
Comparative Microbial Community Analysis of Fur Seals and Aquaculture Salmon Gut Microbiomes in Tasmania
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
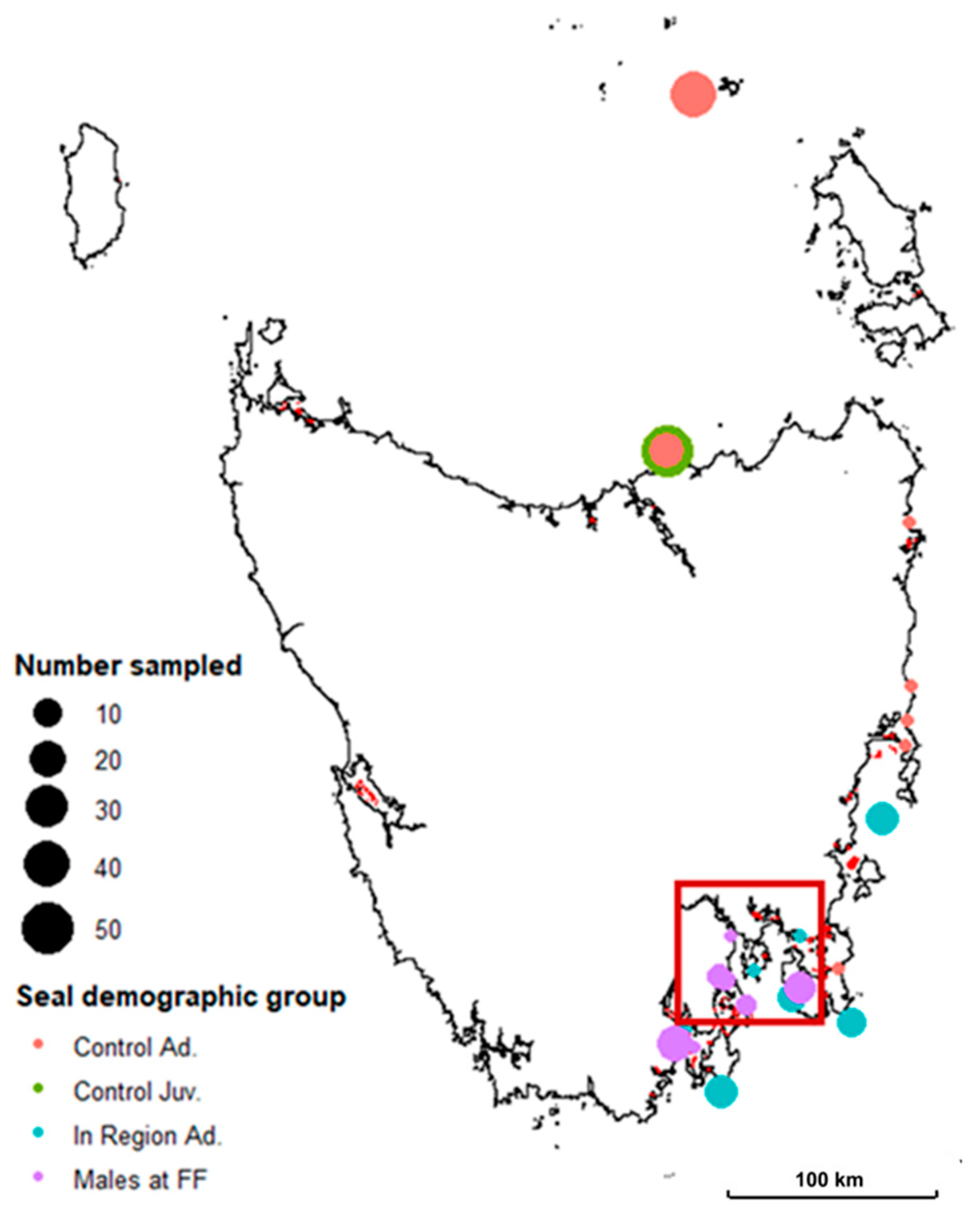
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. PCR and Sanger Sequencing of Enteric Isolates for Comparison to ASV Classification Post-Processing
2.4. Microbiome Analysis
2.5. Compositional Taxonomic Diversity Analysis
2.6. Isolate Sequence Database Construction and Utilization
3. Results
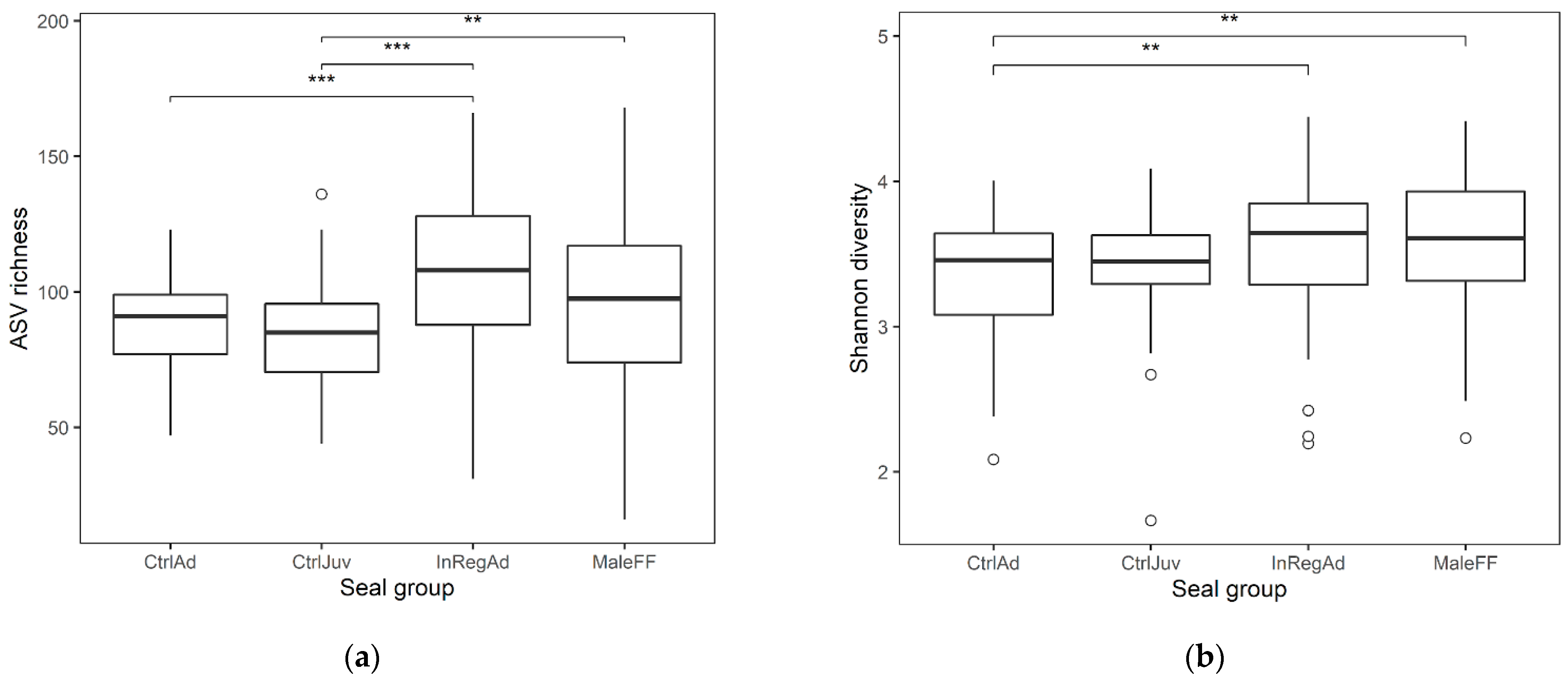
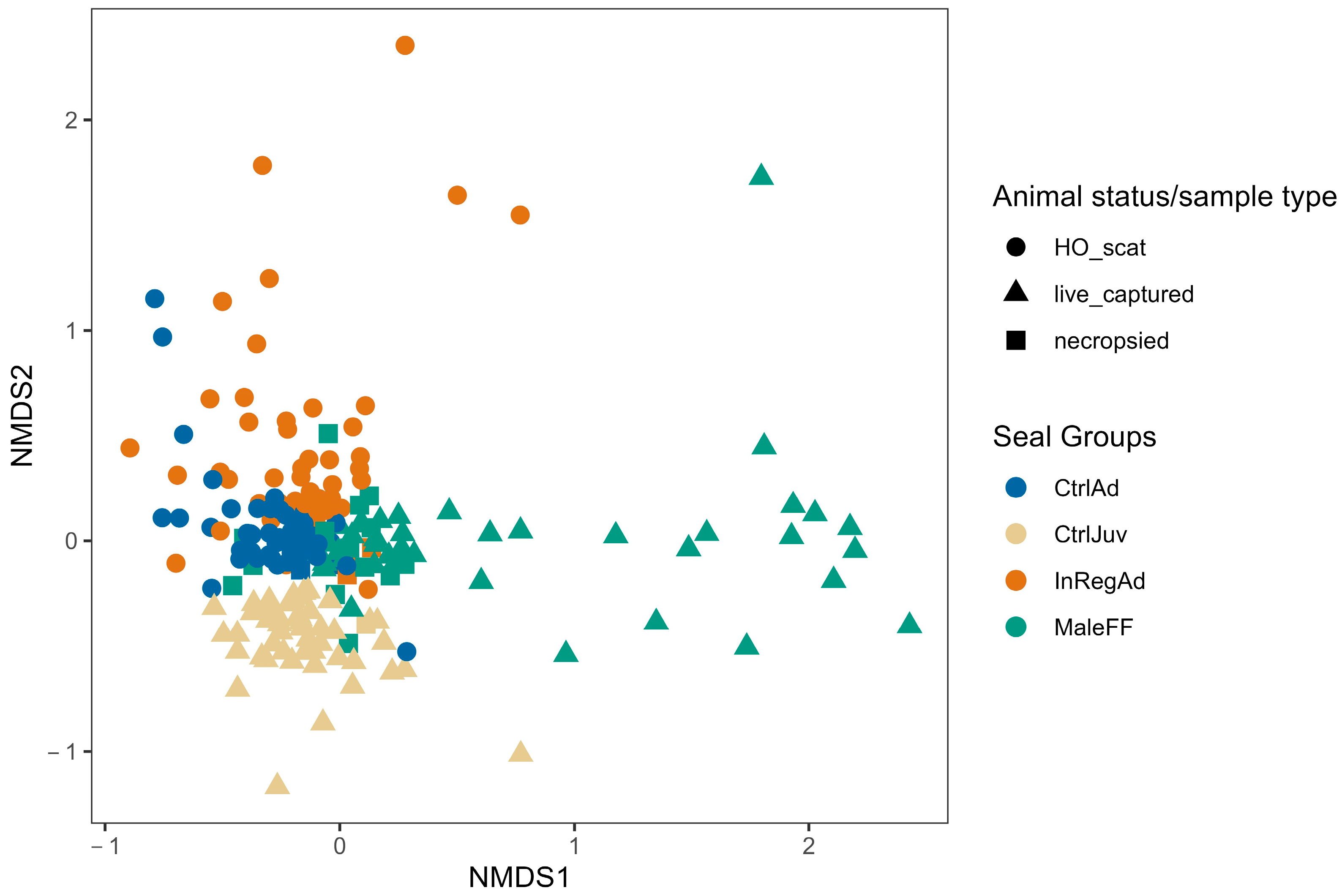
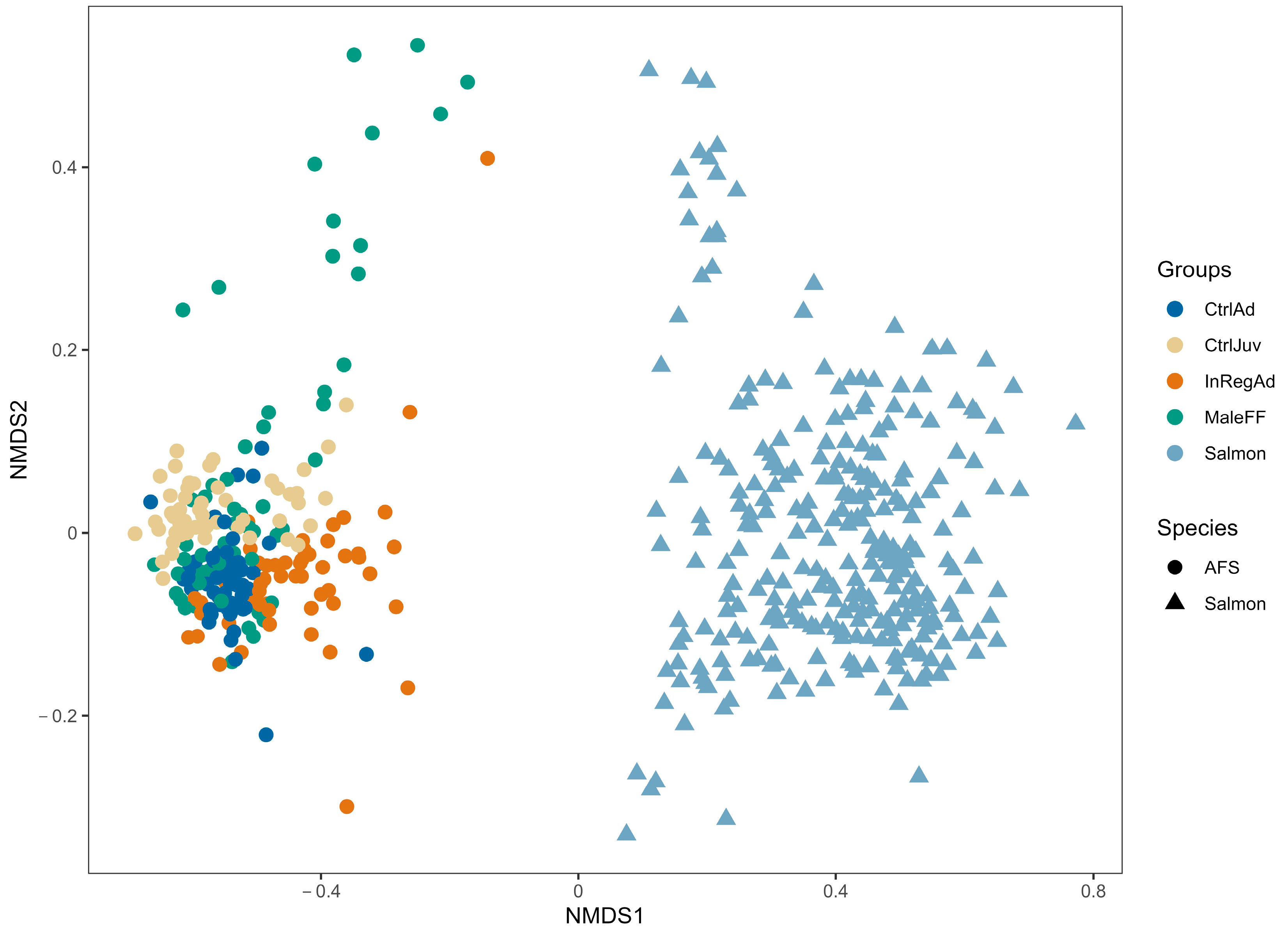
3.1. Seal Sample Alpha and Beta Diversity
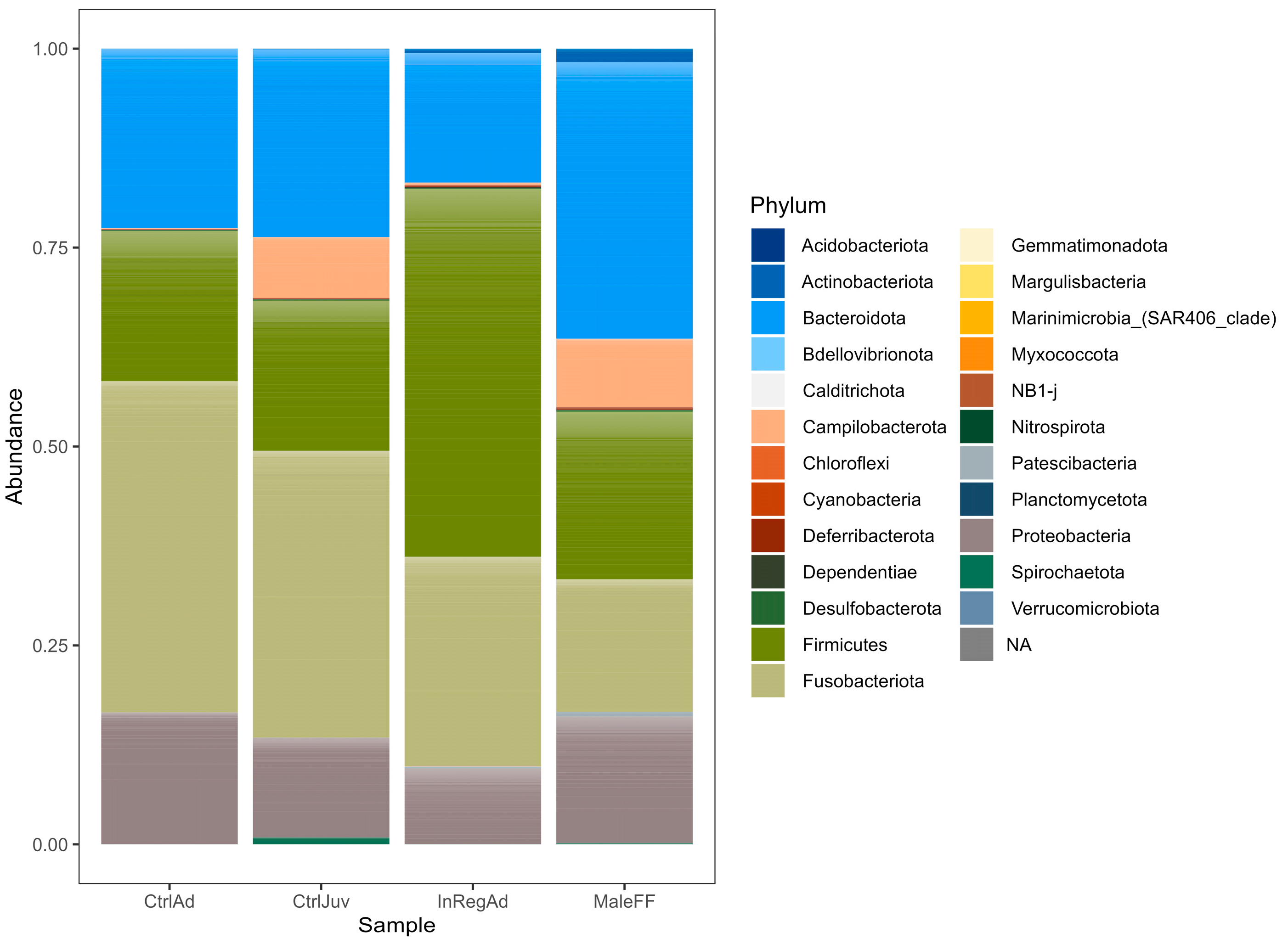
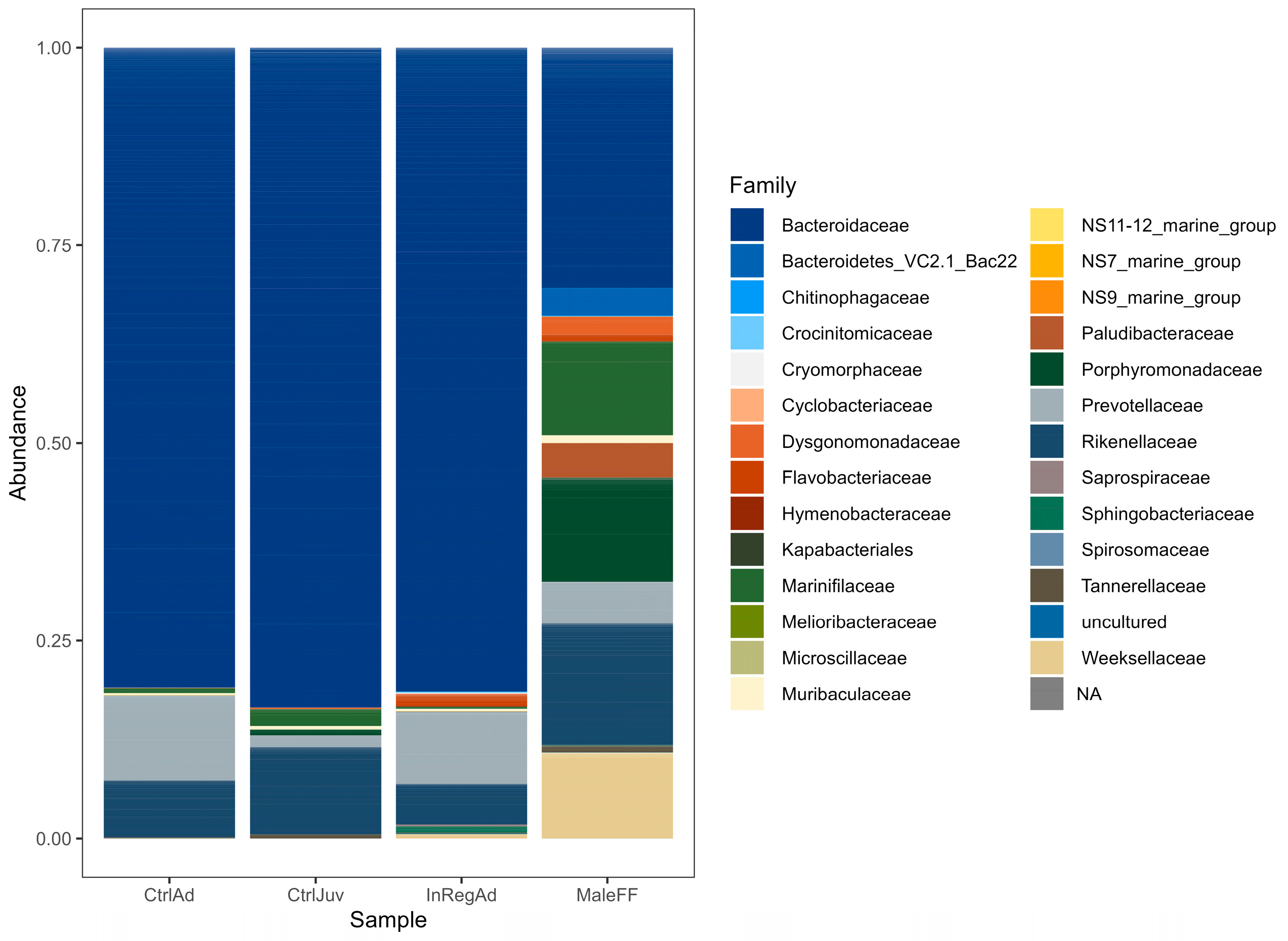
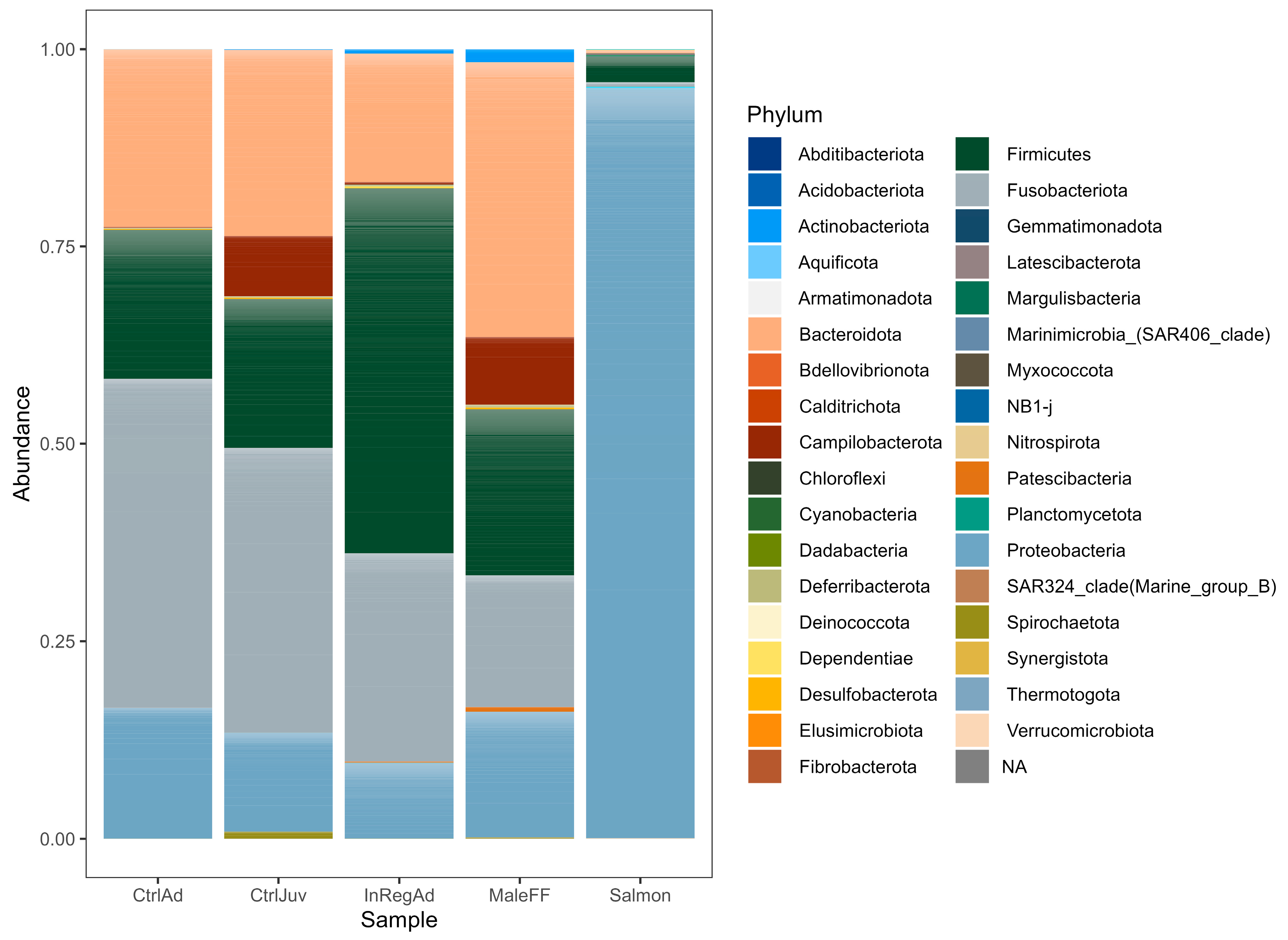
3.2. Taxonomic Diversity Differences within the Seals in Tasmania
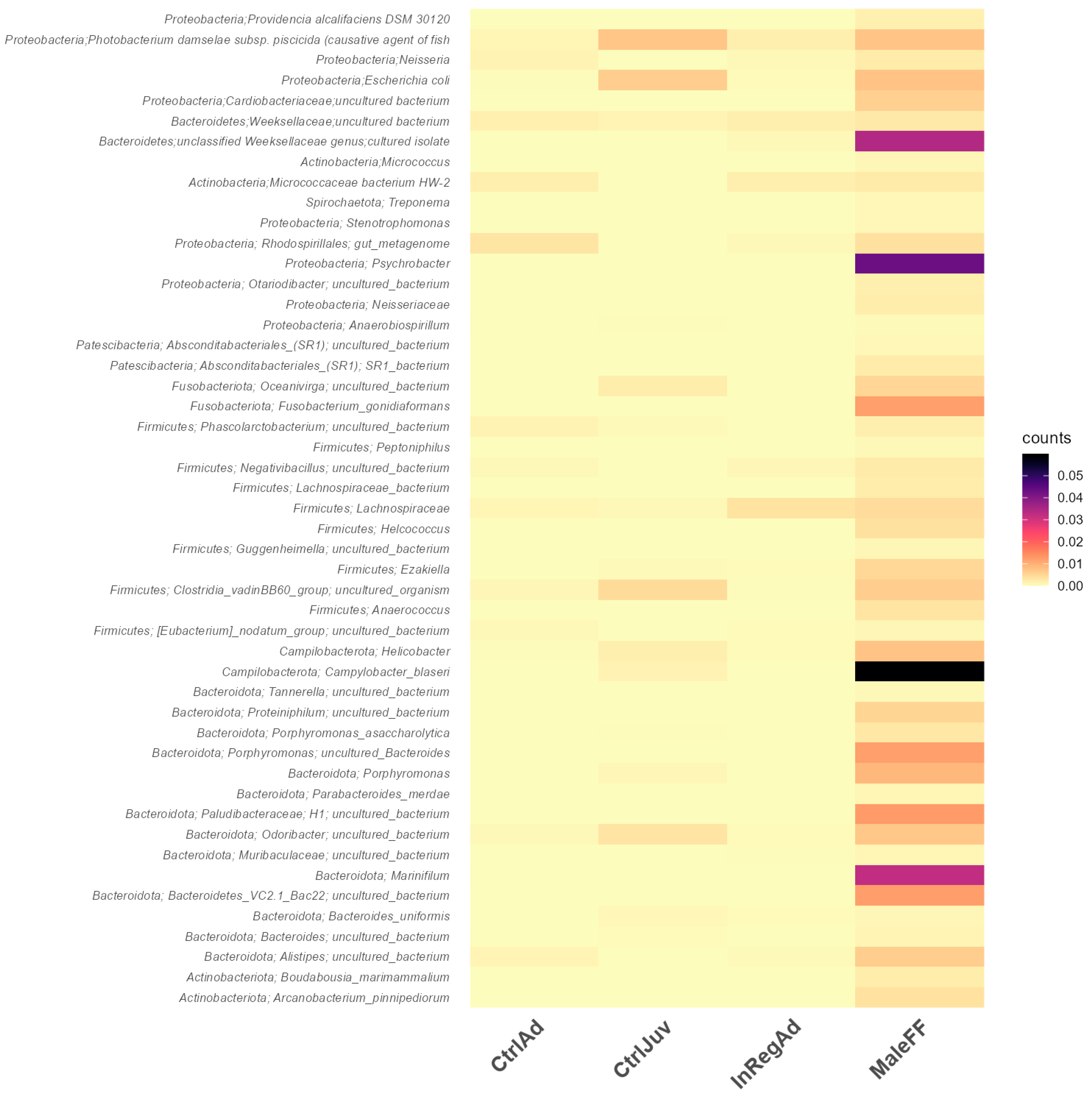
3.3. Differentially Abundant ASVs Also Present in Salmon
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowen, W.; Lidgard, D. Vertebrate Predator Control: Effects on Prey Populations in Terrestrial and Aquatic Ecosystems. 2011. Available online: https://publications.gc.ca/collections/collection_2013/mpo-dfo/Fs70-5-2011-028-eng.pdf (accessed on 28 March 2023).
- Gulland, J.A. Seals and fisheries: A case for predator control? Trends Ecol. Evol. 1987, 2, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Kemper, C.; Pemberton, D.; Cawthorn, M.; Heinrich, S.; Mann, J.; Würsig, B.; Shaughnessy, P. Aquaculture and Marine Mammals: Co-Existence or Conflict? In Marine Mammals: Fisheries, Tourism and Management Issues; CSIRO: Canberra, Australia, 2003. [Google Scholar]
- Australian Bureau of Agricultural Resource Economics and Sciences (ABARES). Australian Fisheries and Aquaculture Statistics 2017; ABARES: Canberra, Australia, 2018. [CrossRef]
- Patterson, H.; Parsa, M.; Larcombe, J.; Noriega, R.; Emery, T.; Georgeson, L.; Marton, N.; Williams, A.; Woodhams, J.; Helidoniotis, F. Fishery Status Reports 2019; ABARES: Canberra, Australia, 2019.
- Kirkwood, R.; Pemberton, D.; Gales, R.; Hoskins, A.J.; Mitchell, T.; Shaughnessy, P.D.; Arnould, J.P.Y.; Kirkwood, R.; Pemberton, D.; Gales, R.; et al. Continued population recovery by Australian fur seals. Mar. Freshw. Res. 2010, 61, 695–701. [Google Scholar] [CrossRef]
- Deagle, B.E.; Kirkwood, R.; Jarman, S.N. Analysis of Australian fur seal diet by pyrosequencing prey DNA in faeces. Mol. Ecol. 2009, 18, 2022–2038. [Google Scholar] [CrossRef]
- Gales, R.; Pemberton, D. Diet of the Australian fur seal in Tasmania. Mar. Freshw. Res. 1994, 45, 653–664. [Google Scholar] [CrossRef]
- Nash, C.E.; Iwamoto, R.N.; Mahnken, C.V.W. Aquaculture risk management and marine mammal interactions in the Pacific Northwest. Aquaculture 2000, 183, 307–323. [Google Scholar] [CrossRef]
- Würsig, B.; Gailey, G.A. Marine mammals and aquaculture: Conflicts and potential resolutions. In Responsible Marine Aquaculture; CABI: Wallingford, UK, 2002; pp. 45–59. [Google Scholar] [CrossRef] [Green Version]
- Pemberton, D.; Shaughnessy, P.D. Interaction between seals and marine fish-farms in Tasmania, and management of the problem. Aquat. Conserv. Mar. Freshw. Ecosyst. 1993, 3, 149–158. [Google Scholar] [CrossRef]
- Robinson, S.; Terauds, A.; Gales, R.; Greenwood, M. Mitigating fur seal interactions: Relocation from Tasmanian aquaculture farms. Aquat. Conserv. 2008, 18, 1180–1188. [Google Scholar] [CrossRef]
- Scott, T.M.; Rose, J.B.; Jenkins, T.M.; Farrah, S.R.; Lukasik, J. Microbial source tracking: Current methodology and future directions. Appl. Environ. Microbiol. 2002, 68, 5796–5803. [Google Scholar] [CrossRef] [Green Version]
- González-Fernández, A.; Symonds, E.M.; Gallard-Gongora, J.F.; Mull, B.; Lukasik, J.O.; Rivera Navarro, P.; Badilla Aguilar, A.; Peraud, J.; Brown, M.L.; Mora Alvarado, D.; et al. Relationships among microbial indicators of fecal pollution, microbial source tracking markers, and pathogens in Costa Rican coastal waters. Water Res. 2021, 188, 116507. [Google Scholar] [CrossRef]
- Geldreich, E.E.; Litsky, W. Fecal coliform and fecal streptococcus density relationships in waste discharges and receiving waters. C R C Crit. Rev. Environ. Control 1976, 6, 349–369. [Google Scholar] [CrossRef]
- Murillo, T.; Schneider, D.; Heistermann, M.; Daniel, R.; Fichtel, C. Assessing the drivers of gut microbiome composition in wild redfronted lemurs via longitudinal metacommunity analysis. Sci. Rep. 2022, 12, 21462. [Google Scholar] [CrossRef]
- Mathai, P.P.; Staley, C.; Sadowsky, M.J. Sequence-enabled community-based microbial source tracking in surface waters using machine learning classification: A review. J. Microbiol. Methods 2020, 177, 106050. [Google Scholar] [CrossRef]
- Paruch, L.; Paruch, A.M. An overview of microbial source tracking using host-specific genetic markers to identify origins of fecal contamination in different water environments. Water 2022, 14, 1809. [Google Scholar] [CrossRef]
- Ahmed, W. Limitations of library-dependent microbial source tracking methods. Water (AWA) 2007, 34, 96–101. [Google Scholar]
- Bourne, D.G.; Dennis, P.G.; Uthicke, S.; Soo, R.M.; Tyson, G.W.; Webster, N. Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J. 2013, 7, 1452–1458. [Google Scholar] [CrossRef]
- Kembel, S.W.; Jones, E.; Kline, J.; Northcutt, D.; Stenson, J.; Womack, A.M.; Bohannan, B.J.; Brown, G.Z.; Green, J.L. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 2012, 6, 1469–1479. [Google Scholar] [CrossRef] [Green Version]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukerb, A.M.; Noël, C.; Quenot, E.; Cadiou, B.; Chevé, J.; Quintric, L.; Cormier, A.; Dantan, L.; Gourmelon, M. Comparative analysis of fecal microbiomes from wild waterbirds to poultry, cattle, pigs, and wastewater treatment plants for a microbial source tracking approach. Front. Microbiol. 2021, 12, 697553. [Google Scholar] [CrossRef] [PubMed]
- Holyoak, M.; Leibold, M.A.; Holt, R.D. Metacommunities: Spatial Dynamics and Ecological Communities; University of Chicago Press: Chicago, IL, USA, 2005. [Google Scholar]
- Miller, E.T.; Svanbäck, R.; Bohannan, B.J.M. Microbiomes as metacommunities: Understanding host-associated microbes through metacommunity ecology. Trends Ecol. Evol. 2018, 33, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Unno, T.; Jang, J.; Han, D.; Kim, J.H.; Sadowsky, M.J.; Kim, O.-S.; Chun, J.; Hur, H.-G. Use of barcoded pyrosequencing and shared OTUs to determine sources of fecal bacteria in watersheds. Environ. Sci. Technol. 2010, 44, 7777–7782. [Google Scholar] [CrossRef] [PubMed]
- Unno, T.; Di, D.Y.W.; Jang, J.; Suh, Y.S.; Sadowsky, M.J.; Hur, H.-G. Integrated online system for a pyrosequencing-based microbial source tracking method that targets Bacteroidetes 16S rDNA. Environ. Sci. Technol. 2012, 46, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Couch, C.; Sanders, J.; Sweitzer, D.; Deignan, K.; Cohen, L.; Broughton, H.; Steingass, S.; Beechler, B. The relationship between dietary trophic level, parasites and the microbiome of Pacific walrus (Odobenus rosmarus divergens). Proc. Biol. Sci. 2022, 289, 20220079. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Kim, J.; Sadowsky, M.J.; Unno, T. Microbial source tracking using metagenomics and other new technologies. J. Microbiol. 2021, 59, 259–269. [Google Scholar] [CrossRef]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The gastrointestinal microbiome: A review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Christian, N.; Whitaker, B.K.; Clay, K. Microbiomes: Unifying animal and plant systems through the lens of community ecology theory. Front. Microbiol. 2015, 6, 869. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. Br. J. Nutr. 2015, 113, S1–S5. [Google Scholar] [CrossRef] [Green Version]
- Bornbusch, S.L.; Greene, L.K.; Rahobilalaina, S.; Calkins, S.; Rothman, R.S.; Clarke, T.A.; LaFleur, M.; Drea, C.M. Gut microbiota of ring-tailed lemurs (Lemur catta) vary across natural and captive populations and correlate with environmental microbiota. Anim. Microbiome 2022, 4, 29. [Google Scholar] [CrossRef]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.P.; Sun, X.; Patel, V.H.; Sanz, C.; Morgan, D.; Dantas, G. The microbiome and resistome of chimpanzees, gorillas, and humans across host lifestyle and geography. ISME J. 2020, 14, 1584–1599. [Google Scholar] [CrossRef]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hills, R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut microbiome: Profound implications for diet and disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco-Sandoval, A.; Schramm, Y.; Heckel, G.; Brassea-Pérez, E.; Martínez-Porchas, M.; Lago-Lestón, A. The Pacific harbor seal gut microbiota in Mexico: Its relationship with diet and functional inferences. PLoS ONE 2019, 14, e0221770. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Chalker, A.; Dewar, M.L.; Arnould, J.P.Y. Age-related differences revealed in Australian fur seal Arctocephalus pusillus doriferus gut microbiota. FEMS Microbiol. Ecol. 2013, 86, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [Green Version]
- De Leaniz, C.G.; Gajardo, G.; Consuegra, S. From Best to Pest: Changing perspectives on the impact of exotic salmonids in the southern hemisphere. Syst. Biodivers. 2010, 8, 447–459. [Google Scholar] [CrossRef]
- Buschmann, A.H.; Cabello, F.; Young, K.; Carvajal, J.; Varela, D.A.; Henríquez, L. Salmon aquaculture and coastal ecosystem health in Chile: Analysis of regulations, environmental impacts and bioremediation systems. Ocean Coast. Manag. 2009, 52, 243–249. [Google Scholar] [CrossRef]
- Allen, H.K.; Bayles, D.O.; Looft, T.; Trachsel, J.; Bass, B.E.; Alt, D.P.; Bearson, S.M.D.; Nicholson, T.; Casey, T.A. Pipeline for amplifying and analyzing amplicons of the V1–V3 region of the 16S rRNA gene. BMC Res. Notes 2016, 9, 380. [Google Scholar] [CrossRef] [Green Version]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef] [Green Version]
- Zarkasi, K.Z.; Taylor, R.S.; Abell, G.C.J.; Tamplin, M.L.; Glencross, B.D.; Bowman, J.P. Atlantic salmon (Salmo salar L.) gastrointestinal microbial community dynamics in relation to digesta properties and diet. Microb. Ecol. 2016, 71, 589–603. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S rRNA Gene Analysis with QIIME2. In Microbiome Analysis; Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2018; Volume 1849, pp. 113–129. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; Stevenes, M.; Wagner, H. Vegan: Community Ecology Package, R Package Version 2.0-2; 2014. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 28 March 2023).
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Grosser, S.; Sauer, J.; Paijmans, A.J.; Caspers, B.A.; Forcada, J.; Wolf, J.B.W.; Hoffman, J.I. Fur seal microbiota are shaped by the social and physical environment, show mother-offspring similarities and are associated with host genetic quality. Mol. Ecol. 2019, 28, 2406–2422. [Google Scholar] [CrossRef] [Green Version]
- Pemberton, D.; Kirkwood, R.J. Pup production and distribution of the Australian fur seal, Arctocephalus pusillus doriferus, in Tasmania. Wildl. Res. 1994, 21, 341–351. [Google Scholar] [CrossRef]
- Kirkwood, R.; Lynch, M.; Gales, N.; Dann, P.; Sumner, M. At-sea movements and habitat use of adult male Australian fur seals (Arctocephalus pusillus doriferus). Can. J. Zool. 2006, 84, 1781–1788. [Google Scholar] [CrossRef]
- Schwarz, D.; Spitzer, S.M.; Thomas, A.C.; Kohnert, C.M.; Keates, T.R.; Acevedo-Gutiérrez, A. Large-scale molecular diet analysis in a generalist marine mammal reveals male preference for prey of conservation concern. Ecol. Evol. 2018, 8, 9889–9905. [Google Scholar] [CrossRef]
- Zarkasi, K.Z.; Abell, G.C.J.; Taylor, R.S.; Neuman, C.; Hatje, E.; Tamplin, M.L.; Katouli, M.; Bowman, J.P. Pyrosequencing-based characterization of gastrointestinal bacteria of Atlantic salmon (Salmo salar L.) within a commercial mariculture system. J. Appl. Microbiol. 2014, 117, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, C.; Hatje, E.; Zarkasi, K.Z.; Smullen, R.; Bowman, J.P.; Katouli, M. The effect of diet and environmental temperature on the faecal microbiota of farmed Tasmanian Atlantic Salmon (Salmo salar L.). Aquac. Res. 2016, 47, 660–672. [Google Scholar] [CrossRef]
- Shah, M.M.; Odoyo, E.; Ichinose, Y. Epidemiology and pathogenesis of Providencia alcalifaciens infections. Am. J. Trop. Med. Hyg. 2019, 101, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Bozal, N.; Montes, M.J.; Tudela, E.; Guinea, J. Characterization of several Psychrobacter strains isolated from Antarctic environments and description of Psychrobacter luti sp. nov. and Psychrobacter fozii sp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1093–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.P. The Genus Psychrobacter. In The Prokaryotes: A Handbook on the Biology of Bacteria Volume 6: Pseudomonadota: Gamma Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 920–930. [Google Scholar]
- Hisar, O.; Yanik, T.; Hisar, S.A. Clinical and pathological investigation of Psychrobacter immobilis infection in rainbow trout (Oncorhynchus mykiss, Walbaum). Isr. J. Aquac. 2002, 54, 189–196. [Google Scholar]
- Romanenko, L.A.; Lysenko, A.M.; Rohde, M.; Mikhailov, V.V.; Stackebrandt, E. Psychrobacter maritimus sp. nov. and Psychrobacter arenosus sp. nov., isolated from coastal sea ice and sediments of the Sea of Japan. Int. J. Syst. Evol. Microbiol. 2004, 54, 1741–1745. [Google Scholar] [CrossRef] [Green Version]
- Romalde, J.L. Photobacterium damselae subsp. piscicida: An integrated view of a bacterial fish pathogen. Int. Microbiol. 2002, 5, 3–9. [Google Scholar] [CrossRef]
- Austin, H.K.; Austin, D.A. Bacterial Fish Pathogens; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Di Francesco, G.; Cammà, C.; Curini, V.; Mazzariol, S.; Proietto, U.; Di Francesco, C.E.; Ferri, N.; Di Provvido, A.; Di Guardo, G. Coinfection by Ureaplasma spp., Photobacterium damselae and an Actinomyces-like microorganism in a bottlenose dolphin (Tursiops truncatus) with pleuropneumonia stranded along the Adriatic coast of Italy. Res. Vet. Sci. 2016, 105, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Dryden, M.; Legarde, M.; Gottlieb, T.; Brady, L.; Ghosh, H.K. Vibrio damsela wound infections in Australia. Med. J. Aust. 1989, 151, 540–541. [Google Scholar] [CrossRef]
- Fouz, B.; Larsen, J.; Nielsen, B.; Barja, J.; Toranzo, A. Characterization of Vibrio damsela strains isolated from turbot Scophthalmus maximus in Spain. Dis. Aquat. Organ. 1992, 12, 155–166. [Google Scholar] [CrossRef]
- Rivas, A.J.; Lemos, M.L.; Osorio, C.R. Photobacterium damselae subsp. damselae, a bacterium pathogenic for marine animals and humans. Front. Microbiol. 2013, 4, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.M.; Wong, J.W. Necrotizing fasciitis caused by Vibrio damsela. Orthopedics 1999, 22, 443–444. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum; Taxon | W * | CtrlAd | CtrlJuv | InRegAd | MaleFF | High Group | Salmon Reads |
|---|---|---|---|---|---|---|---|
| Pseudomonadota; Photobacterium damselae subsp. damselae | 1073 | 44,141 | 3 | 5673 | 152 | CtrlAd | 664 |
| Fusobacteriota; Cetobacterium ceti | 1054 | 10,070 | 0 | 3029 | 252 | CtrlAd | 401 |
| Pseudomonadota; Neisseria;uncultured bacterium | 1033 | 244 | 212 | 3436 | 926 | InRegAd | 1751 |
| Pseudomonadota; Escherichia-Shigella | 1013 | 0 | 2776 | 580 | 18 | CtrlJuv | 161 |
| Bacillota; Bacillus | 1008 | 1381 | 719 | 1679 | 203 | InRegAd | 206 |
| Bacillota; Clostridium sensu stricto 1 | 1007 | 0 | 16 | 1285 | 282 | InRegAd | 477 |
| Bacillota; Bacilli | 991 | 375 | 0 | 2757 | 28 | InRegAd | 41 |
| Pseudomonadota; Plesiomonas | 956 | 0 | 0 | 1568 | 72 | InRegAd | 179 |
| Bacillota; Bacillus | 932 | 296 | 365 | 4247 | 337 | InRegAd | 183 |
| Pseudomonadota; Providencia alcalifaciens DSM 30120 | 926 | 32 | 26 | 1460 | 64 | InRegAd | 38 |
| Bacillota; Peptostreptococcaceae | 889 | 0 | 0 | 2723 | 0 | InRegAd | 707 |
| Pseudomonadota; Providencia alcalifaciens DSM 30120 | 889 | 0 | 0 | 0 | 900 | MaleFF | 32 |
| Bacillota; Clostridium moniliforme | 819 | 0 | 0 | 884 | 207 | InRegAd | 63 |
| Pseudomonadota; Escherichia coli UMEA 4075-1 | 767 | 0 | 118 | 0 | 0 | CtrlJuv | 29 |
| Pseudomonadota; Psychrobacter | 692 | 0 | 0 | 992 | 0 | InRegAd | 42 |
| Pseudomonadota; Escherichia-Shigella | 690 | 0 | 153 | 0 | 7 | CtrlJuv | 10 |
| Pseudomonadota; Providencia alcalifaciens DSM 30120 | 665 | 3 | 0 | 0 | 410 | MaleFF | 74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Agnese, E.; McLaughlin, R.J.; Lea, M.-A.; Soto, E.; Smith, W.A.; Bowman, J.P. Comparative Microbial Community Analysis of Fur Seals and Aquaculture Salmon Gut Microbiomes in Tasmania. Oceans 2023, 4, 200-219. https://doi.org/10.3390/oceans4020014
D’Agnese E, McLaughlin RJ, Lea M-A, Soto E, Smith WA, Bowman JP. Comparative Microbial Community Analysis of Fur Seals and Aquaculture Salmon Gut Microbiomes in Tasmania. Oceans. 2023; 4(2):200-219. https://doi.org/10.3390/oceans4020014
Chicago/Turabian StyleD’Agnese, Erin, Ryan J. McLaughlin, Mary-Anne Lea, Esteban Soto, Woutrina A. Smith, and John P. Bowman. 2023. "Comparative Microbial Community Analysis of Fur Seals and Aquaculture Salmon Gut Microbiomes in Tasmania" Oceans 4, no. 2: 200-219. https://doi.org/10.3390/oceans4020014