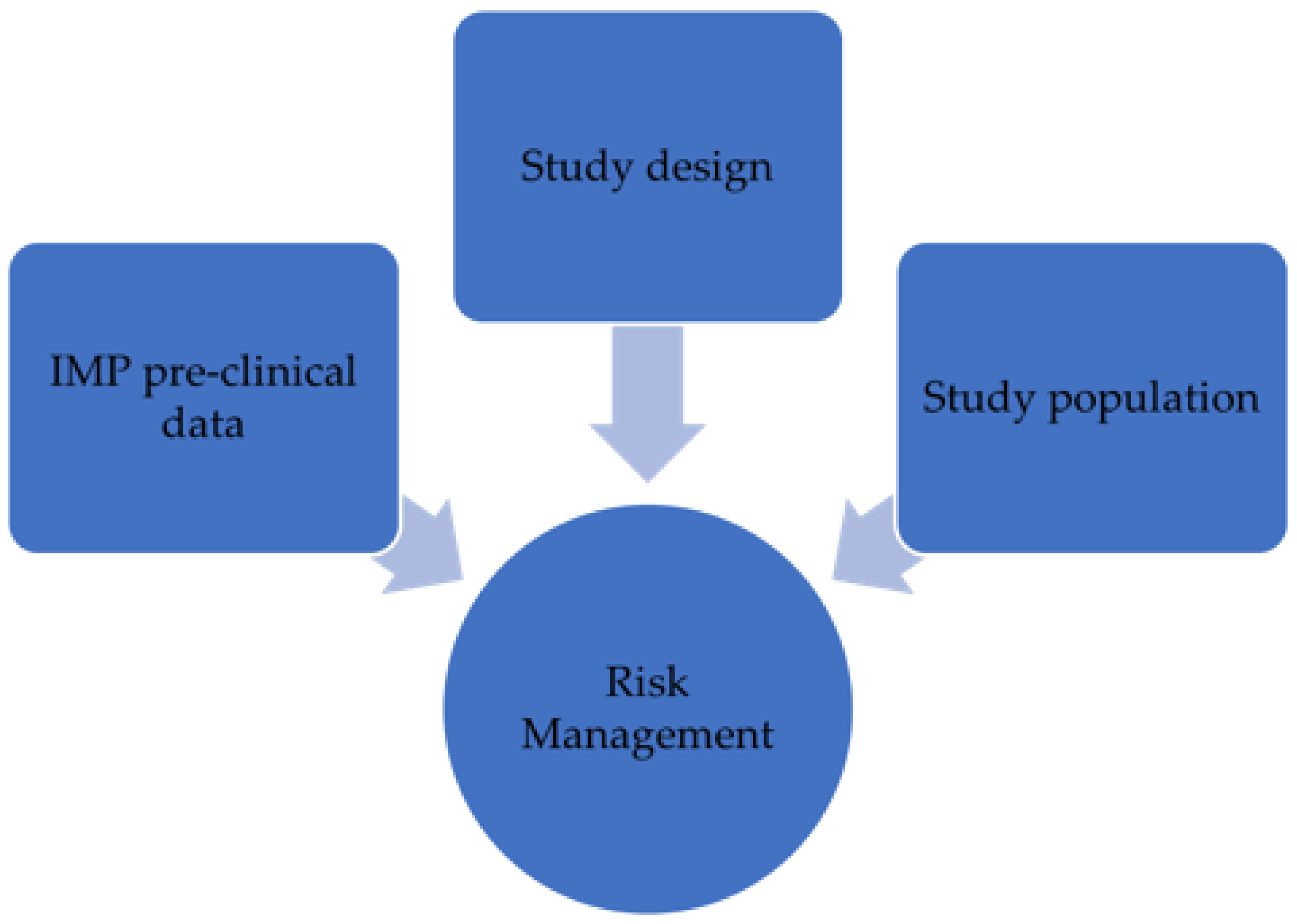{kind=link}
{kind=link}
Abstract
:1. Phase I Clinical Trials: Shaping the Future of Medicine
2. The European Regulatory Framework
- Quality aspects of the IMP (e.g., determination of potency, strengths, materials used, etc.);
- Pharmacokinetics, pharmacodynamics, and toxicology of the therapeutic agent candidate;
- Dosing selection during the phase I clinical trial;
- Phase I clinical studies management (planning, design, conduct).
- Precautions to apply between treating subjects within a cohort as well as between cohorts and study parts;
- Stopping rules;
- Sponsor and investigator responsibilities.
- Pharmacological studies on the IMP;
- Pharmacokinetics, pharmacodynamics, and toxicology;
- Dose selection;
- Safety tests.
- Immunogenicity, safety, and efficacy studies of vaccines;
- Selection of the dose to be administered;
- Concomitant medications;
- Duration of the follow-up period;
- Endpoint analysis;
- Consideration of the particular population (e.g., pregnant women, immunodeficient subjects, etc.).
- The presence of a formal risk assessment/mitigation process of phase I clinical trials in place at the experimental center (SOPs, forms, etc.);
- The presence of trained personnel for medical emergencies;
- The structural requirements of phase I clinical units located in the hospitals.
3. MHRA Phase I Accreditation Scheme: The British Case Study
- A completed application declaration form;
- A phase I accreditation compliance checklist;
- Any associated documents.
- Relocation of the unit or change in facilities;
- Significant changes to procedures that impact on key aspects of the accreditation scheme;
- Changes in key personnel (e.g., the medical director, any PIs authorized for FIH, senior nurses, the clinic manager, the pharmacist, and the Quality Assurance (QA) manager);
- Significant contractual changes in agreements with local hospitals;
- Significant changes in the clinical trial unit systems.
4. Regulatory Requirements of Managing Risks of Phase I Clinical Trials: The Italian Case Study
- The presence of a medical director responsible for the activities carried out at the phase I unit;
- Structural requirements of the clinical site;
- Presence of a robust Quality Management System (QMS) to conduct phase I clinical studies.
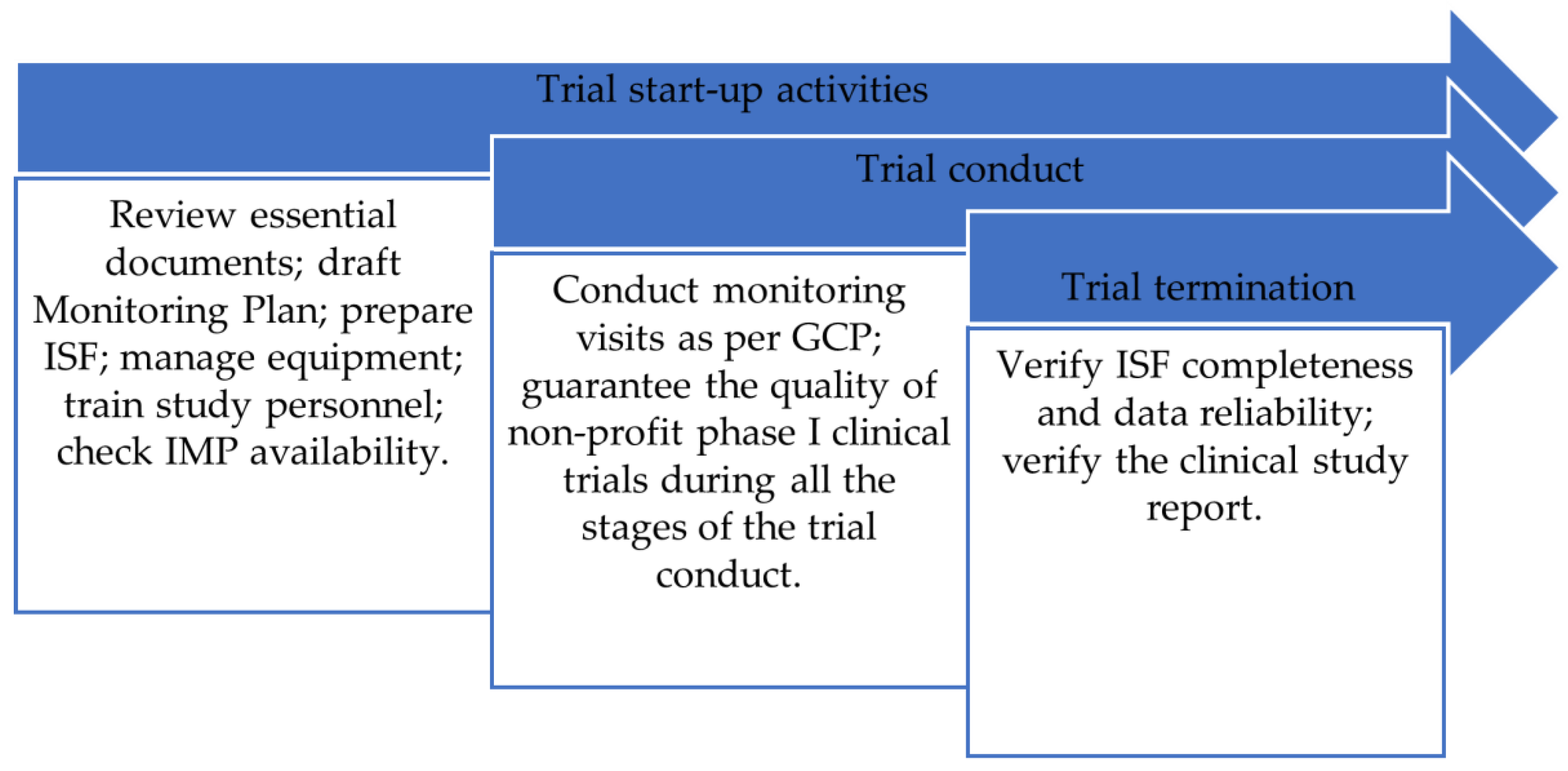
- Trial start-up activities;
- Clinical trial monitoring;
- Clinical trial termination.
5. Main Characteristics of the Clinical Trial Quality Team
- An organizational chart with enough qualified staff members;
- An internal regulation that explains the CTQT functions and roles;
- Standard Operating Procedures (SOPs) that cover every aspect of clinical trial management.
- A CTQT quality system including internal regulations and the hospital SOPs which the quality team refers to;
- A CTQT quality system with dedicated internal regulations and SOPs;
- A CTQT quality system with dedicated internal regulations and dedicated/hospital SOPs to refer to. In this case, if no hospital SOPs describe the processes required by the CTQT document, new SOPs are set up.
- Phase I unit-dedicated CTQT. In this type of organization, the quality team is exclusively dedicated to that specific phase I clinical unit, and it is responsible for the organization and for carrying out non-profit phase I clinical trials.
- Hospital-derived CTQT. In this case, the quality team may supervise the organization of multiple phase I clinical trials within the same hospital but conducted at different clinical trial units.
6. Challenges and Opportunities of the Clinical Trial Quality Team
7. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of Early Drug Discovery: Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.A.; Zyzanski, S.; Baker, J.N.; Drotar, D.; Kodish, E. Communication about the Risks and Benefits of Phase I Pediatric Oncology Trials. Contemp. Clin. Trials 2015, 41, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.A.; Rid, A.; Emanuel, E.; Wendler, D. Risks of Phase I Research with Healthy Participants: A Systematic Review. Clin. Trials 2016, 13, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.E.; Pedroza, C.; Wallace, D.; D’Angio, C.; Bell, E.F.; Das, A. Stopping Guidelines for an Effectiveness Trial: What Should the Protocol Specify? Trials 2016, 17, 240. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Nguyen, D. Risks and Benefits of Phase 1 Clinical Trial Participation. Cancer Control 2014, 21, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.G. Trends in the Risks and Benefits to Patients with Cancer Participating in Phase 1 Clinical Trials. JAMA 2004, 292, 2130. [Google Scholar] [CrossRef] [PubMed]
- Coates, S.; Täubel, J.; Lorch, U. Practical Risk Management in Early Phase Clinical Trials. Eur. J. Clin. Pharmacol. 2019, 75, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Rid, A.; Emanuel, E.J.; Wendler, D. Evaluating the Risks of Clinical Research. JAMA 2010, 304, 1472. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.B. Cancer Clinical Trials: Risks and Benefits. J. Pediatr. Hematol. Oncol. 2001, 23, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.J.; Singh, S.; Sidhu, P.; Sharma, P.K. TGN-1412 and BIA-2474 Trials with Tragic End: Lessons Learnt to Make Clinical Trials Safer. Rev. Recent Clin. Trials 2018, 13, 252–256. [Google Scholar] [CrossRef] [PubMed]
- EMEA/CHMP/SWP/28367/07 Rev. 1 Guideline on Strategies to Identify and Mitigate Risks for First-Inhuman and Early Clinical Trials with Investigational Medicinal Products. Committee for Medicinal Products for Human Use (CHMP) 2016. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-and-mitigate-risks-first-human-and-early-clinical-trials-investigational-medicinal-products-revision-1_en.pdf (accessed on 19 December 2023).
- Brussels, 10.10.2019 C(2019) 7140 final Detailed Guidelines on Good Clinical Practice Specific to Advanced Therapy Medicinal Products. Available online: https://health.ec.europa.eu/system/files/2019-10/atmp_guidelines_en_0.pdf (accessed on 19 December 2023).
- Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation (EC) No 726/2004. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF (accessed on 19 December 2023).
- EMA/CPMP/ICH/286/1995 ICH Guideline M3(R2) on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorisation for Pharmaceuticals. December 2009. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-and-marketing-authorisation-pharmaceuticals-step-5_en.pdf (accessed on 19 December 2023).
- EMEA/CHMP/VWP/164653/05 Rev. 1 Guideline on Clinical Evaluation of Vaccines. Committee for Medicinal Products for Human Use (CHMP) 2023. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-evaluation-vaccines-revision-1_en.pdf (accessed on 19 December 2023).
- Suprin, M.; Chow, A.; Pillwein, M.; Rowe, J.; Ryan, M.; Rygiel-Zbikowska, B.; Wilson, K.J.; Tomlin, I. Quality Risk Management Framework: Guidance for Successful Implementation of Risk Management in Clinical Development. Ther. Innov. Regul. Sci. 2019, 53, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Guidance: MHRA Phase I Accreditation Scheme Requirements. Date: 12 August 2022. Available online: https://assets.publishing.service.gov.uk/media/631612de8fa8f502256a956e/1._MHRA_Phase_I_Accreditation_Scheme_Requirements_v4.1_12_August_2022.pdf (accessed on 19 December 2023).
- EMA/CHMP/ICH/135/1995 Guideline for Good Clinical Practice E6(R2). Committee for Human Medicinal Products /CHMP) 2016. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e6r2-step-5_en.pdf (accessed on 19 December 2023).
- Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001. 2001. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0020-20090807 (accessed on 19 December 2023).
- Regulation (EU) No 536/2014 of the European Parliament and the Council of 16 April 2014 on Clinical Trials on Medicinal Products for Human Use, and Repealing Directive 2001/20/EC. 2014. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32014R0536 (accessed on 19 December 2023).
- Agenzia Italiana Del Farmaco. Determina n. 809/2015 (“Determination on Minimal Requirements for Conducting Phase I Trials in Health Institutions Article 11 of the Presidential Decree 21 September 2011, n. 439 Article 31, Comma 3 of the Legislative Decree 6 November 2007 n. 200”). Gazzetta Ufficiale. 2015, 158 Del 10 Luglio 2015. Available online: https://www.gazzettaufficiale.it/atto/stampa/serie_generale/originario (accessed on 19 December 2023).
- Greco, E.; Ciliberto, E.; Verdura, P.D.; Lo Giudice, E.; Navarra, G. Nanoparticle-Based Concretes for the Restoration of Historical and Contemporary Buildings: New Way for CO2 Reduction in Architecture. Appl. Phys. A Mater. Sci. Process. 2016, 146, 122. [Google Scholar] [CrossRef]
- Marcianò, A.; Chen, D.; Fabrocini, F.; Fields, C.; Greco, E.; Gresnigt, N.; Jinklub, K.; Lulli, M.; Terzidis, K.; Zappala, E. Quantum Neural Networks and Topological Quantum Field Theories. Neural Netw. 2022, 153, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Ciliberto, E.; Cirino, A.M.E.; Capitani, D.; Di Tullio, V. A New Preparation of Doped Photocatalytic TiO2 Anatase Nanoparticles: A Preliminary Study for the Removal of Pollutants in Confined Museum Areas. Appl. Phys. A Mater. Sci. Process. 2016, 122, 530. [Google Scholar] [CrossRef]
- Greco, E.; Shang, J.; Zhu, J.; Zhu, T. Synthesis of Polyacetylene-like Modified Graphene Oxide Aerogel and Its Enhanced Electrical Properties. ACS Omega 2019, 4, 20948–20954. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Tonno, D.; Martena, L.; Taurisano, M.; Perlin, C.; Loiacono, A.C.; Lagravinese, S.; Marsigliante, S.; Maffia, M.; Esposito, S.; Villa, G.; et al. The Requirements of Managing Phase I Clinical Trials Risks: The British and Italian Case Studies. Epidemiologia 2024, 5, 137-145. https://doi.org/10.3390/epidemiologia5010009
Di Tonno D, Martena L, Taurisano M, Perlin C, Loiacono AC, Lagravinese S, Marsigliante S, Maffia M, Esposito S, Villa G, et al. The Requirements of Managing Phase I Clinical Trials Risks: The British and Italian Case Studies. Epidemiologia. 2024; 5(1):137-145. https://doi.org/10.3390/epidemiologia5010009
Chicago/Turabian StyleDi Tonno, Davide, Laura Martena, Manuela Taurisano, Caterina Perlin, Anna Chiara Loiacono, Stefano Lagravinese, Santo Marsigliante, Michele Maffia, Susanna Esposito, Gianluca Villa, and et al. 2024. "The Requirements of Managing Phase I Clinical Trials Risks: The British and Italian Case Studies" Epidemiologia 5, no. 1: 137-145. https://doi.org/10.3390/epidemiologia5010009