
Deciphering the Involvement of the Epicardium in Cardiac Diseases

 , , , , ,
, , , , ,  and
and 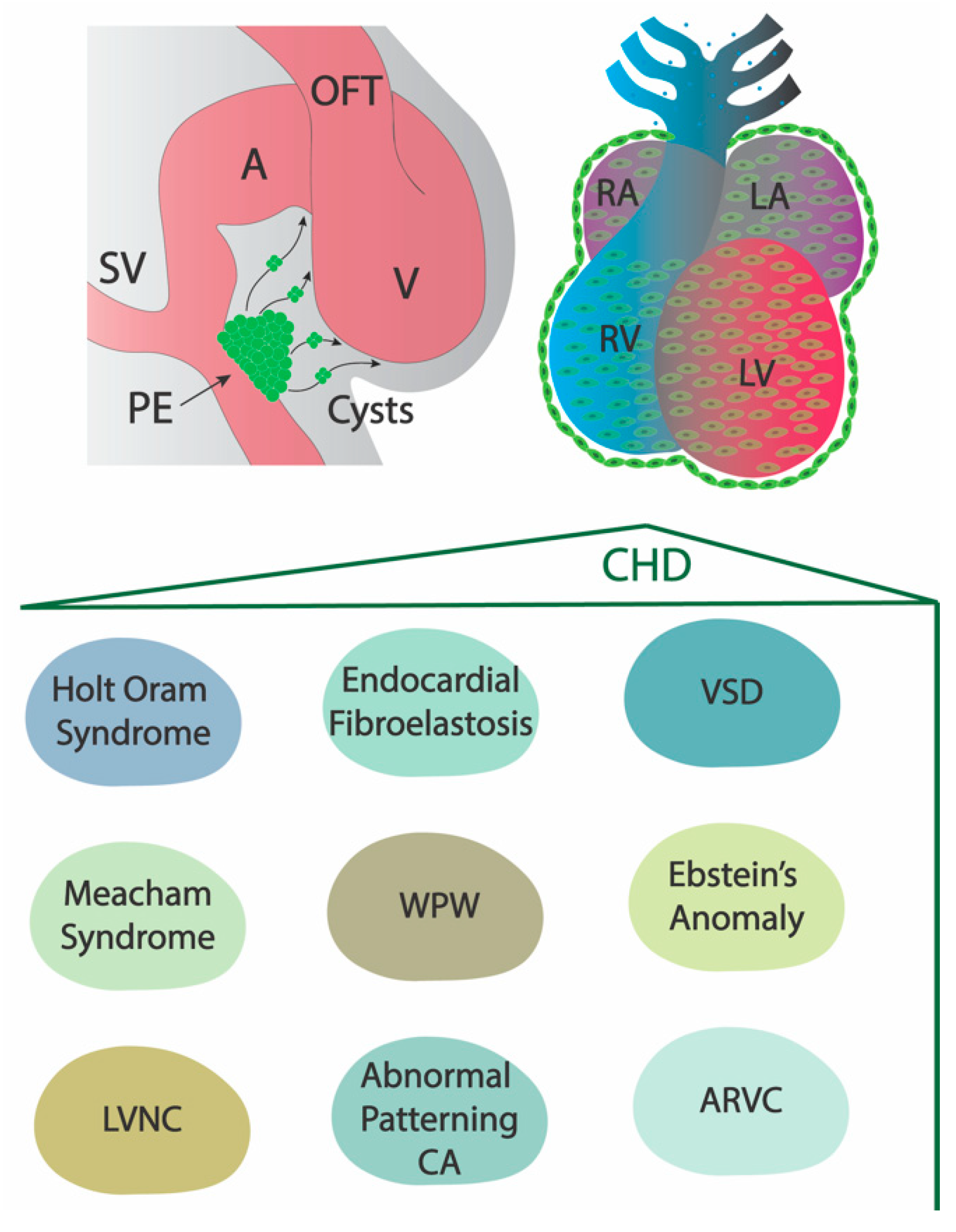{kind=link}
{kind=link}
Abstract
:1. Origin and Derivatives of the Epicardium
2. The Role of the Embryonic Epicardium in Adult Structural Heart Diseases
2.1. Holt–Oram Syndrome
2.2. Meacham Syndrome
2.3. Left Ventricular Non-Compaction
2.4. Endocardial Fibroelastosis
2.5. Abnormal Patterning of the Coronary Arteries
2.6. Arrhythmogenic Right Ventricular Cardiomyopathy
2.7. Ventricular Septal Defect
2.8. Ebstein’s Anomaly and Wolff–Parkinson–White (WPW) Syndrome
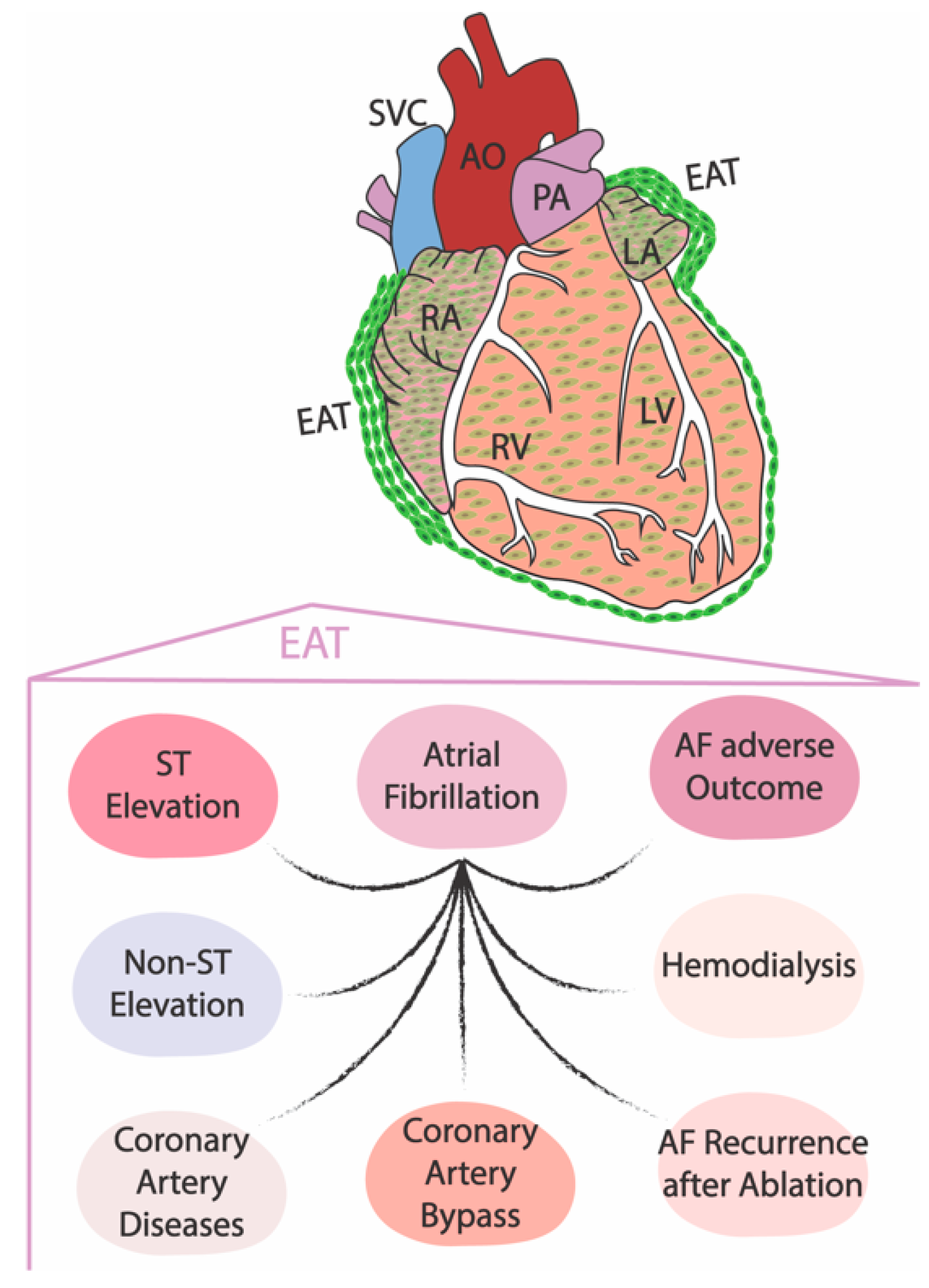
3. The Role of the Embryonic Epicardium in Adult Electrophysiological Heart Diseases
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hoit, B.D. Anatomy and Physiology of the Pericardium. Cardiol. Clin. 2017, 35, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, M.; Ito, K.; Shimada, Y. Origin and development of the epicardium in the mouse embryo. Anat. Embryol. 1987, 176, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Männer, J.; Perez-Pomares, J.M.; Macias, D.; and Munoz-Chapuli, R. The origin, formation and developmental significance of the epicardium: A review. Cells Tissues Organs 2001, 169, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Lie-Venema, H.; van den Akker, N.M.; Bax, N.A.; Winter, E.M.; Maas, S.; Kekarainen, T.; Hoeben, R.C.; deRuiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Sci. World J. 2007, 7, 1777–1798. [Google Scholar] [CrossRef] [PubMed]
- Carmona, R.; Guadix, J.A.; Cano, E.; Ruiz-Villalba, A.; Portillo-Sánchez, V.; Pérez-Pomares, J.M.; Muñoz-Chápuli, R. The embryonic epicardium: An essential element of cardiac development. J. Cell Mol. Med. 2010, 14, 2066–2072. [Google Scholar] [CrossRef] [PubMed]
- Niderla-BieliŃska, J.; Jankowska-Steifer, E.; Flaht-Zabost, A.; Gula, G.; Czarnowska, E.; Ratajska, A. Proepicardium: Current Understanding of its Structure, Induction, and Fate. Anat. Rec. 2019, 302, 893–903. [Google Scholar] [CrossRef]
- Muñoz-Chápuli, R.; Macías, D.; González-Iriarte, M.; Carmona, R.; Atencia, G.; Pérez-Pomares, J.M. The epicardium and epicardial-derived cells: Multiple functions in cardiac development. Rev. Esp. Cardiol. 2002, 55, 1070–1082. [Google Scholar] [CrossRef]
- Mommersteeg, M.T.; Domínguez, J.N.; Wiese, C.; Norden, J.; de Gier-de Vries, C.; Burch, J.B.; Kispert, A.; Brown, N.A.; Moorman, A.F.; Christoffels, V.M. The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc. Res. 2010, 87, 92–101. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell. 2012, 22, 639–650. [Google Scholar] [CrossRef]
- Plavicki, J.S.; Hofsteen, P.; Yue, M.S.; Lanham, K.A.; Peterson, R.E.; Heideman, W. Multiple modes of proepicardial cell migration require heartbeat. BMC Dev. Biol. 2014, 14, 18. [Google Scholar] [CrossRef]
- Cao, Y.; Duca, S.; Cao, J. Epicardium in Heart Development. Cold Spring Harb. Perspect. Biol. 2020, 12, a037192. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Miao, L.; Zhao, C.; Shaikh Qureshi, W.M.; Shieh, D.; Guo, H.; Lu, Y.; Hu, S.; Huang, A.; Zhang, L.; et al. CDC42 is required for epicardial and pro-epicardial development by mediating FGF receptor trafficking to the plasma membrane. Development 2017, 144, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Fernandez, C.; Rodriguez-Outeiriño, L.; Matias-Valiente, L.; Ramirez de Acuña, F.; Hernandez-Torres, F.; Lozano-Velasco, E.; Dominguez, J.N.; Franco, D.; Aranega, A.E. Regulation of Epicardial Cell Fate during Cardiac Development and Disease: An Overview. Int. J. Mol. Sci. 2022, 23, 3220. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.M.; Dronkers, E.; Goumans, M.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Vrancken Peeters, M.-P.F.M.; Gittenberger-de Groot, A.C.; Mentink, M.M.T.; and Poelmann, R.E. Smooth muscle cells and fibroblasts of the coronary arteries derive from epithelial-mesenchymal transformation of the epicardium. Anat. Embryol. 1999, 199, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lie-Venema, H.; Eralp, I.; Maas, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; and DeRuiter, M.C. Myocardial heterogeneity in permissiveness for epicardium-derived cells and endothelial precursor cells along the developing heart tube at the onset of coronary vascularization. Anat. Rec. 2005, 282A, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Risebro, C.A.; Vieira, J.M.; Klotz, L.; Riley, P.R. Characterisation of the human embryonic and foetal epicardium during heart development. Development 2015, 142, 3630–3636. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.-P.F.M.; Mentink, M.M.T.; Gourdie, R.G.; and Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9, discussion E9–E10. [Google Scholar] [CrossRef]
- Rudat, C.; Kispert, A. Wt1 and epicardial fate mapping. Circ. Res. 2012, 111, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Carmona, R.; Barrena, S.; López Gambero, A.J.; Rojas, A.; Muñoz-Chápuli, R. Epicardial cell lineages and the origin of the coronary endothelium. FASEB J. 2020, 43, 5223–5239. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytesfrom within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Braitsch, C.M.; Combs, M.D.; Quaggin, S.E.; Yutzey, K.E. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 2012, 368, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yuan, X.; Deng, H.; Huang, R.; Liu, B.; Xiong, T.; Long, X.; Zhang, L.; Li, Y.; She, Q. Single-cell and spatial heterogeneity landscapes of mature epicardial cells. J. Pharm. Anal. 2023, 13, 894–907. [Google Scholar] [CrossRef]
- Knight-Schrijver, V.R.; Davaapil, H.; Bayraktar, S.; Ross, A.D.B.; Kanemaru, K.; Cranley, J.; Dabrowska, M.; Patel, M.; Polanski, K.; He, X.; et al. A single-cell comparison of adult and fetal human epicardium defines the age-associated changes in epicardial activity. Nat. Cardiovasc. Res. 2022, 1, 1215–1229. [Google Scholar] [CrossRef]
- Lupu, I.E.; Redpath, A.N.; Smart, N. Spatiotemporal Analysis Reveals Overlap of Key Proepicardial Markers in the Developing Murine Heart. Stem Cell Rep. 2020, 14, 770–787. [Google Scholar] [CrossRef]
- Lavine, K.J.; Yu, K.; White, A.C.; Zhang, X.; Smith, C.; Partanen, J.; Ornitz, D.M. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell. 2005, 8, 85–95. [Google Scholar] [CrossRef]
- Vega-Hernández, M.; Kovacs, A.; De Langhe, S.; Ornitz, D.M. FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development 2011, 138, 3331–3340. [Google Scholar] [CrossRef]
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.; Hughes, J.; Hassan, A.B.; Brüning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef]
- Shen, H.; Cavallero, S.; Estrada, K.D.; Sandovici, I.; Kumar, S.R.; Makita, T.; Lien, C.L.; Constancia, M.; Sucov, H.M. Extracardiac control of embryonic cardiomyocyte proliferation and ventricular wall expansion. Cardiovasc. Res. 2015, 105, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Muñoz-Chápuli, R.; Hastie, N.D.; Pérez-Pomares, J.M.; Martínez-Estrada, O.M. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 2011, 138, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Ridge, L.A.; Mitchell, K.; Al-Anbaki, A.; Qureshi, W.M.S.; Stephen, L.A.; Tenin, G.; Lu, Y.; Lupu, I.E.; Clowes, C.; Robertson, A.; et al. Non-muscle myosin IIB (Myh10) is required for epicardial function and coronary vessel formation during mammalian development. PLOS Genet. 2017, 30, e1007068. [Google Scholar] [CrossRef] [PubMed]
- Marques, I.J.; Ernst, A.; Arora, P.; Vianin, A.; Hetke, T.; Sanz-Morejón, A.; Naumann, U.; Odriozola, A.; Langa, X.; Andrés-Delgado, L.; et al. Wt1 transcription factor impairs cardiomyocyte specification and drives a phenotypic switch from myocardium to epicardium. Development. 2022, 149, dev200375. [Google Scholar] [CrossRef] [PubMed]
- Boezio, G.L.M.; Zhao, S.; Gollin, J.; Priya, R.; Mansingh, S.; Guenther, S.; Fukuda, N.; Gunawan, F.; Stainer, D.Y.R. The developing epicardium regulates cardiac chamber morphogenesis by promoting cardiomyocyte growth. Dis. Model. Mech. 2023, 16, dmm049571. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Zuhlke, L.; Black, G.C.; Choy, M.K.; Li, N.; Keavney, B.D. Global birth prevalence of congenital heart defects 1970-2017: Updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 2019, 48, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Basson, C.T.; Cowley, G.D.; Solomon, S.D.; Weissman, B.; Poznanski, A.K.; Traill, T.A.; Seidman, J.G.; Seidman, C.E. The clinical and genetic spectrum of the Holt-Oram syndrome. N. Engl. J. Med. 1994, 330, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Sletten, L.J.; Pierpont, M.E. Variation in severity of cardiac disease in Holt-Oram syndrome. Am. J. Med. Genet. 1996, 65, 128–132. [Google Scholar] [CrossRef]
- Diman, N.Y.S.G.; Brooks, G.; Kruithof, B.P.T.; Elemento, O.; Seidman, J.G.; Seidman, C.E.; Basson, C.T.; Hatcher, C.J. Tbx5 is required for avian and Mammalian epicardial formation and coronary vasculogenesis. Circ. Res. 2014, 115, 834–844. [Google Scholar] [CrossRef]
- Suri, M.; Kelehan, P.; O’neill, D.; Vadeyar, S.; Grant, J.; Ahmed, S.F.; Tolmie, J.; McCann, E.; Lam, W.; Smith, S.; et al. WT1 mutations in Meacham syndrome suggest a coelomic mesothelial origin of the cardiac and diaphragmatic malformations. Am. J. Med. Genet. 2007, 143A, 2312–2320. [Google Scholar] [CrossRef]
- Zamora, M.; Männer, J.; Ruiz-Lozano, P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc. Natl. Acad. Sci. USA 2007, 104, 18109–18114. [Google Scholar] [CrossRef]
- Mahtab, E.A.F.; Vicente-Steij, R.; Hahurij, N.D.; Jongbloed, M.R.M.; Wisse, L.J.; DeRuiter, M.C.; Uhrin, P.; Zaujec, J.; Binder, B.R.; Schalij, M.J.; et al. Podoplanin Deficient Mice Show a RhoA-Related Hypoplasia of the Sinus VenosusMyocardium Including the Sinoatrial Node. Dev. Dyn. 2009, 238, 183–193. [Google Scholar] [CrossRef]
- Arora, H.; Boulberdaa, M.; Qureshi, R.; Bitirim, V.; Gasser, A.; Messaddeq, N.; Dolle, P.; Nebigil, C.G. Prokineticin receptor-1 signaling promotes Epicardial to Mesenchymal Transition during heart development. Sci. Rep. 2016, 6, 25541. [Google Scholar] [CrossRef] [PubMed]
- Bonet, F.; Añez, S.B.; Inácio, J.M.; Futschik, M.E.; Belo, J.A. CCBE1 Is Essential for Epicardial Function during Myocardium Development. Int. J. Mol. Sci. 2022, 23, 12642. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Braitsch, C.M.; Lange, A.W.; James, J.F.; Yutzey, K.E. NFATC1 promotes epicardium-derived cell invasion into myocardium. Development 2011, 138, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, X.; Liu, K.; Tang, J.; He, L.; Pu, W.; Liu, Q.; Li, Y.; Tian, X.; Wang, Y.; et al. Fibroblasts in an endocardial fibroelastosis disease model mainly originate from mesenchymal derivatives of epicardium. Cell Res. 2017, 27, 1157–1177. [Google Scholar] [CrossRef] [PubMed]
- Withana, M.; Uribe, C.; Gregoric, I.D.; Angelini, P. Low Origin of the Coronary Arteries and a Small Aortic Annulus. Tex. Heart Inst. J. 2019, 46, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Matthes, S.A.; Taffet, S.; Delmar, M. Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun. Adhes. 2011, 18, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Cheedipudi, S.M.; Rouhi, L.; Fan, S.; Simon, L.; Zhao, Z.; Hong, K.; Gurha, P.; Marian, A.J. Single-Cell RNA Sequencing Uncovers Paracrine Functions of the Epicardial-Derived Cells in Arrhythmogenic Cardiomyopathy. Circulation 2021, 143, 2169–2187. [Google Scholar] [CrossRef]
- Maione, A.S.; Stadiotti, I.; Pilato, C.A.; Perrucci, G.L.; Saverio, V.; Catto, V.; Vettor, G.; Casella, M.; Guarino, A.; Polvani, G.; et al. Excess TGF-β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 2673. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Gittenberger-de Groot, A.C.; Vicente-Steijn, R.; Wisse, L.J.; Bartelings, M.M.; Everts, S.; Hoppenbrouwers, T.; Kruithof, B.P.T.; Jensen, B.; de Bruin, P.W.; et al. Evolution and Development of Ventricular Septation in the Amniote Heart. PLoS ONE 2014, 9, e106569. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Jensen, B.; Bartelings, M.M.; Richardson, M.K.; Gittenberger-de Groot, A.C.; Nakanishi, T.; Markwald, R.R.; Baldwin, H.S.; Keller, B.B.; Srivastava, D.; et al. The Epicardium in Ventricular Septation During Evolution and Development. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Springer: Tokyo, Japan, 2016; Volume 25. [Google Scholar] [CrossRef]
- Kolditz, D.P.; Wijffels, M.C.E.F.; Blom, N.A.; van der Laarse, A.; Hahurij, N.D.; Lie-Venema, H.; Markwald, R.R.; Poelmann, R.E.; Schalij, M.J.; Gittenberger-de Groot, A.C. Epicardium-Derived Cells in Development of Annulus Fibrosis and Persistence of Accessory Pathways. Circulation 2008, 117, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Tuyls, E.; Gharaviri, A.; Hulsmans, S.; van Hunnik, A.; Kuiper, M.; Serroyen, J.; Zeemering, S.; Kuijpers, N.H.; Schotten, U. Loss of continuity in the thin epicardial layer because of endomysial fibrosis increases the complexity of atrial fibrillatory conduction. Circ. Arrhythm. Electrophysiol. 2013, 6, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Cavallero, S.; Patterson, M.; Shen, H.; Xu, J.; Kumar, S.R.; Sucov, H.M. Adipogenesis and epicardial adipose tissue: A novel fate of the epicardium induced by mesenchymal transformation and PPARγ activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- van Rosendael, A.R.; Smit, J.M.; El’Mahdiui, M.; van Rosendael, P.J.; Leung, M.; Delgado, V.; Bax, J.J. Association between left atrial epicardial fat, left atrial volume, and the severity of atrial fibrillation. Europace 2022, 24, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, N.; Okumura, Y.; Arai, M.; Kurokawa, S.; Nagashima, K.; Watanabe, R.; Wakamatsu, Y.; Yagyu, S.; Ohkubo, K.; Nakai, T.; et al. Effect of obesity and epicardial fat/fatty infiltration on electrical and structural remodeling associated with atrial fibrillation in a novel canine model of obesity and atrial fibrillation: A comparative study. J. Cardiovasc. Electrophysiol. 2021, 32, 889–899. [Google Scholar] [CrossRef]
- Nakamori, S.; Nezafat, M.; Ngo, L.H.; Manning, W.J.; Nezafat, R. Left Atrial Epicardial Fat Volume Is Associated With Atrial Fibrillation: A Prospective Cardiovascular Magnetic Resonance 3D Dixon Study. J. Am. Heart Assoc. 2018, 7, e008232. [Google Scholar] [CrossRef] [PubMed]
- Yorgun, H.; Canpolat, U.; Aytemir, K.; Hazırolan, T.; Şahiner, L.; Kaya, E.B.; Kabakci, G.; Tokgözoğlu, L.; Özer, N.; Oto, A. Association of epicardial and peri-atrial adiposity with the presence and severity of non-valvular atrial fibrillation. Int. J. Cardiovasc. Imaging 2015, 31, 649–657. [Google Scholar] [CrossRef]
- Zhao, L.; Harrop, D.L.; Ng, A.C.T.; Wang, W.Y.S. Epicardial Adipose Tissue Is Associated with Left Atrial Dysfunction in People Without Obstructive Coronary Artery Disease or Atrial Fibrillation. Can. J. Cardiol. 2018, 34, 1019–1025. [Google Scholar] [CrossRef]
- Bos, D.; Vernooij, M.W.; Shahzad, R.; Kavousi, M.; Hofman, A.; van Walsum, T.; Deckers, J.W.; Ikram, M.A.; Heeringa, J.; Franco, O.H.; et al. Epicardial Fat Volume and the Risk of Atrial Fibrillation in the General Population Free of Cardiovascular Disease. JACC Cardiovasc. Imaging. 2017, 10, 1405–1407. [Google Scholar] [CrossRef]
- Oba, K.; Maeda, M.; Maimaituxun, G.; Yamaguchi, S.; Arasaki, O.; Fukuda, D.; Yagi, S.; Hirata, Y.; Nishio, S.; Iwase, T.; et al. Effect of the Epicardial Adipose Tissue Volume on the Prevalence of Paroxysmal and Persistent Atrial Fibrillation. Circ. J. 2018, 82, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Eren, H.; Omar, M.B.; Kaya, Ü.; Öcal, L.; Yilmaz, M.F.; Akkan, S. Epicardial adipose tissue may predict new-onset atrial fibrillation in patients with ST-segment elevation myocardial infarction. J. Cardiovasc. Med. 2021, 22, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Eren, H.; Omar, M.B.; Öcal, L. Epicardial fat tissue may predict new-onset atrial fibrillation in patients with non-ST-segment elevation myocardial infarction. Turk. Kardiyol. Dern. Ars. 2021, 49, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, H.; Yoshida, K.; Nogami, A.; Ieda, M. Difference in epicardial adipose tissue distribution between paroxysmal atrial fibrillation and coronary artery disease. Heart Vessel. 2020, 35, 1070–1078. [Google Scholar] [CrossRef]
- Sevinc, D.; Pasaoglu, L.; Coskun, R.; Atci, N.; Alimli, A.; Ucar, O. Relationships between left atrial pericardial fat and permanent atrial fibrillation: Results of a case-control study. Diagn. Interv. Imaging 2016, 97, 307–313, Erratum in: Diagn. Interv. Imaging 2017, 98, 283. Ucar, O.. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Park, J.; Park, J.K.; Uhm, J.S.; Joung, B.; Lee, M.H.; Pak, H.N. Pericardial fat volume is associated with clinical recurrence after catheter ablation for persistent atrial fibrillation, but not paroxysmal atrial fibrillation: An analysis of over 600-patients. Int. J. Cardiol. 2014, 176, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.F.; Hung, C.L.; Tsao, H.M.; Lin, Y.J.; Yun, C.H.; Lai, Y.H.; Chang, S.L.; Lo, L.W.; Hu, Y.F.; Tuan, T.C.; et al. Epicardial adipose tissue thickness and ablation outcome of atrial fibrillation. PLoS ONE 2013, 8, e74926. [Google Scholar] [CrossRef]
- Drossos, G.; Koutsogiannidis, C.P.; Ananiadou, O.; Kapsas, G.; Ampatzidou, F.; Madesis, A.; Bismpa, K.; Palladas, P.; Karagounis, L. Pericardial fat is strongly associated with atrial fibrillation after coronary artery bypass graft surgery. Eur. J. Cardiothorac. Surg. 2014, 46, 1014–1020, discussion 1020. [Google Scholar] [CrossRef]
- Guzel, F.B.; Altunoren, O.; Gunes, H.; Seyithanoglu, M.; Kerkutluoglu, M.; Sezal, D.T.; Gungor, O. The relation between epicardial fat tissue thickness and atrial fibrillation ın hemodialysis patients. Semin. Dial. 2020, 33, 428–434. [Google Scholar] [CrossRef]
- Chu, C.Y.; Lee, W.H.; Hsu, P.C.; Lee, M.K.; Lee, H.H.; Chiu, C.A.; Lin, T.H.; Lee, C.S.; Yen, H.W.; Voon, W.C.; et al. Association of Increased Epicardial Adipose Tissue Thickness with Adverse Cardiovascular Outcomes in Patients With Atrial Fibrillation. Medicine 2016, 95, e2874. [Google Scholar] [CrossRef]
- Nalliah, C.J.; Bell, J.R.; Raaijmakers, A.J.A.; Waddell, H.M.; Wells, S.P.; Bernasochi, G.B.; Montgomery, M.K.; Binny, S.; Watts, T.; Joshi, S.B.; et al. Epicardial Adipose Tissue Accumulation Confers Atrial Conduction Abnormality. J. Am. Coll. Cardiol. 2020, 76, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Wang, N.; Meigs, J.B.; Hoffmann, U.; Massaro, J.M.; Fox, C.S.; Magnani, J.W. Pericardial fat is associated with atrial conduction: The Framingham Heart Study. J. Am. Heart Assoc. 2014, 3, e000477. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm. 2018, 15, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Kira, S.; Abe, I.; Ishii, Y.; Miyoshi, M.; Oniki, T.; Arakane, M.; Daa, T.; Teshima, Y.; Yufu, K.; Shimada, T.; et al. Role of angiopoietin-like protein 2 in atrial fibrosis induced by human epicardial adipose tissue: Analysis using an organo-culture system. Heart Rhythm. 2020, 17, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xi, W.; Yin, L.; Wang, J.; Shen, H.; Gao, Y.; Min, J.; Zhang, Y.; Wang, Z. Human Epicardial Adipose Tissue cTGF Expression is an Independent Risk Factor for Atrial Fibrillation and Highly Associated with Atrial Fibrosis. Sci. Rep. 2018, 8, 3585. [Google Scholar] [CrossRef] [PubMed]
- Suffee, N.; Moore-Morris, T.; Jagla, B.; Mougenot, N.; Dilanian, G.; Berthet, M.; Proukhnitzky, J.; Le Prince, P.; Tregouet, D.A.; Pucéat, M.; et al. Reactivation of the Epicardium at the Origin of Myocardial Fibro-Fatty Infiltration During the Atrial Cardiomyopathy. Circ. Res. 2020, 126, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, E.R.; Al-Shama, R.F.M.; Kawasaki, M.; Fabrizi, B.; Neefs, J.; Wesselink, R.; Ernault, A.C.; Piersma, S.; Pham, T.V.; Jimenez, C.R.; et al. Atrial epicardial adipose tissue abundantly secretes myeloperoxidase and activates atrial fibroblasts in patients with atrial fibrillation. J. Transl. Med. 2023, 21, 366. [Google Scholar] [CrossRef] [PubMed]
- Kusayama, T.; Furusho, H.; Kashiwagi, H.; Kato, T.; Murai, H.; Usui, S.; Kaneko, S.; Takamura, M. Inflammation of left atrial epicardial adipose tissue is associated with paroxysmal atrial fibrillation. J. Cardiol. 2016, 68, 406–411. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, F.; Yang, M.; Zhong, J. Increasing Level of Interleukin-1β in Epicardial Adipose Tissue Is Associated with Persistent Atrial Fibrillation. J. Interferon Cytokine Res. 2020, 40, 64–69. [Google Scholar] [CrossRef]
- Li, B.; Po, S.S.; Zhang, B.; Bai, F.; Li, J.; Qin, F.; Liu, N.; Sun, C.; Xiao, Y.; Tu, T.; et al. Metformin regulates adiponectin signalling in epicardial adipose tissue and reduces atrial fibrillation vulnerability. J. Cell Mol. Med. 2020, 24, 7751–7766. [Google Scholar] [CrossRef]
- Girerd, N.; Scridon, A.; Bessière, F.; Chauveau, S.; Geloen, A.; Boussel, L.; Morel, E.; Chevalier, P. Periatrial epicardial fat is associated with markers of endothelial dysfunction in patients with atrial fibrillation. PLoS ONE 2013, 8, e77167. [Google Scholar] [CrossRef] [PubMed]
- Shaihov-Teper, O.; Ram, E.; Ballan, N.; Brzezinski, R.Y.; Naftali-Shani, N.; Masoud, R.; Ziv, T.; Lewis, N.; Schary, Y.; Levin-Kotler, L.P.; et al. Extracellular Vesicles From Epicardial Fat Facilitate Atrial Fibrillation. Circulation 2021, 143, 2475–2493. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Luo, F.; Lei, K. Exosomes Containing LINC00636 Inhibit MAPK1 through the miR-450a-2-3p Overexpression in Human Pericardial Fluid and Improve Cardiac Fibrosis in Patients with Atrial Fibrillation. Mediat. Inflamm. 2021, 2021, 9960241. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Shao, X.; Liu, B.; Lv, M.; Pandey, P.; Guo, C.; Zhang, R.; Zhang, Y. Genome-wide screening of functional long noncoding RNAs in the epicardial adipose tissues of atrial fibrillation. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165757. [Google Scholar] [CrossRef] [PubMed]
- Pokushalov, E.; Kozlov, B.; Romanov, A.; Strelnikov, A.; Bayramova, S.; Sergeevichev, D.; Bogachev-Prokophiev, A.; Zheleznev, S.; Shipulin, V.; Salakhutdinov, N.; et al. Botulinum toxin injection in epicardial fat pads can prevent recurrences of atrial fibrillation after cardiac surgery: Results of a randomized pilot study. J. Am. Coll. Cardiol. 2014, 64, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Kim, B.J.; Cha, T.J.; Heo, J.H.; Kim, H.S.; Lee, J.W. Impact of duration and dosage of statin treatment and epicardial fat thickness on the recurrence of atrial fibrillation after electrical cardioversion. Heart Vessel. 2015, 30, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Carmona, R.; López-Sánchez, C.; Garcia-Martinez, V.; Garcia-López, V.; Muñoz-Chápuli, R.; Lozano-Velasco, E.; Franco, D. Novel Insights into the Molecular Mechanisms Governing Embryonic Epicardium Formation. J. Cardiovasc. Dev. Dis. 2023, 10, 440. [Google Scholar] [CrossRef]
- Streef, T.J.; Groeneveld, E.J.; van Herwaarden, T.; Hjortnaes, J.; Goumans, M.J.; Smits, A.M. Single-cell analysis of human fetal epicardium reveals its cellular composition and identifies CRIP1 as a modulator of EMT. Stem Cell Rep. 2023, 18, 1421–1435. [Google Scholar] [CrossRef]
- Tyser, R.C.V.; Ibarra-Soria, X.; McDole, K.; Arcot Jayaram, S.; Godwin, J.; van den Brand, T.A.H.; Miranda, A.M.A.; Scialdone, A.; Keller, P.J.; Marioni, J.C.; et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science 2021, 371, eabb2986. [Google Scholar] [CrossRef]
- Quijada, P.; Trembley, M.A.; Misra, A.; Myers, J.A.; Baker, C.D.; Pérez-Hernández, M.; Myers, J.R.; Dirkx, R.A., Jr.; Cohen, E.D.; Delmar, M.; et al. Coordination of endothelial cell positioning and fate specification by the epicardium. Nat. Commun. 2021, 12, 4155. [Google Scholar] [CrossRef]
- Meier, A.B.; Zawada, D.; De Angelis, M.T.; Martens, L.D.; Santamaria, G.; Zengerle, S.; Nowak-Imialek, M.; Kornherr, J.; Zhang, F.; Tian, Q.; et al. Epicardioid single-cell genomics uncovers principles of human epicardium biology in heart development and disease. Nat. Biotechnol. 2023, 3, 1–14. [Google Scholar] [CrossRef]
- Zou, R.; Zhang, M.; Zou, Z.; Shi, W.; Tan, S.; Wang, C.; Xu, W.; Jin, J.; Milton, S.; Chen, Y.; et al. Single-cell transcriptomics reveals zinc and copper ions homeostasis in epicardial adipose tissue of heart failure. Int. J. Biol. Sci. 2023, 19, 4036–4051. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona, R.; López-Sánchez, C.; García-Martinez, V.; García-López, V.; Muñoz-Chápuli, R.; Lozano-Velasco, E.; Franco, D. Deciphering the Involvement of the Epicardium in Cardiac Diseases. Hearts 2023, 4, 81-93. https://doi.org/10.3390/hearts4040011
Carmona R, López-Sánchez C, García-Martinez V, García-López V, Muñoz-Chápuli R, Lozano-Velasco E, Franco D. Deciphering the Involvement of the Epicardium in Cardiac Diseases. Hearts. 2023; 4(4):81-93. https://doi.org/10.3390/hearts4040011
Chicago/Turabian StyleCarmona, Rita, Carmen López-Sánchez, Virginio García-Martinez, Virginio García-López, Ramón Muñoz-Chápuli, Estefanía Lozano-Velasco, and Diego Franco. 2023. "Deciphering the Involvement of the Epicardium in Cardiac Diseases" Hearts 4, no. 4: 81-93. https://doi.org/10.3390/hearts4040011