
Genetic Diversity and Epidemic Types of Porcine Reproductive and Respiratory Syndrome (PRRS) Virus in Japan from 2018 to 2020



Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Extraction and Sequencing
2.2. Phylogenetic Tree Construction and Cluster Classification
2.3. Identity of PRRSV Genome Sequences Detected in This Study with Reference Strains
2.4. Statistical Analysis
3. Results
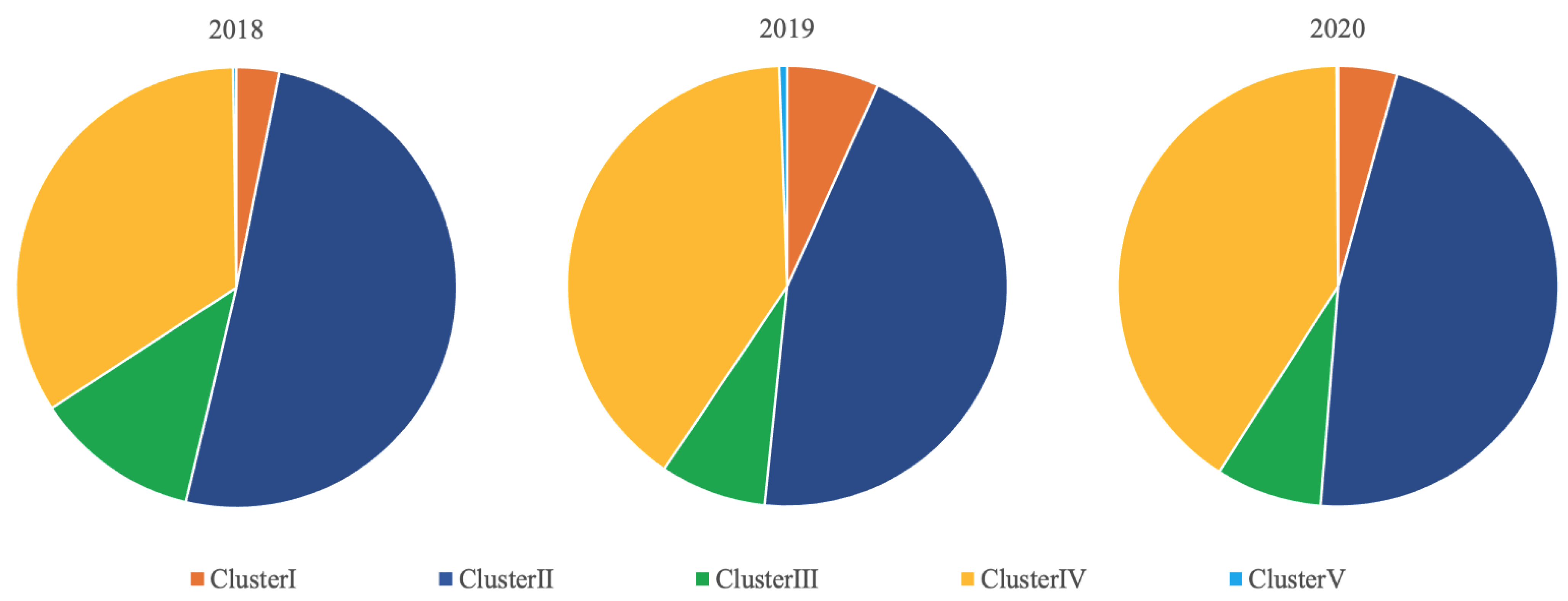
3.1. Predominant Type and Cluster
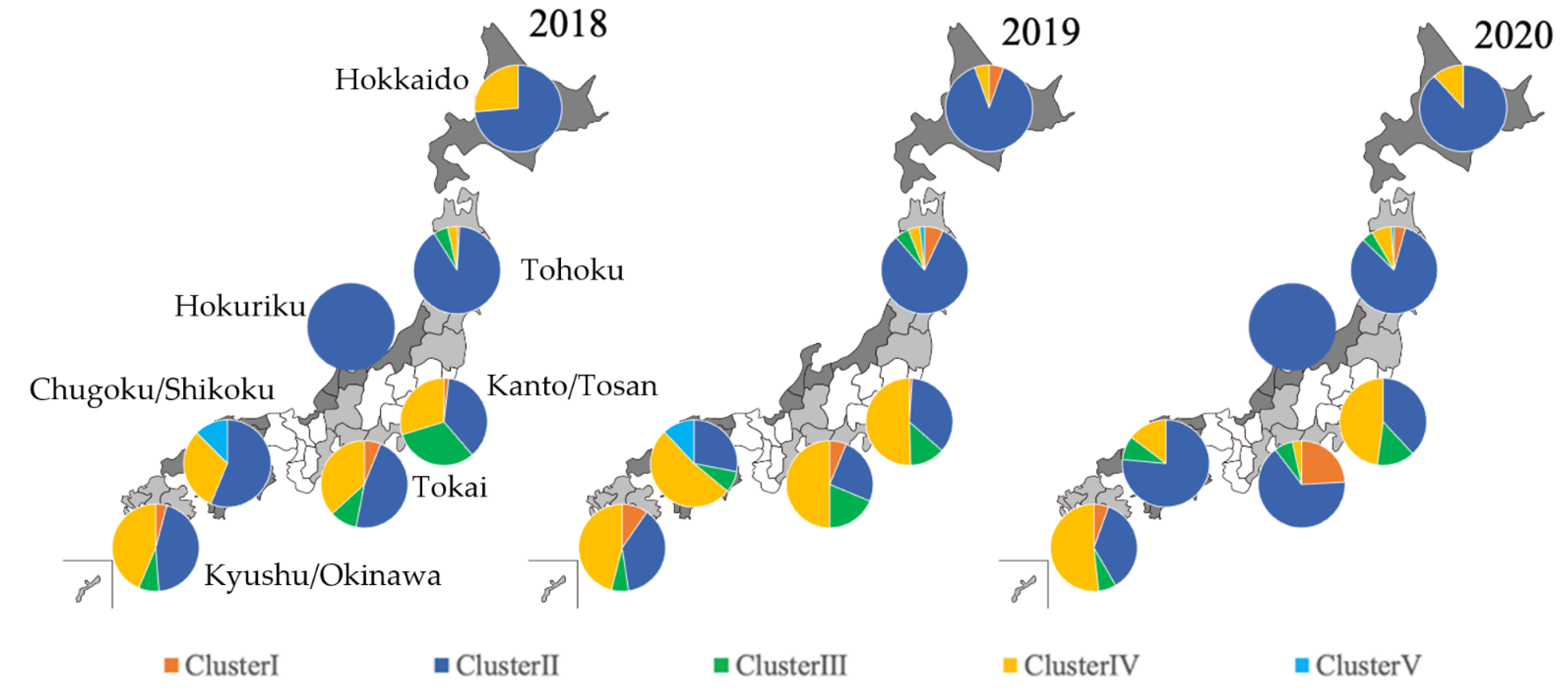
3.2. Distribution of Predominant Cluster by Region
3.3. Genetic Diversity of Each Cluster to the Reference Strain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PRRS | porcine reproductive and respiratory syndrome virus |
| PRRSV | porcine reproductive and respiratory syndrome virus |
| PRDC | porcine respiratory disease complex |
| ELISA | enzyme-linked immune-sorbent assay |
| RT-PCR | reverse transcription-polymerase chain reaction test |
| ORFs | open reading frames |
| RNA | ribonucleic acid |
| MLV | modified live vaccine |
| MEGA | molecular evolutionary genetic analysis |
| BMCMC | Bayesian Markov chain Monte Carlo |
References
- Rossow, K.D. Porcine reproductive and respiratory syndrome. Vet. Pathol. 1998, 38, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D. Nidovirales: A new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 1997, 142, 629–633. [Google Scholar] [PubMed]
- Mateu, E.; Martín, M.; Vidal, D. Genetic diversity and phylogenetic analysis of glycoprotein 5 of European-type porcine reproductive and respiratory virus strains in Spain. J. Gen. Virol. 2003, 84, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Keffaber, K.K. Reproductive failure of unknown etiology. Am. Assoc. Swine Pract. Newsl. 1989, 1, 1–10. [Google Scholar]
- Done, S.H.; Paton, D.J.; White, M.E.C. Porcine reproductive and respiratory syndrome (PRRS): A review, with emphasis on pathological, virological and diagnostic aspects. Br. Vet. J. 1996, 152, 153–174. [Google Scholar] [CrossRef]
- Wensvoort, G.; Terpstra, C.; Pol, J.M.; ter Laak, E.A.; Bloemraad, M.; de Kluyver, E.P.; Kragten, C.; van Buiten, L.; den Besten, A.; Wagenaar, F. Mystery swine disease in the Netherlands: The isolation of Lelystad virus. Vet. Q. 1991, 13, 121–130. [Google Scholar] [CrossRef]
- Collins, J.E.; Benfield, D.A.; Christianson, W.T.; Harris, L.; Hennings, J.C.; Shaw, D.P.; Goyal, S.M.; McCullough, S.; Morrison, R.B.; Joo, H.S. Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J. Vet. Diagn. Investig. 1992, 4, 117–126. [Google Scholar] [CrossRef]
- Forsberg, R.; Storgaard, T.; Nielsen, H.S.; Oleksiewicz, M.B.; Cordioli, P.; Sala, G.; Hein, J.; Bøtner, A. The genetic diversity of European type PRRSV is similar to that of the North American type but is geographically skewed within Europe. Virology 2002, 299, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, T.L.; Hahn, E.C.; Weigel, R.M.; Scherba, G. Genetic, geographical and temporal variation of porcine reproductive and respiratory syndrome virus in Illinois. J. Gen. Virol. 2000, 81, 171–179. [Google Scholar] [CrossRef]
- Kapur, V.; Elam, M.R.; Pawlovich, T.M.; Murtaugh, M.P. Genetic variation in porcine reproductive and respiratory syndrome virus isolates in the midwestern United States. J. Gen. Virol. 1996, 77, 1271–1276. [Google Scholar] [CrossRef]
- Ropp, S.L.; Wees, C.E.; Fang, Y.; Nelson, E.A.; Rossow, K.D.; Bien, M.; Arndt, B.; Preszler, S.; Steen, P.; Christopher-Hennings, J.; et al. Characterization of emerging European-like porcine reproductive and respiratory syndrome virus isolates in the United States. J. Virol. 2004, 78, 3684–3703. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Yamada, S.; Murakami, Y.; Morozumi, T.; Kobayashi, H.; Mitani, K.; Ito, N.; Kubo, M.; Kimura, K.; Kobayashi, M. Isolation of porcine reproductive and respiratory syndrome (PRRS) virus from Heko-Heko disease of pigs. J. Vet. Med. Sci. 1994, 56, 389–391. [Google Scholar] [CrossRef] [Green Version]
- Eddicks, M.; Eddicks, L.; Stadler, J.; Hermanns, W.; Ritzmann, M. Der Porcine Respiratory Disease Complex (PRDC)—Eine klinische Übersicht [The porcine respiratory disease complex (PRDC)—A clinical review]. Tierärztliche Prax. Ausg. G Großtiere/Nutztiere 2021, 49, 120–132. [Google Scholar]
- Saade, G.; Deblanc, C.; Bougon, J.; Marois-Créhan, C.; Fablet, C.; Auray, G.; Belloc, C.; Leblanc-Maridor, M.; Gagnon, C.A.; Zhu, J.; et al. Coinfections and their molecular consequences in the porcine respiratory tract. Vet. Res. 2020, 51, 80. [Google Scholar] [CrossRef]
- Zhu, H.; Chang, X.; Zhou, J.; Wang, D.; Zhou, J.; Fan, B.; Ni, Y.; Yin, J.; Lv, L.; Zhao, Y.; et al. Co-infection analysis of bacterial and viral respiratory pathogens from clinically healthy swine in Eastern China. Vet. Med. Sci. 2021, 7, 1815–1819. [Google Scholar] [CrossRef]
- Yamane, I. Evaluation of the economic losses due to the porcine reproductive and respiratory syndrome. Proc. Jpn. Pig Vet. Soc. 2013, 61, 1–4. [Google Scholar]
- Bøtner, A. Diagnosis of PRRS. Vet. Microbiol. 1997, 55, 295–301. [Google Scholar] [CrossRef]
- Halbur, P.G.; Andrews, J.J.; Huffman, E.L.; Paul, P.S.; Meng, X.J.; Niyo, Y. Development of a streptavidin-biotin immunoperoxidase procedure for the detection of porcine reproductive and respiratory syndrome virus antigen in porcine lung. J. Vet. Diagn. Investig. 1993, 6, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; He, Y.; Xu, X.; Leng, X.; Li, S.; Wen, Y.; Wang, F.; Xia, M.; Cheng, S.; Wu, H. Pathological and immunological characteristics of piglets infected experimentally with a HP-PRRSV TJ strain. BMC Vet. Res. 2016, 12, 230. [Google Scholar] [CrossRef] [Green Version]
- Terpstra, C.; Wensvoort, G.; Pol, J.M. Experimental reproduction of porcine epidemic abortion and respiratory syndrome (mystery swine disease) by infection with Lelystad virus: Koch’s postulates fulfilled. Vet. Q. 1991, 13, 131–136. [Google Scholar] [CrossRef]
- Yoon, I.J.; Joo, H.S.; Christianson, W.T.; Kim, H.S.; Collins, J.E.; Morrison, R.B.; Dial, G.D. An indirect fluorescent antibody test for the detection of antibody to swine infertility and respiratory syndrome virus in swine sera. J. Vet. Diagn. Investig. 1992, 4, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittawornrat, A.; Prickett, J.; Chittick, W.; Wang, C.; Engle, M.; Johnson, J.; Patnayak, D.; Schwartz, T.; Whitney, D.; Olsen, C.; et al. Porcine reproductive and respiratory syndrome virus (PRRSV) in serum and oral fluid samples from individual boars: Will oral fluid replace serum for PRRSV surveillance? Virus Res. 2010, 154, 170–176. [Google Scholar] [CrossRef]
- Lopez, W.A.; Angulo, J.; Zimmerman, J.J.; Linhares, D.C.L. Porcine reproductive and respiratory syndrome monitoring in breeding herds using processing fluids. J. Swine Health Prod. 2018, 26, 146–150. [Google Scholar]
- Dea, S.; Gagnon, C.A.; Mardassi, H.; Pirzadeh, B.; Rogan, D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: Comparison of the North American and European isolates. Arch. Virol. 2000, 145, 659–688. [Google Scholar] [CrossRef] [PubMed]
- Christopher-Hennings, J.; Nelson, E.A.; Nelson, J.K.; Hines, R.J.; Swenson, S.L.; Hill, H.T.; Zimmerman, J.J.; Katz, J.B.; Yaeger, M.J.; Chase, C.C. Detection of porcine reproductive and respiratory syndrome virus in boar semen by PCR. J. Clin. Microbiol. 1995, 33, 1730–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, Y.; Kanno, T.; Shimizu, M.; Yamada, S.; Ohashi, S.; Nakamine, M.; Shirai, J. Nested PCR for detection and typing of porcine reproductive and respiratory syndrome (PRRS) virus in pigs. J. Vet. Med. Sci. 1996, 8, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; O’Connell, C.M.; Costa, A.; Pan, Y.; Smyth, J.A.; Verardi, P.H.; Burgess, D.J.; Van Kruiningen, H.J.; Garmendia, A.E. A PRRSV GP5-Mosaic vaccine: Protection of pigs from challenge and ex vivo detection of IFNγ responses against several genotype 2 strains. PLoS ONE 2019, 14, e0208801. [Google Scholar]
- Andreyev, V.G.; Wesley, R.D.; Mengeling, W.L.; Vorwald, A.C.; Lager, K.M. Genetic variation and phylogenetic relationships of 22 porcine reproductive and respiratory syndrome virus (PRRSV) field strains based on sequence analysis of open reading frame 5. Arch. Virol. 1997, 142, 993–1001. [Google Scholar] [CrossRef]
- Kappes, M.A.; Faaberg, K.S. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.È.; Delisle, B.; Arsenault, J.; Poljak, Z.; D’Allaire, S. Positioning Quebec ORF5 sequences of porcine reproductive and respiratory syndrome virus (PRRSV) within Canada and worldwide diversity. Infect. Genet. Eevol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2019, 74, 103999. [Google Scholar] [CrossRef]
- Takagi, M. The situation and latest scientific information of porcine reproductive and respiratory syndrome (PRRS). Proc. Jpn. Pig Vet. Soc. 2014, 63, 1–5. [Google Scholar]
- Iseki, H.; Takagi, M.; Miyazaki, A.; Katsuda, K.; Mikami, O.; Tsunemitsu, H. Genetic analysis of ORF5 in porcine reproductive and respiratory syndrome virus in Japan. Microbiol. Immunol. 2011, 55, 211–216. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, M.; Kaku, Y.; Murakami, Y.; Shimizu, M.; Kato, K.; Ikeda, H. Polymerase chain reaction-based genetic typing of Japanese porcine reproductive and respiratory syndrome viruses. J. Vet. Diagn. Investig. 2004, 16, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 2013, 48, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Iseki, H.; Morozumi, T.; Takagi, M.; Kawashima, K.; Shibahara, T.; Uenishi, H.; Tsunemitsu, H. Genomic sequence and virulence evaluation of MN184A-like porcine reproductive and respiratory syndrome virus in Japan. Microbiol. Immunol. 2016, 60, 824–834. [Google Scholar] [CrossRef]
- Kikuti, M.; Sanhueza, J.; Vilalta, C.; Paploski, I.A.D.; VanderWaal, K.; Corzo, C.A. Porcine reproductive and respiratory syndrome virus 2 (PRRSV-2) genetic diversity and occurrence of wild type and vaccine-like strains in the United States swine industry. PLoS ONE 2021, 16, e0259531. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.; Hon, C.C.; Hui, R.K.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C. Molecular epidemiology of PRRSV: A phylogenetic perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.; Hon, C.C.; Murtaugh, M.P.; Davies, P.R.; Hui, R.K.; Li, J.; Wong, L.T.; Yip, C.W.; Jiang, J.W.; et al. Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J. Virol. 2010, 84, 8700–8711. [Google Scholar] [CrossRef] [Green Version]
- Lalonde, C.; Provost, C.; Gagnon, C.A. Whole-genome sequencing of porcine reproductive and respiratory syndrome virus from field clinical samples improves the genomic surveillance of the virus. J. Clin. Microbiol. 2020, 58, e00097-20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Administrative Region | 2018 | 2019 | 2020 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Prefectures | Farms | Detects * | Prefectures | Farms | Detects * | Prefectures | Farms | Detects * | |
| Hokkaido | 1 | 6 | 19 | 1 | 9 | 37 | 1 | 8 | 34 |
| Tohoku | 5 | 30 | 112 | 6 | 48 | 114 | 6 | 38 | 95 |
| Kanto/Tosan | 6 | 53 | 168 | 5 | 49 | 200 | 7 | 52 | 184 |
| Hokuriku | 1 | 1 | 2 | 0 | 0 | 0 | 1 | 2 | 3 |
| Tokai | 4 | 21 | 49 | 3 | 9 | 16 | 2 | 12 | 29 |
| Kinki | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chugoku/Shikoku | 3 | 6 | 16 | 3 | 7 | 25 | 4 | 7 | 34 |
| Kyusyu/Okinawa | 8 | 138 | 435 | 8 | 146 | 456 | 7 | 139 | 454 |
| Total | 28 | 255 | 801 | 26 | 268 | 848 | 28 | 258 | 833 |
| Origin | Specimen | 2018 | 2019 | 2020 |
|---|---|---|---|---|
| Live pig | Blood/serum | 600 | 694 | 644 |
| Oral fluid | 166 | 125 | 148 | |
| Processing fluid | 8 | 18 | 15 | |
| Sperm | 1 | 0 | 0 | |
| Sub-total | 775 | 837 | 807 | |
| Dead pig | Lung/Hilar lymph node | 23 | 9 | 20 |
| Tonsil | 1 | 0 | 0 | |
| Lymph node | 1 | 0 | 4 | |
| Aborted fetus | 1 | 2 | 1 | |
| Pleural effusion | 0 | 0 | 1 | |
| Sub-total | 26 | 11 | 26 | |
| Total | 801 | 848 | 833 | |
| Name of Reference Strain | Cluster Type | Accession Number | ORF5 Gene Sequence |
|---|---|---|---|
| PrimePac® PRRS | I | AF066384 | atgttggggaaatgcttgaccgcgggttgttgctcgcgattgctttctttttggtgtatcgtgccgttctgttttgctgtgctcgtcaacgccagctacagcagcagctctcatttacagttgatttataacttgacgctatgtgagctgaatggtacagattggctggctaataaatttgattgggcagtggagagttttgtcatctttcctgtgttgacccacatcgtttcctatggtgcactaaccaccagccacttccttgacacagttggtctggttactgtgtctaccgccgggtttgttcatgggcggtatgtcctgagtagcatctacgcggtctgtgccctggctgcgttaatttgcttcgtcattaggttggcgaagaactgtatgtcctggcgctactcatgcaccagatacaccaactttcttctggacactaagggcagactctatcgttggcggtcgcctgtcatcatagagaaagggggtaaggtagaggtcgaaggccatctgatcgacctcaaaagagttgtgcttgatggttccgcggcaacccctttaaccagagtttcagcggaacaatggggtcgtccctag |
| Fostera® PRRS | I | MK820650 | atgttggggaaatgcttgaccgcgggctgttgctcgcgattgctttctttgtggtgtatcgtgccgttctggtttgctgtgctcggcaacgccaacagcagcagcagctctcatttccagttgatttataacttgacgctatgtgagctgaatggcacagattggctggcagaaaaatttgattgggcggtggagacttttgtcatctttcccgtgttgactcacattgtttcctattgtgcactcaccaccagccatttccttgacacagttggtctggttactgtgtccaccgccgggttttatcacgggcggtatgtcttgagtagcatctacgcggtctgtgctctggctgcgttgatttgcttcgttattaggcttgcgaagaactgcatgtcctggcgctactcttgtaccagatataccaacttccttctggacactaagggcagactctatcgttggcggtcgcccgttatcatagaaaaaaggggtaaggttgaggtcgaaggtcatctgatcgacctcaaaagagttgtgcttgatggttccgtggcaacccctttaaccagagtttcagcggaacaatggggtcgtctctag |
| Ingelvac® PRRS MLV | II | AF020048 | atgttggagaaatgcttgaccgcgggctgttgctcgcaattgctttctttgtggtgtatcgtgccgttctgttttgctgtgctcgccaacgccagcaacgacagcagctcccatctacagctgatttacaacttgacgctatgtgagctgaatggcacagattggctagctaacaaatttgattgggcagtggagagttttgtcatctttcccgttttgactcacattgtctcctatggtgccctcactaccagccatttccttgacacagtcgctttagtcactgtgtctaccgccgggtttgttcacgggcggtatgtcctaagtagcatctacgcggtctgtgccctggctgcgttgacttgcttcgtcattaggtttgcaaagaattgcatgtcctggcgctacgcgtgtaccagatataccaactttcttctggacactaagggcggactctatcgttggcggtcgcctgtcatcatagagaaaaggggcaaagttgaggtcgaaggtcatctgatcgacctcaaaagagttgtgcttgatggttccgtggcaacccctataaccagagtttcagcggaacaatggggtcgtccttag |
| EDRD-1 | III | AB288356 | atgttggggaaatgcttgaccgcgggctgttgctcgcgattgccttttttgtggtgtatcgtgccgttctgtcttgctgtgctcgtcaacgccagcgacagcagcagctcccatttacagttgatttataacctgacgctatgtgagctgaatggcacagattggctggctgacaaatttgattgggcagtggagagttttgtcatctttcccgtgttgactcacattgtttcttactgcgccctcactaccagccacttccttgacacagttggtctggtcgctgtgtctaccgccgggttttaccacgggcggtatgttctgagtagcatctatgcggtctgtgccctggctgcgttggtttgcttcgtcatcagattgacgaagaattgcatgtcctggcgctactcatgtaccagatataccaactttcttctggataccaagggcagactctatcgttggcggtcgcccgtcatcatagagaaagggggtaaggttgaggttgaaggtcatctgatcgacctcaagagagttgtgcttgatggttccgcggcaacccctataaccaaagtttcagcggaacaatggggtcatccctag |
| Jpn5-37 | IV | AB546125 | atgttggggaaatgcttgaccgcgggctattgctcgcaattgccttttttgtggtgtatcgtgccgttctgtcttgctgcgctcgtcaacgccaacagcaacagcagctcccatttacagttgatttataacttaacgatatgtgagctgaatggcacagattggctgaacaatcattttagttgggcagtggagactttcgttatctttcctgcgttgactcatattgtttcctacggcgcccttactaccagccacttccttgacacggtcggcctaatcactgtgtccaccgctggatactaccatgggcggtatgtcttgagtagcatctatgctgtctgcgccctggctgcgctgacttgcttcgtcatcaggttgacgaaaaattgtatgtcttggcgctactcatgtaccagatataccaactttcttctggacaccaagggcagactctatcgctggcggtcacccgtcattatagagaaaatgggtaaaattgaggttggaggtgacctgatcgacctcaagagagttgtgcttgatggttccgcggcaacccctgtaaccaaagtttcagcggaacaatggagtcgtccttag |
| Jos1 | V | AB175699 | atgttggggaaatgcttgaccgcgggctgttgctcgcgattgctttttttgtggtgtatcgtgccgtcctgttttgttgcgctcgtcagcgccaacaacagcagcagctctcatttacagttgatctacaacctgacgctatgtgagctgaatggcacagattggctggctaataaattcaattgggcagtggaaagttttgtcatctttcctgtgttgactcacattgtctcttatggtgcccttactaccagccatttccttgacacagtctgcttggtcactgtatctaccgccggtttttaccacgggcggtatgttttgagcagcatctacgcggtttgtgccctggccgcgttggtttgcttcgccgttaggttcgcgaagaattgcatgtcctggcgctattcgtgtaccaggtataccaactttcttctggacactaagggcagactctatcgttggcggtcgcctgtcatcatagagaaaaggggtaaagttgaggtcgaaggtcatctgatcgacctcaagagagttgtgcttgatggttccgcggcaacccctataaccaaaatttcagcggaacaatggggtcatctctag |
| Cluster | Reference Strain | Year | Number of Isolates | Homology Distribution (%) | Mean (%) | SD * | Median (%) |
|---|---|---|---|---|---|---|---|
| Cluster I | PrimePac® | 2018 | 25 | 85.3–93.3 | 88.8 | 2.5 | 88.0 |
| 2019 | 57 | 85.0–93.5 | 90.4 | 3.1 | 92.6 | ||
| 2020 | 36 | 85.3–93.7 | 90.7 | 3.0 | 92.2 | ||
| Cluster I | Fostera® PRRS | 2018 | 25 | 85.7–99.8 | 89.9 a,** | 4.6 | 88.4 |
| 2019 | 57 | 86.0–100.0 | 94.3 b,** | 5.8 | 99.1 | ||
| 2020 | 36 | 85.2–99.8 | 94.6 | 5.6 | 97.8 | ||
| Cluster II | Ingelvac® PRRS MLV | 2018 | 405 | 85.0–100.0 | 98.2 | 2.7 | 99.3 |
| 2019 | 381 | 87.2–100.0 | 98.3 | 2.3 | 99.3 | ||
| 2020 | 391 | 85.0–100.0 | 98.3 | 2.5 | 99.3 | ||
| Cluster III | EDRD-1 | 2018 | 97 | 86.0–91.3 | 88.6 | 1.3 | 88.4 |
| 2019 | 66 | 86.3–92.3 | 88.8 | 1.5 | 88.6 | ||
| 2020 | 65 | 86.3–90.6 | 88.7 | 0.1 | 88.7 | ||
| Cluster IV | Jpn5-37 | 2018 | 272 | 83.6–89.2 | 86.1 a,** | 1.1 | 85.8 |
| 2019 | 339 | 80.4–88.9 | 85.8 b,** | 1.1 | 85.8 | ||
| 2020 | 340 | 82.9–88.4 | 85.8 | 1.1 | 85.8 | ||
| Cluster V | Jos1 | 2018 | 2 | 90.1–90.4 | 90.3 | 0.2 | 90.3 |
| 2019 | 5 | 86.5–90.6 | 88.8 | 1.8 | 88.6 | ||
| 2020 | 1 | 87.7 | 87.7 | NT | 87.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyutoku, F.; Yokoyama, T.; Sugiura, K. Genetic Diversity and Epidemic Types of Porcine Reproductive and Respiratory Syndrome (PRRS) Virus in Japan from 2018 to 2020. Epidemiologia 2022, 3, 285-296. https://doi.org/10.3390/epidemiologia3020022
Kyutoku F, Yokoyama T, Sugiura K. Genetic Diversity and Epidemic Types of Porcine Reproductive and Respiratory Syndrome (PRRS) Virus in Japan from 2018 to 2020. Epidemiologia. 2022; 3(2):285-296. https://doi.org/10.3390/epidemiologia3020022
Chicago/Turabian StyleKyutoku, Fumiaki, Takashi Yokoyama, and Katsuaki Sugiura. 2022. "Genetic Diversity and Epidemic Types of Porcine Reproductive and Respiratory Syndrome (PRRS) Virus in Japan from 2018 to 2020" Epidemiologia 3, no. 2: 285-296. https://doi.org/10.3390/epidemiologia3020022