
Cyclic Six-Atomic Boron-Nitrides: Quantum-Chemical Consideration by Ab Initio CCSD(T) Method


Abstract
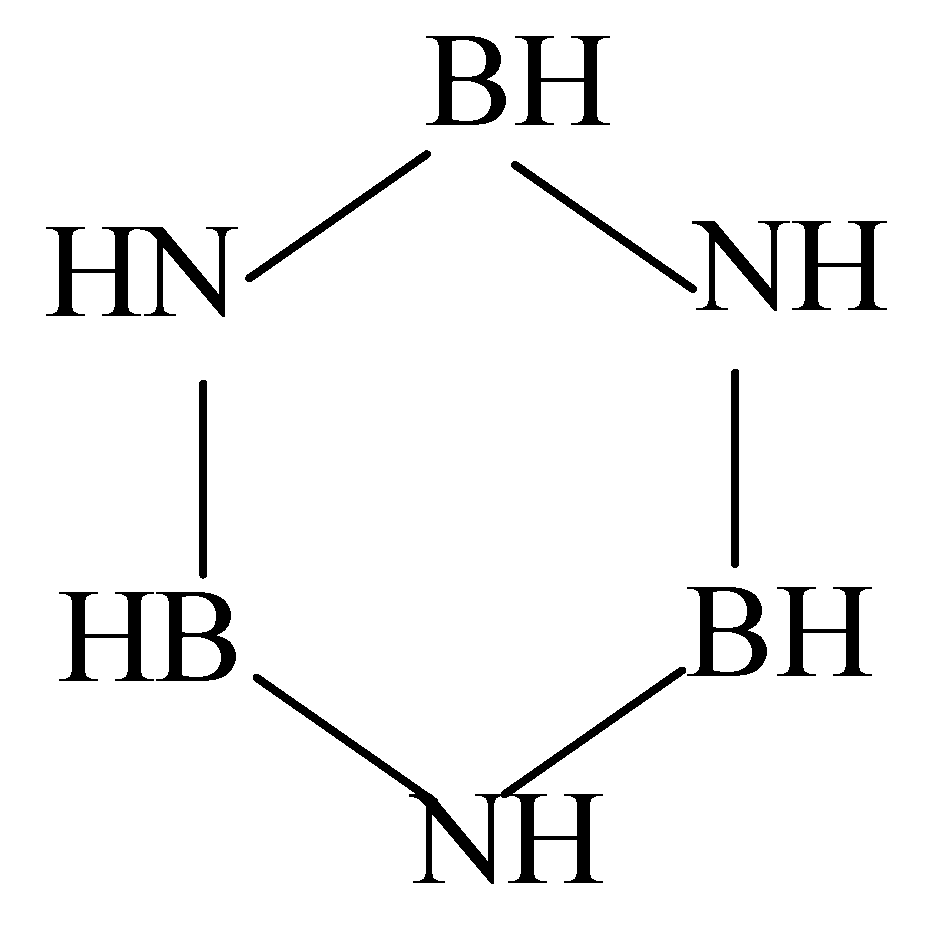
:1. Introduction
2. Method
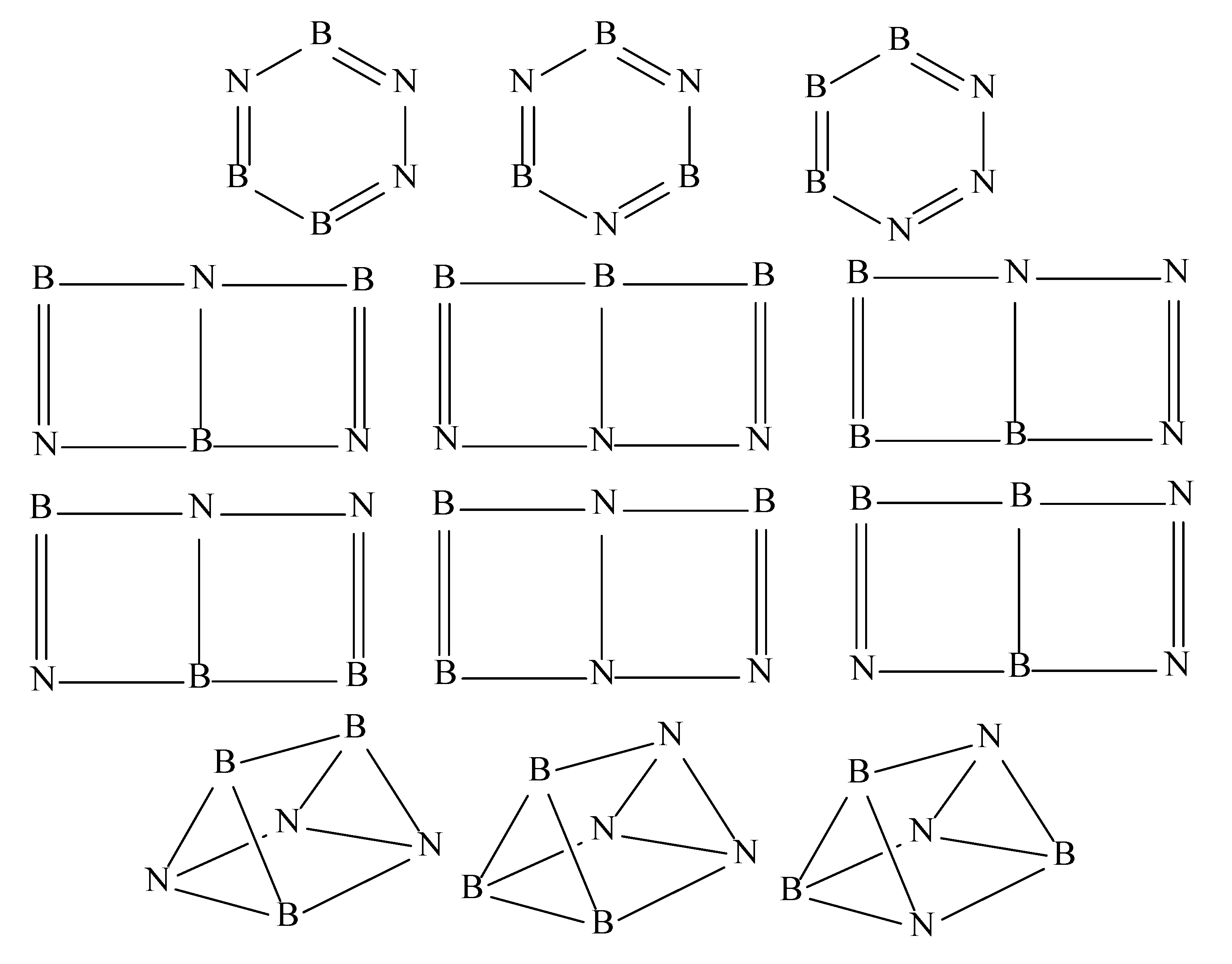
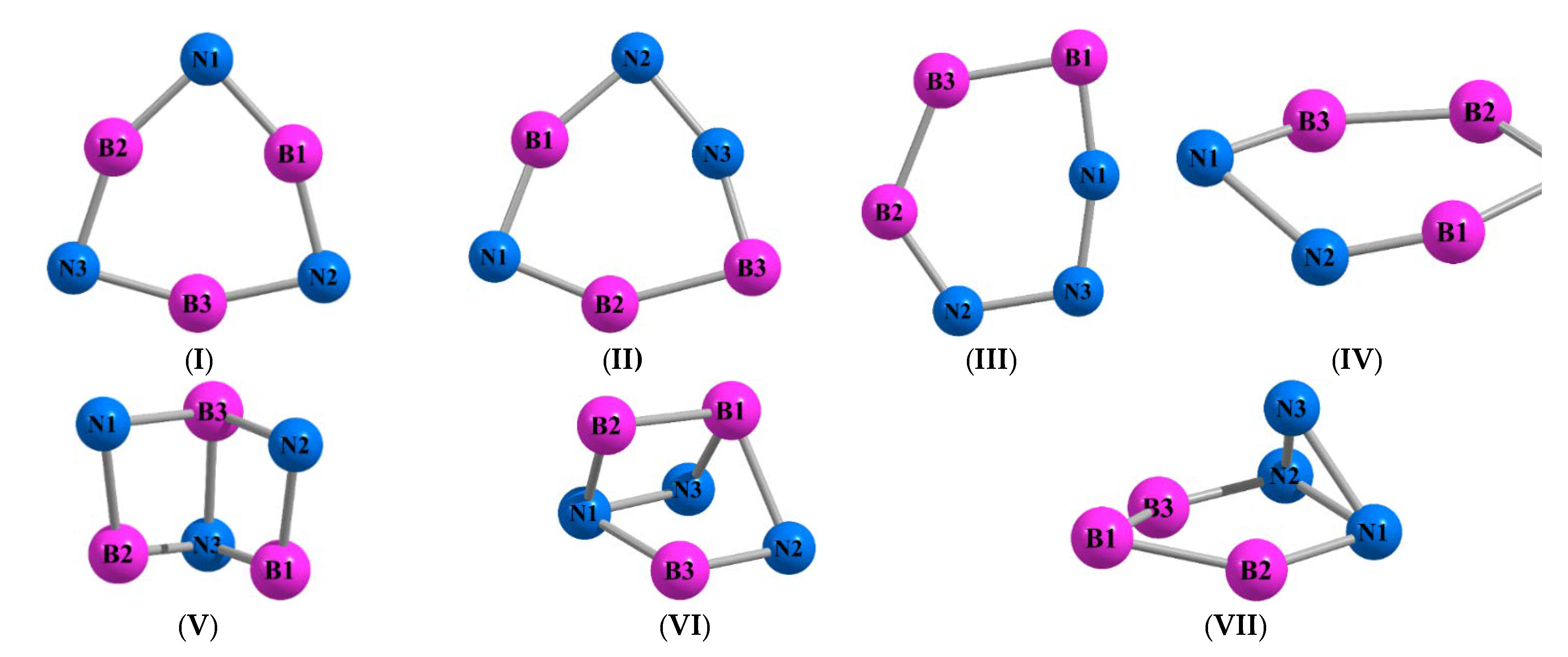
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coursen, D.I.; Hoard, J.L. The Structure of B-Trichloroborazole. J. Am. Chem. Soc. 1952, 74, 1742–1750. [Google Scholar] [CrossRef]
- Hughes, E.W. The crystal structure of ammonia-borane, H3NBH3. J. Am. Chem. Soc. 1956, 78, 502–503. [Google Scholar] [CrossRef]
- Lippert, E.L.; Lipscomb, W.N. The structure of H3NBH3. J. Am. Chem. Soc. 1956, 78, 503–504. [Google Scholar] [CrossRef]
- Harshbarger, W.; Lee, G.H.; Porter, R.F.; Bauer, S.H. Structure of B-monoamino-borazine. J. Am. Chem. Soc. 1969, 91, 551–555. [Google Scholar] [CrossRef]
- Chang, C.-H.; Porter, R.F.; Bauer, S.H. Molecular structure of dimethyl-cyclotetrazenoborane, (CH3)2N4BH. Inorg. Chem. 1969, 8, 1677–1683. [Google Scholar] [CrossRef]
- Harshbarger, W.; Lee, G.H.; Porter, R.F.; Bauer, S.H. Structure of borazine. Inorg. Chem. 1969, 8, 1683–1689. [Google Scholar] [CrossRef]
- Gopinathan, M.S.; Whitehead, M.A.; Coulson, C.A.; Carruthers, J.R.; Rollett, J.S. A reinvestigation of the crystal and molecular structure of hexachloroborazine. Acta Cryst. B 1974, 30, 731–737. [Google Scholar] [CrossRef]
- Kar, T.; Scheiner, S.; Roy, A.K. Structure and Properties of [8]BN-Circulenes: Inorganic Analogues of [8]Circulenes. J. Phys. Chem. C 2015, 119, 15541–15546. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pandey, S.K.; Misra, N. Stability versus aromaticity in mono-hydroxylated borazine, 1,2-azaborine and 1,3,2,4-diazadiborine. Mol. Phys. 2016, 114, 1763–1770. [Google Scholar] [CrossRef]
- Li, D.-Z.; Feng, L.-Y.; Pei, L.; Zhang, L.-J.; Wu, S.-G.; Zhai, H.-J. Pentagonal five-center four-electron π bond in ternary B3N2H5 cluster: An extension of the concept of three-center four-electron ω bond. Phys. Chem. Chem. Phys. 2017, 19, 2479–2486. [Google Scholar] [CrossRef]
- Less, R.J.; Hanf, S.; García-Rodríguez, R.; Bond, A.D.; Wright, D.S. A [HN(BH═NH)2]2– Dianion, Isoelectronic with a β-Diketiminate. Organometallics 2018, 37, 628–631. [Google Scholar] [CrossRef]
- Anafcheh, M.; Ahmadi, E.; Zahedi, M. Addition of borazine to boron nitride nanotubes: [2+2] cycloaddition or bond cleavage. Monatsh. Chem. 2019, 150, 1019–1024. [Google Scholar] [CrossRef]
- Prieschl, D.; Bélanger-Chabot, G.; Guo, X.; Dietz, M.; Müller, M.; Krummenacher, I.; Lin, Z.; Braunschweig, H. Synthesis of Complex Boron–Nitrogen Heterocycles Comprising Borylated Triazenes and Tetrazenes Under Mild Conditions. J. Am. Chem. Soc. 2020, 142, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Kinjo, R. Inorganic Benzene Valence Isomers. Chem. Asian J. 2020, 15, 2558–2574. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Espinosa, Y.; Méndez-Sánchez, R.A.; Sadurní, E. On the electronic structure of benzene and borazine: An algebraic description. J. Phys. B Atomic. Mol. Opt. Phys. 2020, 53, 105101. [Google Scholar] [CrossRef]
- Vel, L.; Demazeau, G.; Etourneau, J. Cubic Boron Nitride: Synthesis, Physicochemical Properties and Applications. Mater. Sci. Eng. B 1991, 10, 149–164. [Google Scholar] [CrossRef]
- Rubio, A.; Corkill, J.L.; Cohen, M.L. Theory of Graphitic Boron Nitride Nanotubes. Phys. Rev. B 1994, 49, 5081–5084. [Google Scholar] [CrossRef]
- Zedlitz, R. Properties of Amorphous Boron Nitride Thin Films. J. Non-Cryst. Solids 1996, 198–200, 403–410. [Google Scholar] [CrossRef]
- Golberg, D.; Bando, Y.; Tang, C.C.; Zhi, C.Y. Boron Nitride Nanotubes. Adv. Mater. 2007, 19, 2413–2432. [Google Scholar] [CrossRef]
- Goriachko, A.; He, Y.; Knapp, M.; Over, H.; Corso, M.; Brugger, T.; Berner, S.; Osterwalder, J.; Greber, T. Self-Assembly of a Hexagonal Boron Nitride Nanomesh on Ru(0001). Langmuir 2007, 23, 2928–2931. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Kuroda, S.; Muramatsu, Y. Electronic Structure and Intercalation Chemistry of Graphite-Like Layered Material with a Composition of BC6N. J. Phys. Chem. Solids 2008, 69, 1171–1178. [Google Scholar] [CrossRef]
- Cai, Q.; Scullion, D.; Gan, W.; Falin, A.; Zhang, S.; Watanabe, K.; Taniguchi, T.; Chen, Y.; Santos, E.J.C. High thermal conductivity of high-quality monolayer boron nitride and its thermal expansion. Sci. Adv. 2019, 5, eaav0129. [Google Scholar] [CrossRef] [PubMed]
- Brazhkin, V.V.; Solozhenko, V.L. Myths about new ultrahard phases: Why materials that are significantly superior to diamond in elastic moduli and hardness are impossible. J. Appl. Phys. 2019, 125, 130901. [Google Scholar] [CrossRef]
- Mirkarimi, P.B.; McCarty, K.F.; Medlin, D.L. Review of Advances in Cubic Boron Nitride Film Synthesis. Mater. Sci. Eng. R Rep. 1997, 21, 47–100. [Google Scholar] [CrossRef]
- Boese, R.; Maulitz, A.H.; Stellberg, P. Solid-State Borazine: Does it Deserve to be Entitled “Inorganic Benzene”? Chem. Ber. 1994, 127, 1887–1889. [Google Scholar] [CrossRef]
- Jeon, J.K.; Uchimaru, Y.; Kim, D.P. Synthesis of novel amorphous boron carbonitride ceramics from the borazine derivative copolymer via hydroboration. Inorg. Chem. 2004, 43, 4796–4798. [Google Scholar] [CrossRef]
- Shen, W.; Li, M.; Li, F.; Wang, S. Theoretical study of borazine and its derivatives. Inorg. Chim. Acta 2007, 360, 619–624. [Google Scholar] [CrossRef]
- Islas, R.; Chamorro, E.; Robles, J.; Heine, T.; Santos, J.C.; Merino, G. Borazine: To be or not to be aromatic. Struct. Chem. 2007, 18, 833–839. [Google Scholar] [CrossRef]
- Dong, S.H.; Zheng, Z.M.; Han, W.J.; Zhao, T.; Xu, C.H. Synthesis of new borazines containing crosslinkable groups. Chin. Chem. Lett. 2008, 19, 1398–1400. [Google Scholar] [CrossRef]
- Lei, Y.P.; Wang, Y.D.; Song, Y.C.; Li, Y.H.; Wang, H.; Deng, C.; Xie, Z.F. Facile synthesis of a melt-spinnable polyborazine from asymmetric alkylaminoborazine. Chin. Chem. Lett. 2010, 21, 1079–1082. [Google Scholar] [CrossRef]
- Kervyn, S.; Fenwick, O.; Di Stasio, F.; Shin, Y.S.; Wouters, J.; Accorsi, G.; Osella, S.; Beljonne, D.; Cacialli, F.; Bonifazi, D. Polymorphism, fluorescence, and optoelectronic properties of a borazine derivative. Chem. A Eur. J. 2013, 19, 7771–7779. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.; Nagnathappa, M.; Kharat, B.; Chaudhari, A. Theoretical study of borazine and substituted borazines using density functional theory method. J. Mol. Liquids 2014, 193, 13–22. [Google Scholar] [CrossRef]
- Neogi, I.; Szpilman, A.M. Synthesis and Reactions of Borazines. Synthesis 2021, 54, 1877–1907. [Google Scholar] [CrossRef]
- Báez-Grez, R.; Pino-Rios, R. The hidden aromaticity in borazine. RSC Adv. 2022, 12, 7906–7910. [Google Scholar] [CrossRef] [PubMed]
- Pople, A.; Krishnan, R.; Schlegel, H.B.; Binkley, J.S. Electron Correlation Theories and Their Application to the Study of Simple Reaction Potential Surfaces. Int. J. Quantum Chem. 1978, 14, 545–560. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D., III. Many-body perturbation-theory, coupled-pair many-electron theory, and importance of quadruple excitations for correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Purvis, G.D., III; Bartlett, R.J. A full coupled-cluster singles and doubles model-the inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Krishnan, R. Quadratic configuration interaction-a general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis-sets for anion calculations. 3. The 3-21+G basis set for 1st-row elements, Li-F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods. 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular structures and thermodynamics of stable N4, N6 and N8 neutral poly-nitrogens according to data of QCISD(T)/TZVP method. Chem. Phys. Lett. 2020, 753, 137594. [Google Scholar] [CrossRef]
- Mikhailov, O.V. Molecular and Electronic Structures of Neutral Polynitrogens: Review on the Theory and Experiment in 21st Century. Int. J. Mol. Sci. 2022, 23, 2841. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Standard Thermodynamic Properties of Chemical Substances. In Handbook of Chemistry and Physics Online; CRC Press: Taylor & Francis Group: London, UK; Available online: https://hbcp.chemnetbase.com/faces/documents/05_02/05_02_0001.xhtml (accessed on 25 August 2022).




{kind=link}
{kind=link}
{kind=link}
| Molecule B3N3 (I) | |||
|---|---|---|---|
| Bond Lengths, pm | Bond Angles, deg | ||
| (N1B1) | 136.5 | (N1B1N2) | 151.8 |
| (B1N2) | 136.5 | (B1N2B3) | 88.2 |
| (N2B3) | 136.5 | (N2B3N3) | 151.8 |
| (B3N3) | 136.5 | (B3N3B2) | 88.2 |
| (N3B2) | 136.5 | (N3B2N1) | 151.8 |
| (B2N1) | 136.5 | (B2N1B1) | 88.2 |
| Selected torsion (dihedral) angles, deg | |||
| (N1B1N2B3) | −0.4 | (B1N2B3N3) | 0.4 |
| (N1B1B3B2) | 0.0 | (B1N1N3N2) | 0.0 |
| Molecule B3N3 (II) | |||
| Bond lengths, pm | Bond angles, deg | ||
| (N1B1) | 136.3 | (N1B1N2) | 151.3 |
| (B1N2) | 138.1 | (B1N2N3) | 89.6 |
| (N2N3) | 135.4 | (N2N3B3) | 156.8 |
| (N3B3) | 129.5 | (N3B3B2) | 88.0 |
| (B3B2) | 158.7 | (B3B2N1) | 142.8 |
| (B2N1) | 134.6 | (B2N1B1) | 91.5 |
| Selected torsion (dihedral) angles, deg | |||
| (N1B1N3B2) | 0.0 | (N1B1N2B2) | 0.0 |
| (N1B2B3B1) | 0.0 | (B1N2N3N1) | 0.0 |
| Molecule B3N3 (III) | |||
| Bond lengths, pm | Bond angles, deg | ||
| (N1B1) | 131.4 | (N1B1B3) | 87.4 |
| (B1B3) | 154.5 | (B1B3B2) | 121.2 |
| (B3B2) | 157.1 | (B3B2N2) | 123.8 |
| (B2N2) | 133.5 | (B2N2N3) | 115.2 |
| (N2N3) | 134.6 | (N2N3N1) | 107.3 |
| (N3N1) | 130.5 | (N3N1B1) | 164.9 |
| Selected torsion (dihedral) angles, deg | |||
| (N1B1N2B2) | −1.7 | (B1N1N2B2) | −2.3 |
| (N1B1B3B2) | −2.9 | (B1N1N3N2) | −9.7 |
| Molecule B3N3 (IV) | |||
| Bond lengths, pm | Bond angles, deg | ||
| (N1B3) | 134.2 | (N1B3B2) | 150.0 |
| (B3B2) | 163.7 | (B3B2N3) | 128.3 |
| (B2N3) | 132.8 | (B2N3B1) | 80.4 |
| (N3B1) | 142.1 | (N3B1N2) | 154.4 |
| (B1N2) | 132.3 | (B1N2N1) | 126.3 |
| (N2N1) | 154.7 | (N2N1B3) | 75.0 |
| Selected torsion (dihedral) angles, deg | |||
| (N1B3B2N3) | 10.4 | (N2B1N3B2) | 7.3 |
| (N1B3B2B1) | 15.7 | (N1N2B1N3) | −33.0 |
| Molecule B3N3 (V) | |||
| Bond lengths, pm | Bond angles, deg | ||
| (N1B2) | 153.1 | (N1B3N3) | 102.1 |
| (B2N3) | 135.6 | (B3N3B2) | 74.6 |
| (N3B1) | 147.4 | (N3B2N1) | 111.3 |
| (B1N2) | 148.4 | (B2N1B3) | 71.6 |
| (N2B3) | 153.9 | (N2B3N3) | 101.7 |
| (B3N1) | 150.5 | (B3N3B1) | 74.5 |
| (B3N3) | 156.0 | (N3B1N2) | 108.7 |
| (B1N2B3) | 74.9 | ||
| (N1B3N2) | 82.2 | ||
| (B2N3B1) | 73.3 | ||
| Selected torsion (dihedral) angles, deg | |||
| (N1B3N3B2) | −5.2 | (N1N2B1B2) | 1.0 |
| (N2B3N3B1) | 2.7 | (N1N3N2B3) | −54.0 |
| (N3N1B3N2) | −100.4 | (B3B2N3B1) | 78.1 |
| (N1B3N2B1) | 98.1 | (N2N3N1B2) | −95.8 |
| Molecule B3N3 (VI) | |||
| Bond lengths, pm | Bond angles, deg | ||
| (N1B2) | 149.8 | (N1B2B1) | 72.1 |
| (B2B1) | 169.2 | (B2B1N3) | 100.7 |
| (B1N3) | 146.6 | (B1N3N1) | 78.4 |
| (N3N1) | 151.3 | (N3N1B2) | 108.0 |
| (N1B3) | 149.7 | (N1B3N2) | 95.8 |
| (B3B2) | 171.8 | (B3N2N3) | 86.8 |
| (N3N2) | 164.9 | (N2N3N1) | 87.2 |
| (N2B1) | 156.6 | (N3N1B3) | 90.1 |
| (N2B3) | 144.5 | (N2B1B2) | 102.8 |
| (B1B2B3) | 69.7 | ||
| (B2B3N2) | 106.9 | ||
| (B3N2B1) | 80.5 | ||
| (N1B3B2) | 55.0 | ||
| (B3B2N1) | 55.0 | ||
| (B2N1B3) | 70.0 | ||
| (N3B1N2) | 65.8 | ||
| (B1N2N3) | 54.2 | ||
| (N2N3B1) | 60.0 | ||
| Selected torsion (dihedral) angles, deg | |||
| (N1B2B1N3) | 6.1 | (B1N1N2N3) | −70.2 |
| (N1B3N2N3) | −2.3 | (B2N1N2N3) | −118.8 |
| (N2B1B2B3) | −2.8 | (B3N1N2N3) | 176.7 |
| Molecule B3N3 (VII) | |||
| Bond lengths, pm | Bond angles, deg | ||
| (N1N3) | 149.2 | (N1N3N2) | 63.1 |
| (N3N2) | 149.2 | (N3N2B3) | 73.3 |
| (N2B3) | 147.9 | (N2B3B1) | 140.9 |
| (B3B1) | 157.4 | (B3B1B2) | 66.4 |
| (B1B2) | 157.4 | (B1B2N1) | 140.9 |
| (B2N1) | 147.9 | (B2N1N3) | 73.3 |
| Selected torsion (dihedral) angles, deg | |||
| (N2B3B1B2) | −24.3 | (N1N2B3B1) | 20.5 |
| (N1B2B1B3) | 24.3 | (N1N2B3B2) | 0.1 |
| (B1N1N3N2) | −90.8 | (N3B1B2B3) | −52.3 |
| (B2N1N3N2) | −104.6 | (B3N2N3N1) | 104.6 |
| Compound | Total Energy, Hartree | Relative Total Energy, kJ∙mole−1 |
|---|---|---|
| B3N3 (I) | −238.347767 | 0.0 |
| B3N3 (II) | −238.192298 | 409.6 |
| B3N3 (III) | −238.062655 | 752.3 |
| B3N3 (IV) | −238.169862 | 470.3 |
| B3N3 (V) | −238.124054 | 588.8 |
| B3N3 (VI) | −238.047742 | 788.7 |
| B3N3 (VII) | −238.005818 | 900.0 |
| Compound | The Charges on the Atoms, in Electron Charge Units (ē) | |||||
|---|---|---|---|---|---|---|
| N1 | N2 | N3 | B1 | B2 | B3 | |
| B3N3 (I) | −1.19049 | −1.19062 | −1.19040 | +1.19069 | +1.19015 | +1.19067 |
| B3N3 (II) | −1.18894 | −0.73637 | −0.44979 | +1.20616 | +0.79351 | +0.37543 |
| B3N3 (III) | −0.45135 | −0.63555 | −0.03730 | +0.48064 | +0.66080 | −0.01723 |
| B3N3 (IV) | −0.53902 | −0.77371 | −1.04131 | +1.05055 | +0.73557 | +0.56792 |
| B3N3 (V) | −0.61776 | −0.61806 | −1.03235 | +0.73432 | +0.73354 | +0.80030 |
| B3N3 (VI) | −0.74180 | −0.75310 | −0.29335 | +0.63671 | +0.35787 | +0.79367 |
| B3N3 (VII) | −0.34059 | −0.34113 | −0.27877 | −0.00824 | +0.48370 | +0.48503 |
| Compound | ΔfH0, kJ∙mol−1 | S0, J∙mol−1∙K−1 | ΔfG0, kJ∙mol−1 |
|---|---|---|---|
| B3N3 (I) | 192.1 | 306.8 | 191.0 |
| B3N3 (II) | 597.6 | 314.2 | 594.2 |
| B3N3 (III) | 929.3 | 326.0 | 922.4 |
| B3N3 (IV) | 662.1 | 329.9 | 654.0 |
| B3N3 (V) | 785.1 | 311.0 | 782.6 |
| B3N3 (VI) | 985.7 | 308.6 | 984.0 |
| B3N3 (VII) | 1092.8 | 318.5 | 1088.1 |
| Compound | ΔfH0, kJ∙mol−1 | S0, J∙mol−1∙K−1 | ΔfG0, kJ∙mol−1 |
|---|---|---|---|
| B3H6N3 (gas) | −513.7 | 329.1 | −404.8 |
| B3H6N3 (liq) | (−541.0) | (199.6) | (−392.7) |
| O2 (gas) | 2.0 (0) | 222.0 (205.2) | −3.1 (0) |
| B3N3 (I) (gas) | 192.1 | 306.8 | 191.0 |
| H2O (gas) | −239.6 (−241.8) | 197.2 (188.8) | −228.9 (−228.6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chachkov, D.V.; Mikhailov, O.V. Cyclic Six-Atomic Boron-Nitrides: Quantum-Chemical Consideration by Ab Initio CCSD(T) Method. Quantum Rep. 2022, 4, 351-361. https://doi.org/10.3390/quantum4030025
Chachkov DV, Mikhailov OV. Cyclic Six-Atomic Boron-Nitrides: Quantum-Chemical Consideration by Ab Initio CCSD(T) Method. Quantum Reports. 2022; 4(3):351-361. https://doi.org/10.3390/quantum4030025
Chicago/Turabian StyleChachkov, Denis V., and Oleg V. Mikhailov. 2022. "Cyclic Six-Atomic Boron-Nitrides: Quantum-Chemical Consideration by Ab Initio CCSD(T) Method" Quantum Reports 4, no. 3: 351-361. https://doi.org/10.3390/quantum4030025