
The Changing Face of Paediatric Human Growth Hormone Therapy

Centre for Endocrinology, William Harvey Research Institute, John Vane Building, Barts and the London School of Medicine & Dentistry, Charterhouse Square, Queen Mary, University of London, London EC1M 6BQ, UK
Endocrines 2022, 3(3), 419-427; https://doi.org/10.3390/endocrines3030033
Submission received: 9 May 2022
/
Revised: 29 June 2022
/
Accepted: 1 July 2022
/
Published: 6 July 2022
(This article belongs to the Special Issue Growth and Growth Disorders)
{kind=link}
{kind=link}
Abstract
:Human growth hormone (hGH) has been used therapeutically to promote growth in children for over 60 years. Pituitary-extracted hGH has demonstrated positive growth promotion since the early 1960s. In 1985, prion-induced contamination of hGH triggered a global epidemic of Creutzfeldt–Jakob disease that was responsible for its discontinuation. Recombinant hGH immediately replaced pituitary hGH and, being available in large amounts, was used and licenced for therapy in GH-deficient children, followed by approval for non-GH deficient disorders such as Turner syndrome, short stature related to birth size small for gestational age, idiopathic short stature, SHOX deficiency, Prader–Willi syndrome and Noonan syndrome. RhGH therapy was refined by the use of growth prediction models; however, unmet needs, such as the variability in response and non-adherence resulted in the development of long-acting rhGH (LArhGH) molecules, which are currently in clinical trials and have shown non-inferiority in comparison with daily rhGH. It is likely that LArhGH will enter clinical practice in 2022 and 2023 and will need to demonstrate safety in terms of immunogenicity, IGF-1 generation, metabolic status and tolerability of potential injection pain and local reactions.
1. Introduction
Human growth hormone (GH) is now a recognized therapy for children with GH deficiency and is approved for the treatment of a number of non-GH-deficient disorders [1]. It is appropriate, after more than 60 years of hGH therapy, to appraise its progress, which has advanced over the years and is still evolving—with several new developments about to make an impact on clinical care [1]. Abnormalities in the physical height of children were first linked to disturbances of GH secretion in the early 20th century. Harvey Cushing was one of the first physicians to link linear growth to the function of the pituitary gland [2] and the concept of treating impaired growth was advanced by the studies of Herbert Evans at the University of California, San Francisco, who demonstrated a growth-promoting effect of pituitary extracts administered to rats with hypopituitarism [3]. The existence of a pituitary hormone linked to linear growth regulation was pursued by C.H. Li, working with Evans, and in 1971 the primary structure of human growth hormone was finally characterised as a protein with 191 amino acids and two disulphide bonds [4].
In terms of the clinical application of a circulating factor capable of stimulating growth in children, Ernst Knobil, while working at Harvard, demonstrated the species-specificity of human GH [5]. This knowledge was important in the process of recovering human cadaver pituitary glands for the extraction of pituitary hGH, which in the USA was organised by the National Pituitary Agency. hGH was thus extracted and purified for the treatment of paediatric GH deficiency. The extraction process was practiced in several countries. Dr Maurice Rabin, working at the Tufts Medical Center in Massachusetts, reported in 1958—and subsequently in 1962—the first clinical descriptions of effective growth promotion in children with hypopituitarism [6,7].
The next key step in clinical practice was the development of a radioimmunoassay (RIA) for the measurement of GH in human subjects with impaired growth. The first RIA was reported by Yalow and Berson in 1963 [8], together with the description of insulin-induced hypoglycaemia tests as the optimal technique for assessing GH secretion [9].
2. The Era of Pituitary-Extracted Human GH
Starting in 1963, the collection, extraction and purification of pituitary-extracted hGH, free from contamination by other pituitary hormones, was organized on a national level. Despite the demonstration of efficacy in terms of human growth-promotion, the processes of extraction and purification remained crude. One example is the method of extraction developed by Dr Philip Lowry in the UK, where the organisation of hGH therapy was carried out under the auspices of the Medical Research Council and the Department of Health [10]. Supplies of hGH were short and limited to treatment of children with severe GH deficiency.
In the late 1960s, following the establishment of GH provocative tests in clinical practice, an empirical peak stimulated GH level of 5–7 μg/L or above was considered the criterion level for the separation of normality from GH deficiency. Many clinicians used a GH cut-off of 10 μg/L to exclude GH deficiency. The cut-off for the definition of GHD being raised to 10 µg/L from the previous 7 µg/L cut-off was largely due to the company which tested and marketed the first rhGH using a cut-off of 10 µg/L in their clinical trials and in their application for FDA approval.
Only recently has a cut-off value of <7 μg/L been accepted as the international definition of GH deficiency. The international reference preparation (IRP) for the hGH assay depended initially on purified pituitary GH and then on purified 22kDa recombinant hGH with a potency of 3 IU/mg [11]. Today, a serum concentration of 20 μIU hGH is equivalent to 6.7 µg/L [1].
The hypothalamic-releasing peptides somatostatin and GHRH—characterized in 1973 and 1982, respectively [1]—regulate pituitary GH secretion, which is pulsatile [12,13]; therefore, pharmacological stimulation tests became established for diagnosis. The range of diagnostic stimuli included arginine, L-DOPA, glucagon, and propranolone [14].
GH deficiency in children is a multi-faceted disorder requiring a combination of clinical and auxological assessments and investigation of the GH-IGF-1 axis and brain MRI [11]. The MRI presentation of severe GH deficiency has been demonstrated to be an ectopic posterior pituitary gland, an indistinct pituitary stalk and a small volume pituitary gland consistent with pituitary hypoplasia [15].
2.1. Treatment with Pituitary-Extracted hGH
The GH-deficient patients treated with pituitary-hGH were usually rather old, eg >10 years with a severely short stature and multiple pituitary hormone deficiencies (Figure 1). The hGH was administered intramuscularly two or three times weekly until 1983, when daily subcutaneous injections were introduced [16] and were equally effective. The dose of pituitary-hGH was 30–100 mIU per kg body weight given three times weekly [17].
2.2. Creutzfeldt–Jakob Disease
In 1984, a 20-year-old patient who had been treated with cadaveric pituitary-hGH for 14 years developed the serious neurodegenerative disorder, Creutzfeldt–Jakob disease [18]. The patient died within six months of the onset of symptoms. Post-mortem examination showed the characteristic spongiform encephalopathy of this condition caused by misfolding of a prion protein (PrP). If the abnormal prion had its origin in an infected cadaver pituitary gland, the infective agent could be passed to patients receiving pituitary-hGH. A global epidemic of CJD developed, with 226 cases reported in patients who had received cadaveric pituitary-hGH (Figure 1). Prescriptions of pituitary-hGH immediately ceased in virtually every country. The era of pituitary-derived hGH was over.
3. Recombinant hGH
In the 1970s, molecular biology permitted the DNA sequence encoding recombinant human GH (rhGH) to be expressed [19]. The recombinant peptide contained an additional methionine residue (meth-hGH), and in 1985, the first hGH produced from E. coli was approved for the treatment of paediatric GH deficiency. Safety and efficacy were documented in obligatory post-marketing surveillance studies organised by pharmaceutical companies and formed the extensive databases of the Kabi International Growth Study (KIGS) in Europe and the National Cooperative Growth Study (NCGS) in the USA. Both have provided a wealth of important scientific information. With the industrial production of rhGH becoming possible and restriction on supplies now removed, hGH therapy moved into a new era. A much wider range of GH deficiency, from severe to mild cases, was now accepted for replacement therapy. As a result, children diagnosed as GH deficient were younger. A rhGH regimen of 33 μg/kg body weight/day was generally adopted as the standard replacement therapy [11]. The modification of this dosage, dependent on patient needs and the prediction of the growth response, will be seen later.
3.1. Treatment of Non-GH Deficiency Disorders
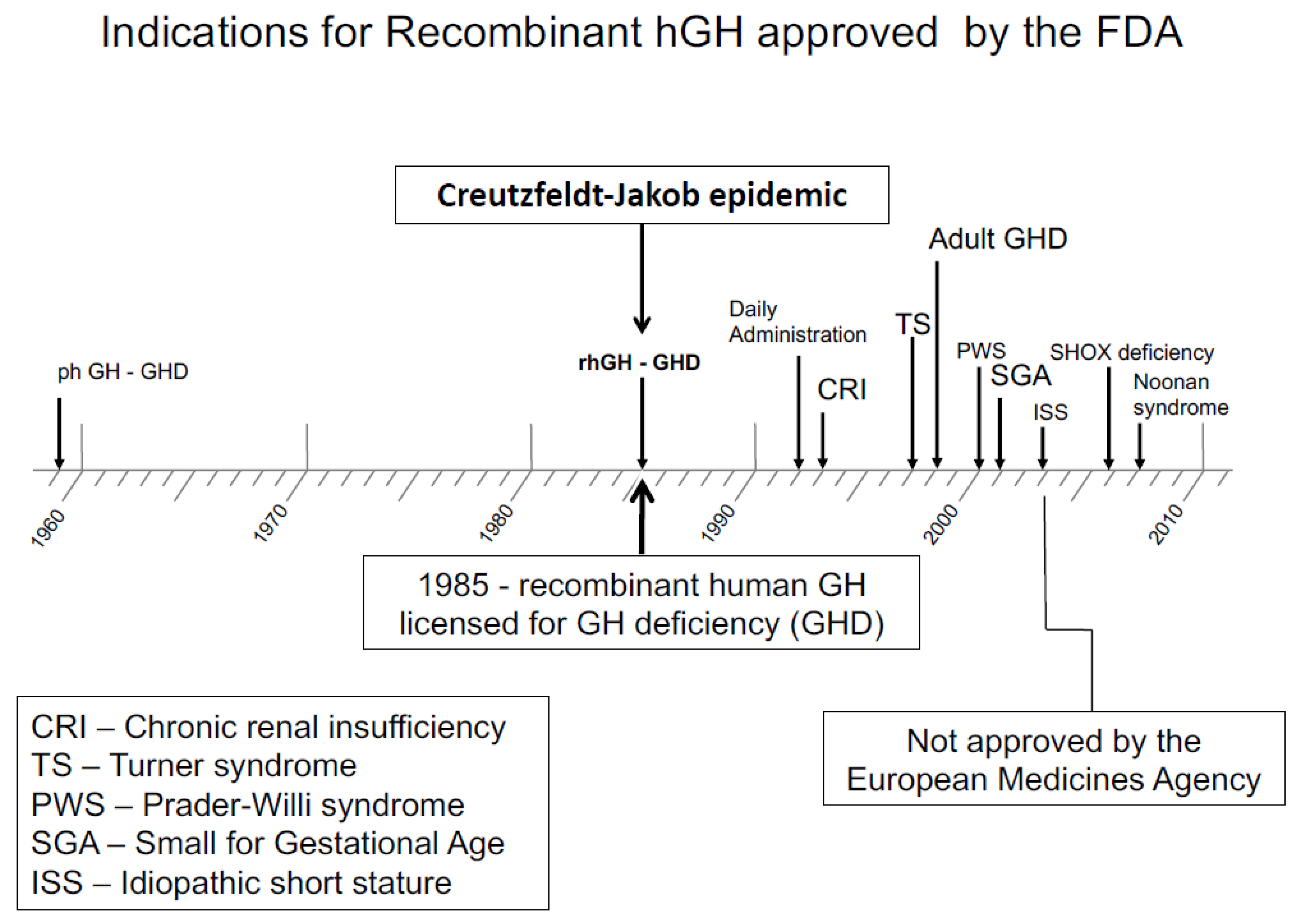
A wide availability of rhGH allowed trials to be started in growth disorders not associated with GH deficiency. The first of these was Turner syndrome. The question was: could rhGH therapy, used in higher pharmacological doses—rather than replacement doses—accelerate growth and lead to height gain, which would increase adult height and provide a measurable clinical benefit? Extensive trials in Turner syndrome demonstrated adult height gain [20], leading to FDA and EMA approval of this condition for rhGH therapy. The approved dose was significantly higher than for GH deficiency, being in the region of 50 μg/kg/day [1]. Other non-GH deficient disorders followed, such as short stature related to birth size small for gestational age (SGA) [21,22], idiopathic short stature (ISS) [23]—which was approved in the USA but not in Europe—Prader–Willi syndrome, SHOX deficiency [24] and Noonan syndrome [25] (Figure 2). Growth failure in chronic renal insufficiency had previously been approved for in 1963.
Short stature related to SGA was approved for rhGH by the FDA in 2001 and the EMA in 2003. rhGH treatment had demonstrated gain of adult height when hGH therapy was initiated more than two years before the onset of puberty [21]. There was debate regarding the optimal hGH dose; a dose of 33 μg/kg/day was proposed by the group from Rotterdam [22], but other groups maintained that a higher dose of 67 μg/kg/day induced better-quality catch-up growth [27]. Both doses had similar effects on long-term growth; however, the higher dose tended to induce supra-physiological levels of serum IGF-1 during therapy. Hence, the lower dose of 33 μg/kg/day is now recommended by the EMA [28,29].
3.2. Safety of rhGH
Due to the collection of extensive data on the rhGH post-marketing databases, rhGH therapy has been demonstrated to be safe. Abnormalities reported during therapy tend to reflect the nature of the primary disorder being treated. For example, benign intracranial hypertension and slipped femoral epiphyses are most likely to occur in children with severe forms of GH deficiency. There are no data which demonstrate that a higher risk of cancer exists in children without an inherent risk. Post-surveillance registries, by recording adverse drug reactions, have contributed significantly to the overall positive reputation of rhGH as regards safety. Adverse events are lowest in patients with idiopathic GH deficiency or ISS [1]. EMA recommendations regarding the dose of rhGH for each indication need to be adhered to, and when prescribed according to these recommendations, the evidence suggests that rhGH therapy is very safe [30]. A comprehensive review of GH therapy in childhood and adult cancer survivors demonstrated that there is no evidence for an association between GH replacement and increased mortality from cancer amongst GH-deficient childhood cancer survivors [31].
3.3. The Continuum Model of GH-IGF-1 Axis Defects
The so-called continuum model of GH-IGF-1 axis defects [32] should be described in the context of the development of rhGH therapy. The model consists of two axes: the X axis describes GH sensitivity and the Y axis, GH secretion. Disorders of GH and IGF-1 secretion and action are positioned on the graph and are represented by a continuum ranging from severe to mild GH deficiency, through isolated short stature to mild and severe GH resistance [33].
These abnormalities present clinically with short stature and the clinician needs to address the challenge of growth-promotion with a therapy that is safe and has demonstrated efficacy. The two poles of the continuum—namely GH deficiency and GH resistance—can be treated with licensed rhGH and rhIGF-1, respectively [33]. Intermediate defects, where the degree of short stature is less severe, are more challenging. As will be discussed below in the context of growth prediction models, the more severe the GH deficiency is—based on the peak GH concentration in the diagnostic GH provocation test—the more responsive the patient is to rhGH. A patient with severe GH deficiency will usually respond to a small dose of 20 µg/kg/day, whereas in patients with mild GH deficiency, a higher rhGH dose of ~35 μg/kg/day would be more likely to induce an expected growth response. A patient with isolated short stature without GH deficiency requires an even higher rhGH dose, e.g., 50 μg/kg/day. When patients with GH resistance—also known as primary IGF-1 deficiency—are encountered, they will be unresponsive to rhGH, and the best option for their management is replacement with rhIGF-1—which is the logical approach, as their primary defect is by causing a deficiency in IGF-1 that is not responsive to rhGH [33,34].
4. Growth Prediction Models
Major progress in the use of rhGH has been made since 1985; however, a number of unmet clinical needs remain. One of these relates to the variability in growth response, with many patients not achieving optimal catch-up growth or adult height gain [29]. This is the case across the range of approved growth disorders, including GH deficiency [35,36]. In order to address this issue, mathematical models have been defined which incorporate different variables influencing growth. Use of this model can predict the growth response to rhGH therapy in different indications [37,38,39,40,41]. An increase in height velocity of 2 cm/year in a child with GH deficiency is predicted using a standard dose of rhGH 0.3 mg/kg/week, compared with SGA and Turner syndrome patients. The severity of the GH deficiency is the highest-rated variable in GH-deficient patients compared to the dose of rhGH per kg body weight per week, which has the highest predictive power in patients with non-GH-deficient short stature [38].
The use of mathematical models in the clinical setting has proven to be problematic, although their use has led to a smaller number of poor responders to rhGH therapy [42]. However, the development of prediction models represents a major milestone in the story of rhGH therapy. Their use underlines the principles of precision medicine, i.e., the individualization of care compared with the approach of standard care for all patients.
5. Poor Adherence to rhGH Therapy
A further unmet need related to long-standing rhGH therapy is the need to maintain high levels of patient adherence to the prescribed rhGH regimen. Long-term therapy with rhGH injections is demanding for the child and family, and there is good evidence that adherence decreases over time and that there is a direct relationship between the rate of adherence—in terms of the percentage of prescribed injections that are successfully administered—and the degree of short and long-term catch-up growth during therapy [43,44]. The detection of poor adherence can also be challenging in the clinical setting. Trained medical and nursing personnel are required to sensitively question the child and family. There are multiple potential reasons for poor adherence which can be seen in the context of the behavioural COM-B model [45,46].
Effective management of poor adherence to rhGH also requires the paediatric endocrinologist or specialist nurse to learn techniques of non-judgemental motivational interviewing [46]. Organisation, time, knowledge of common issues affecting adherence and the ability to build a close relationship with the family with open questions and an emphasis on pre-hGH treatment education are key components of this type of healthcare professional–patient interaction. The same healthcare professional should discuss adherence at each outpatient visit. Patient choice in the brand of rhGH and its injection device has been shown to be a key determinant factor in future adherence and response [47].
Electronic monitoring of injections is improving the rate of adherence and provides important feedback data on the evidence of sub-optimal administration of injections [44]. Self-reported data usually underestimate the degree of poor adherence and electronic monitoring has been shown to give a more accurate account [48]. The easypod™ injection device and Easypod Connect© system provide data on adherence that is related to the quality of the growth response [49]. The device facilitates the administration of a pre-set dose of rhGH, records injection times and doses, and provides the patient with information such as number of doses remaining [50]. The injection data can be collected and downloaded by healthcare personnel and patients, which enables distance monitoring. Thus, healthcare personnel can address issues of non-adherence to rhGH treatments at an early stage. Studies have, to date, indicated good tolerability of the device and overall high levels of adherence over several years [49,51].
6. The Future: Long-Acting rhGH and Oral GH Secretagogue Therapy
The burden on the child and family of daily injections of rhGH for many years has led to the development of long-acting rhGH molecules (LArhGH). The first of these was developed in 1999 but did not prove clinically advantageous, and it is in the last 5 years that new and improved preparations have been produced. The technology varies, with various options available. GH pharmacokinetic curves can be extended by the manipulation of drug release from subcutaneous depots and the manipulation of in vivo clearance from the circulation [52].
Prolonged biological action of rhGH can be obtained using a range of techniques including: (1) PEGylation, which combines polyethylene glycol moieties with rhGH, causing an increase in the size of the rhGH molecule and thereby reducing renal clearance and immunogenicity (Gene Sciences, Shanghai, China); (2) rhGH is conjugated to a fatty acid that can reversibly bind serum albumin, slowing drug elimination (Novo Nordisk, Bagsvaerd, Denmark); (3) TransCon technology: a ternary complex formed between an unmodified rhGH molecule, an inert carrier that shields the drug and a temporary linker (Ascendis, San Fransisco, CA, USA); and (4) the fusion of three copies of a naturally occurring C-terminal peptide of human chorionic gonadotropin (hCG) to the coding sequence of rhGH (OPKO-Pfizer, New York, NY, USA) [52,53].
Phase III trials have currently been published for the Ascendis and OPKO-Pfizer products and for Jintrolong—the pegylated rhGH that is well established in China. The non-inferiority of their efficacy in comparison with daily rhGH has been demonstrated in each case. Registration of these products for reimbursement is awaited from the FDA and EMA; however, it is very likely that they will be entering clinical practice in 2022 and 2023. The impact that this will have on prescriptions of daily rhGH is difficult to predict. It is likely that non-adherent patients will be identified first for LArhGH therapy, followed by naïve GH-deficient subjects and those treated during puberty and undergoing transitional care for transfer to adult services.
In terms of safety, a number of factors will need careful scrutiny and documentation in national, international and company-specific registries. Immunogenicity will need monitoring with assays for neutralising anti-rhGH antibodies. Serum IGF-1 is generally higher during LArhGH therapy than with daily injections [53]. The day of IGF-1 monitoring post-LArhGH injection is important, and guidance will need to be given to clinicians if dose-reduction is necessary. Finally, long-term metabolic status and injection pain and discomfort will require monitoring and scoring.
Oral GH Secretagogue Therapy
Lumos Pharma (Austin, TX, USA) is a US biotech company which has developed LUM-201, an oral GH secretagogue that increases GH pulsatility by stimulation of the GHSR1a receptor (ghrelin receptor) in the hypothalamus and pituitary. Predictive enrichment markers for the positive effect of LUM-201 have been defined as a peak GH response of ≥5 ng/mL to a single LUM-201 dose and a baseline serum IGF-1 concentration of >30 ng/mL. These positive predictive markers will be present in moderate or mild cases of paediatric GH deficiency, but not in severe hypopituitarism. LUM-201 has therefore been designed as a potential therapy for non-severe cases of GH deficiency. The data on increases in GH pulsatility are impressive and a phase II therapeutic trial is currently in progress [54,55].
7. Conclusions
Human GH has been used in various formulations for over 60 years to induce growth acceleration in GH-deficient children and, more recently, in non-GH-deficient disorders. Since 1985, rhGH has been used extensively—although until recently, no significant change in its development or formulation has occurred. However, we are now on the edge of a major era of therapeutic evolution with the change from daily to weekly rhGH administration. Transparency in efficacy and safety is essential as real-world experience accumulates with the introduction of LArhGH. It is hoped that adherence and efficacy will improve; however, the new formulations will need to be able to adjust to the nuances of growth prediction models. Acceptability by patients and families will need to be shown and safety demonstrated in terms of immunogenicity, metabolic status and GH biomarkers such as IGF-1 concentrations.
Funding
There is no relevant funding of this review article.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author M.O.S. has had consultancy contracts with Pfizer, Merck Healthcare KGaA, Darmstadt and Sandoz and has received honoraria for lectures from Ipsen, Gene Sciences and Novo Nordisk.
References
- Ranke, M.B.; Wit, J.M. Growth hormone—Past present and future. Nat. Rev. Endocrinol. 2018, 14, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Cushing, H. The Pituitary Body and Its Disorders. Clinical States Produced by Disorders of the Hypophysis Cerebri; J. B. Lippencott: Philadelphia, PA, USA, 1912. [Google Scholar]
- Evans, H.M.; Long, J.A. The effect of the anterior lobe administered intraperitoneally upon growth, maturity and oestrus cycles of the rat. Anat. Res. 1921, 6, 62–63. [Google Scholar]
- Li, C.H.; Dixon, J.S. Human pituitary growth hormone: 32. The primary structure of the hormone: Revision. Arch. Biochem. Biophys. 1971, 146, 233–236. [Google Scholar] [CrossRef]
- Knobil, E.; Greep, R.O. The physiology of growth hormone with particular reference to its action in the rhesus monkey and the “species specificity” problem. Recent Prog. Horm. Res. 1959, 15, 1–58. [Google Scholar]
- Raben, M.S. Treatment of a pituitary dwarf with growth hormone. J. Clin. Endocrinol. Metab. 1958, 18, 901–903. [Google Scholar] [CrossRef]
- Raben, M.S. Growth hormone—Clinical use of human growth hormone. N. Engl. J. Med. 1962, 266, 82–86. [Google Scholar] [CrossRef]
- Glick, S.M.; Roth, J.; Yalow, R.S.; Berson, S.A. Immunoassay of human growth hormone in plasma. Nature 1963, 199, 784–787. [Google Scholar] [CrossRef]
- Roth, J.; Glick, S.M.; Yalow, R.S.; Berson, S.A. Hypoglycaemia: A potent stimulus to secretion of human growth hormone. Science 1963, 140, 987–988. [Google Scholar] [CrossRef]
- Jones, R.; Benker, G.; Salacinsky, P.; Lloyd, T.; Lowry, P.J. Large-scale preparation of highly purified pyrogen-free human growth hormone for clinical use. J. Endocrinol. 1979, 82, 77–86. [Google Scholar] [CrossRef]
- GH Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: Summary statement of the GH Research Society. J. Clin. Endocrinol. Metab. 2000, 85, 3990–3993. [Google Scholar]
- Miller, J.D.; Tannenbaum, G.S.; Colle, E.; Guyda, H.J. Daytime pulsatile growth hormone secretion during childhood and adolescence. J. Clin. Endocrinol. Metab. 1982, 55, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Albertsson Wikland, K.; Rosberg, S. Analyses of 24-hour growth hormone profiles in children: Relation to growth. J. Clin. Endocrinol. Metab. 1988, 67, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B. Diagnostics of Endocrine Function in Children and Adolescents; Ranke, M.B., Mullis, P.E., Eds.; Karger: Basel, Switzerland, 2011; pp. 103–137. [Google Scholar]
- Maghnie, M.; Lindberg, A.; Koltowska-Haggstrom, M.; Ranke, M.B. Magnetic resonance imaging in 15,053 children with GH deficiency in KIGS (Pfizer international database). Eur. J. Endocrinol. 2013, 68, 2011–2017. [Google Scholar]
- Kastrup, D.W.; Christiansen, J.S.; Anderson, J.K.; Orskov, H. Increased growth rate following transfer to daily sc. administration from three weekly im. injection of hGH in growth hormone deficient children. Acta Endocrinol. 1983, 104, 148–152. [Google Scholar] [CrossRef]
- Aceto, T., Jr.; Frasier, S.D.; Hayles, A.B.; Meyer-Bahlburg, H.F.; Parker, M.L.; Munschauer, R.; di Chiro, G. Collaborative study of the effects of human growth hormone in growth hormone deficiency. I. first year of therapy. J. Clin. Endocrinol. Metab. 1972, 35, 483–496. [Google Scholar] [CrossRef]
- Hintz, R.L. The prismatic case of Creutzfeldt-Jakob disease associated with pituitary growth hormone treatment. J. Clin. Endocrinol. Metab. 1995, 80, 2298–2301. [Google Scholar]
- Goeddel, D.V.; Heyneker, H.L.; Hozumi, T.; Arentzen, R.; Itakura, K.; Yansura, D.G.; Ross, M.J.; Miozzari, G.; Crea, R.; Seeburg, P.H. Direct expression in Escherichia coli of a DNA sequence coding for human growth hormone. Nature 1979, 281, 544–549. [Google Scholar] [CrossRef]
- Stephure, D.K. Canadian growth hormone advisory committee. Impact of growth hormone supplementation on adult height in turner syndrome: Results of the Canadian randomized controlled trial. J. Clin. Endocrinol. Metab. 2005, 90, 3360–3366. [Google Scholar]
- Dahlgren, J.; Albertsson Wikland, K.; Swedish Study Group for Growth Hormone Treatment. Final height in short children born small for gestational age treated with growth hormone. Pediatr. Res. 2005, 57, 216–222. [Google Scholar] [CrossRef]
- Van Pareren, Y.; de Muinck Keizer-Schrama, S.M.P.F.; Stijnen, T.; Sas, T.C.J.; Jansen, M.; Otten, B.J.; Hoorweg-Nijman, J.J.; Vulsma, T.; Stokvis-Brantsma, W.H.; Rouwé, C.W.; et al. Final height in girls with Turner syndrome after long-term growth hormone.e treatment in three dosages and low dose estrogens. J. Clin. Endocrinol. Metab. 2003, 88, 1119–1125. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Rogol, A.D.; Deal, C.L.; Saenger, P.; Reiter, E.O.; Ross, J.L.; Chernausek, S.D.; Savage, M.O.; Wit, J.M.; on behalf of the 2007 ISS Consensus Workshop Participants. Consensus statement on diagnosis and treatment of children with idiopathic short stature. A summary of the GRS/LWPES/ESPE workshop. J. Clin. Endocrinol. Metab. 2008, 93, 4210–4217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, N.C.B.; Funari, M.F.A.; Vasques, G.A.; Andrade, N.L.; Rezende, R.C.; Brito, V.; Scalco, R.C.; Arnh, I.J.P.; Mendonca, B.B.; Jorge, A.A.L. Adult height of patients with SHOX haploinsufficiency with or without GH therapy: A real-world single-center study. Horm. Res. Paediatr. 2022. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, T.R.; Abuzzahab, J.; Backeljauw, P.; Birkegård, A.C.; Blair, J.; Dahlgren, J.; Júlíusson, P.B.; Ostrow, V.; Pietropoli, A.; Polak, M.; et al. Long-term effectiveness and safety of childhood growth hormone treatment in Noonan Syndrome. Horm. Res. Paediatr. 2020, 93, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.O.; Alherbish, A. Growth hormone therapy for paediatric growth disorders: The past, present and future. Dr. Sulaiman Al Habib Med. J. 2020, 2, 4–9. [Google Scholar] [CrossRef] [Green Version]
- de Zegher, F.; Francois, I.; van Helvoirt, M.; Beckers, D.; Ibáñez, L.; Chatelain, P. Growth hormone treatment of short children born small for gestational age. Trends Endocrinol. Metab. 1998, 9, 233–237. [Google Scholar] [CrossRef]
- Finken, M.J.J.; van der Steen, M.; Smeets, C.C.J.; Walenkamp, M.J.E.; de Bruin, C.; Hokken-Koelega, A.C.S.; Wit, M.J. Children born small for gestational age: Differential diagnosis, molecular genetic evaluation, and implications. Endocr. Rev. 2018, 39, 851–894. [Google Scholar] [CrossRef] [Green Version]
- Wit, J.M.; Deeb, A.; Bin-Abbas, B.; Al Mutair, A.; Koledova, E.; Savage, M.O. Achieving optimal short- and long-term responses to paediatric growth hormone therapy. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 329–340. [Google Scholar] [CrossRef]
- Deodati, A.; Ferroli, B.B.; Cianfarani, S. Association between growth hormone therapy and mortality, cancer and cardiovascular risk: Systematic review and meta-analysis. Growth Horm. IGF Res. 2014, 24, 105–111. [Google Scholar] [CrossRef]
- Boguszewski, M.C.S.; Boguszewski, C.L.; Chemaitilly, W.; Cohen, L.E.; Gebauer, J.; Higham, C.; Hoffman, A.R.; Polak, M.; Yuen, K.C.J.; Nathalie Alos, N.; et al. Safety of growth hormone replacement in survivors of cancer and intracranial and pituitary tumours: A consensus statement. Eur. J. Endocrinol. 2022, 186, P35–P52. [Google Scholar] [CrossRef]
- Cohen, P. Controversy in clinical endocrinology: Problems with reclassification of insulin-like growth factor I production and action disorders. J. Clin. Endocrinol. Metab. 2006, 91, 4235–4236. [Google Scholar] [CrossRef]
- Savage, M.O.; Burren, C.P.; Rosenfeld, R.G. The continuum of growth hormone-IGF-I axis defects causing short stature: Diagnostic and therapeutic challenges. Clin. Endocrinol. 2010, 72, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Chernausek, S.D.; Backeljauw, P.F.; Frane, J.; Kuntze, J.; Underwood, L.E.; GH Insensitivity Syndrome Collaborative Group. Long-term treatment with recombinant insulin-like growth factor (IGF)-I in children with severe IGF-I deficiency due to growth hormone insensitivity. J. Clin. Endocrinol. Metab. 2007, 92, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, E.O.; Price, D.A.; Wilton, P.; Albertsson-Wikland, K.; Ranke, M.B. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: Analysis of a large international database. J. Clin. Endocrinol. Metab. 2006, 91, 2047–2054. [Google Scholar] [CrossRef] [Green Version]
- Bang, P.; Bjerknes, R.; Dahlgren, J.; Dunkel, L.; Gustafsson, J.; Juul, A.; Kriström, B.; Tapanainen, P.; Aberg, V. A comparison of different definitions of growth response in short prepubertal children treated with growth hormone. Horm. Res. Paediatr. 2011, 75, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Ranke, M.B.; Albertsson-Wikland, K.; Carrascosa, A.; Rosenfeld, R.G.; Van Buuren, S.; Kriström, B.; Schoenau, E.; Audi, L.; Hokken-Koelega, A.C.; et al. Personalized approach to growth hormone treatment: Clinical use of growth prediction models. Horm. Res. Paediatr. 2013, 79, 257–270. [Google Scholar] [CrossRef]
- Ranke, M.B.; Lindberg, A.; KIGS International Board. Observed and predicted growth responses in prepubertal children with growth disorders: Guidance of growth hormone treatment by empirical variables. J. Clin. Endocrinol. Metab. 2010, 95, 1229–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspers, S.; Ranke, M.B.; Han, D.; Loftus, J.; Wollmann, H.; Lindberg, A.; Roelants, M.; Kleintjens, J. Implications of a data-driven approach to treatment with growth hormone in children with growth hormone deficiency and Turner syndrome. Appl. Health Econ. Health Policy 2013, 11, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Schonau, E.; Westermann, F.; Rauch, F.; Stabrey, A.; Wassmer, G.; Keller, E.; Bramswig, J.; Blum, W.F.; German Lilly Growth Response Study Group. A new and accurate prediction model for growth response to growth hormone treatment in children with growth hormone deficiency. Eur. J. Endocrinol. 2001, 144, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Wikland, K.A.; Kriström, B.; Rosberg, S.; Svensson, B.; Nierop, A.E. Validated multivariate models predicting the growth response to growth hormone treatment in individual short children with a broad range in GH secretion capacities. Pediatr. Res. 2000, 48, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Kriström, B.; Aronson, A.S.; Dahlgren, J.; Gustafsson, J.; Halldin, M.; Ivarsson, S.A.; Nilsson, N.O.; Svensson, J.; Tuvemo, T.; Albertsson-Wikland, K. Growth hormone (GH) dosing during catch-up growth guided by individual responsiveness decreases growth response variability in prepubertal children with GH deficiency or idiopathic short stature. J. Clin. Endocrinol. Metab. 2009, 94, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Van Dommelen, P.; Koledova, E.; Wit, J.M. Effect of adherence to growth hormone treatment on 0–2 year catch-up growth in children with growth hormone deficiency. PLoS ONE 2018, 13, e0206009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutfield, W.S.; Derraik, J.G.B.; Gunn, A.J.; Reid, K.; Delany, T.; Hofman, P.L. Non-compliance with growth hormone treatment in children is common and impairs linear growth. PLoS ONE 2011, 6, e16223. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Eliasson, L.; Barber, N.; Weinman, J. Applying COM-B to medication adherence. Eur. Health Psychol. 2014, 16, 7–17. [Google Scholar]
- Child, J.; Davies, C.; Frost, K.; McDermid, E.; Pidcock, R.; Weinman, J.; Savage, M.O. Managing paediatric growth disorders: Integrating technology into a personalised approach. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 225–232. [Google Scholar] [CrossRef]
- Fisher, B.G.; Acerini, C.L. Understanding the growth hormone therapy adherence paradigm: A systematic review. Horm. Res. Pediatr. 2013, 79, 189–196. [Google Scholar] [CrossRef]
- Loche, S.; Salerno, M.; Garofalo, P.; Cardinale, G.M.; Licenziati, M.R.; Citro, G.; Caruso Nicoletti, M.; Cappa, M.; Longobardi, S.; Maghnie, M.; et al. Adherence in children with growth hormone deficiency treated with r-hGH and the easypod™ device. J. Endocrinol. Investig. 2016, 39, 1419–1424. [Google Scholar] [CrossRef] [Green Version]
- Koledova, E.; Stoyanov, G.; Ovbude, L.; Davies, P.S.W. Adherence and long-term growth outcomes: Results from the easypod™ connect observational study (ECOS) in paediatric patients with growth disorders. Endocr. Connect. 2018, 7, 914–923. [Google Scholar] [CrossRef]
- Dahlgren, J. Easypod: A new electronic injection device for growth hormone. Expert Rev. Med. Devices 2008, 5, 297–304. [Google Scholar] [CrossRef]
- Deeb, A.; Al Yarubi, S.; Bin Abbas, B.; Al Jubeh, J.; Chaturvedi, D.; Al Hassani, N.; Mutair, A.; Al Masri, N.; Al Sanad, Y.; Al Shidhani, A.; et al. Patients’ perception of the use of the EasyPod® growth hormone injector device and impact on injection adherence: A multi-center regional study. Front. Paediatr. 2021, 10, 839278. [Google Scholar] [CrossRef]
- Pampanini, V.; Deodati, A.; Inzaghi, E.; Cianfarani, S. Long-acting growth hormone preparations and their use in children with growth hormone deficiency. Horm. Res. Paediatr. 2022. [Google Scholar] [CrossRef]
- Miller, B.S.; Velazquez, E.; Yuen, K.C. Long-Acting growth hormone preparations—Current status and future considerations. J. Clin. Endocrinol. Metab. 2020, 105, e2121–e2133. [Google Scholar] [CrossRef] [PubMed]
- Bright, G.M.; Do, M.T.; McKew, J.C.; Blum, W.F.; Thorner, M.O. Development of a predictive enrichment marker for the oral GH secretagogue LUM-201 in pediatric growth hormone deficiency. J. Endocr. Soc. 2021, 5, bvab030. [Google Scholar] [CrossRef] [PubMed]
- Bright, G.M.; Thorner, M.O. A GH secretagogue receptor agonist (LUM-201) elicits greater GH responses than standard GH secretagogues in subjects of a pediatric GH deficiency trial. Horm. Res. Paediatr. 2022, 95, 76–81. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Treatment of hypopituitarism in children with pituitary-extracted human growth hormone. Some of the pioneering paediatric endocrinologists who prescribed this therapy.
Figure 1.
Treatment of hypopituitarism in children with pituitary-extracted human growth hormone. Some of the pioneering paediatric endocrinologists who prescribed this therapy.

Figure 2.
Indications for recombinant hGH therapy approved by the FDA after 1985 [26].
Figure 2.
Indications for recombinant hGH therapy approved by the FDA after 1985 [26].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Savage, M.O. The Changing Face of Paediatric Human Growth Hormone Therapy. Endocrines 2022, 3, 419-427. https://doi.org/10.3390/endocrines3030033
AMA Style
Savage MO. The Changing Face of Paediatric Human Growth Hormone Therapy. Endocrines. 2022; 3(3):419-427. https://doi.org/10.3390/endocrines3030033
Chicago/Turabian StyleSavage, Martin O. 2022. "The Changing Face of Paediatric Human Growth Hormone Therapy" Endocrines 3, no. 3: 419-427. https://doi.org/10.3390/endocrines3030033