

Pure Hydrolysis of Polyamides: A Comparative Study

 , and
, and
Abstract
:1. Introduction
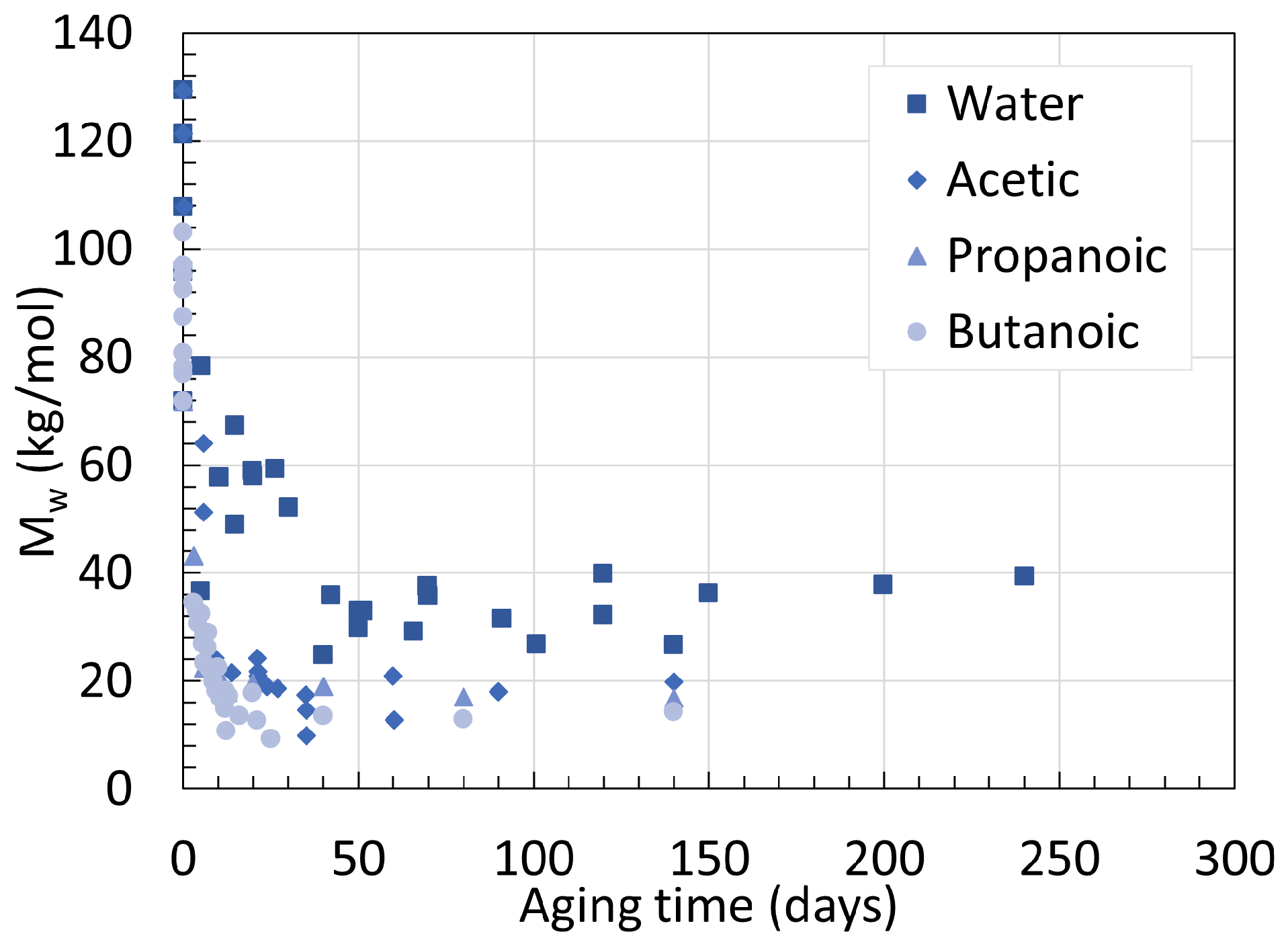
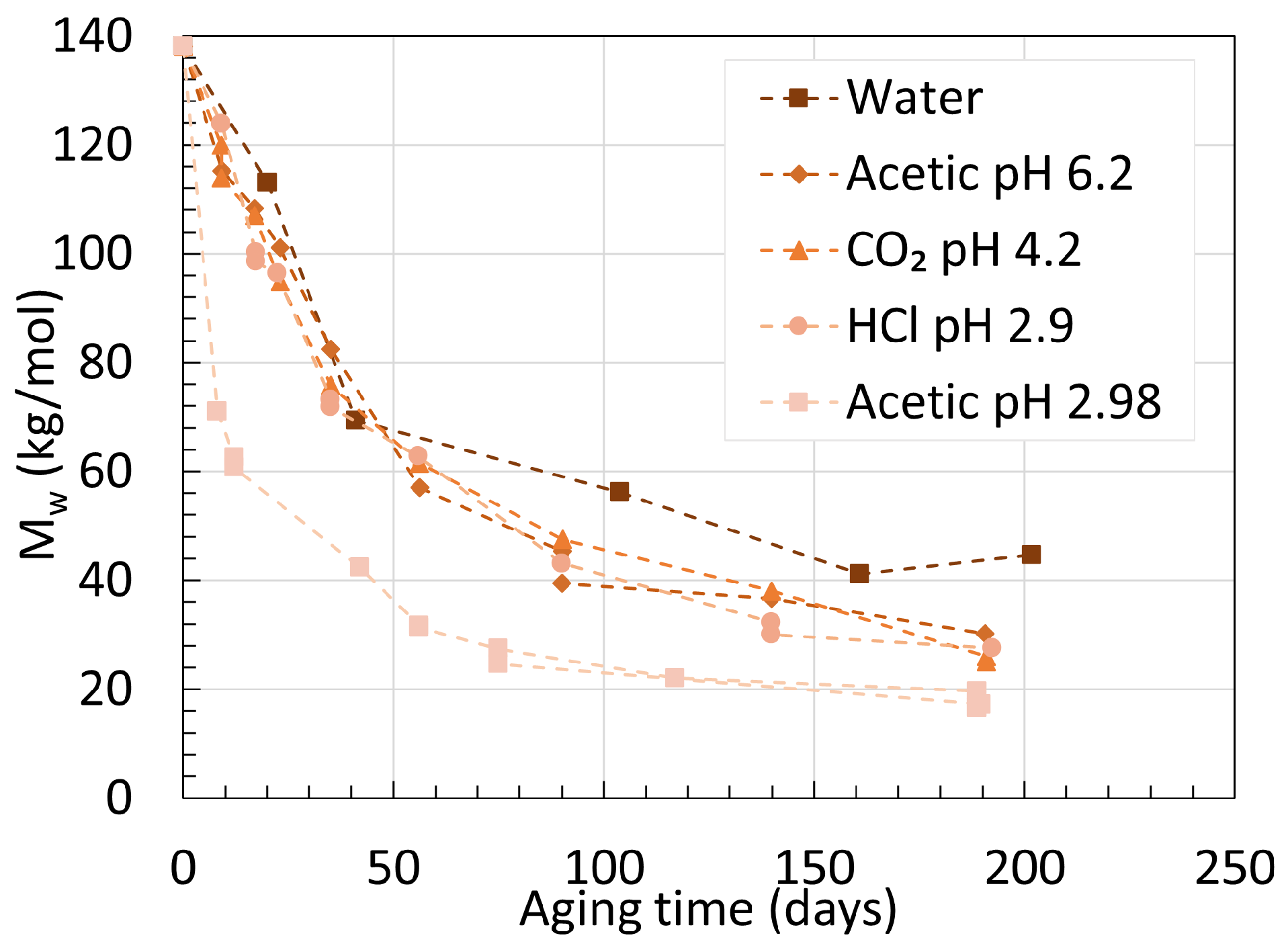
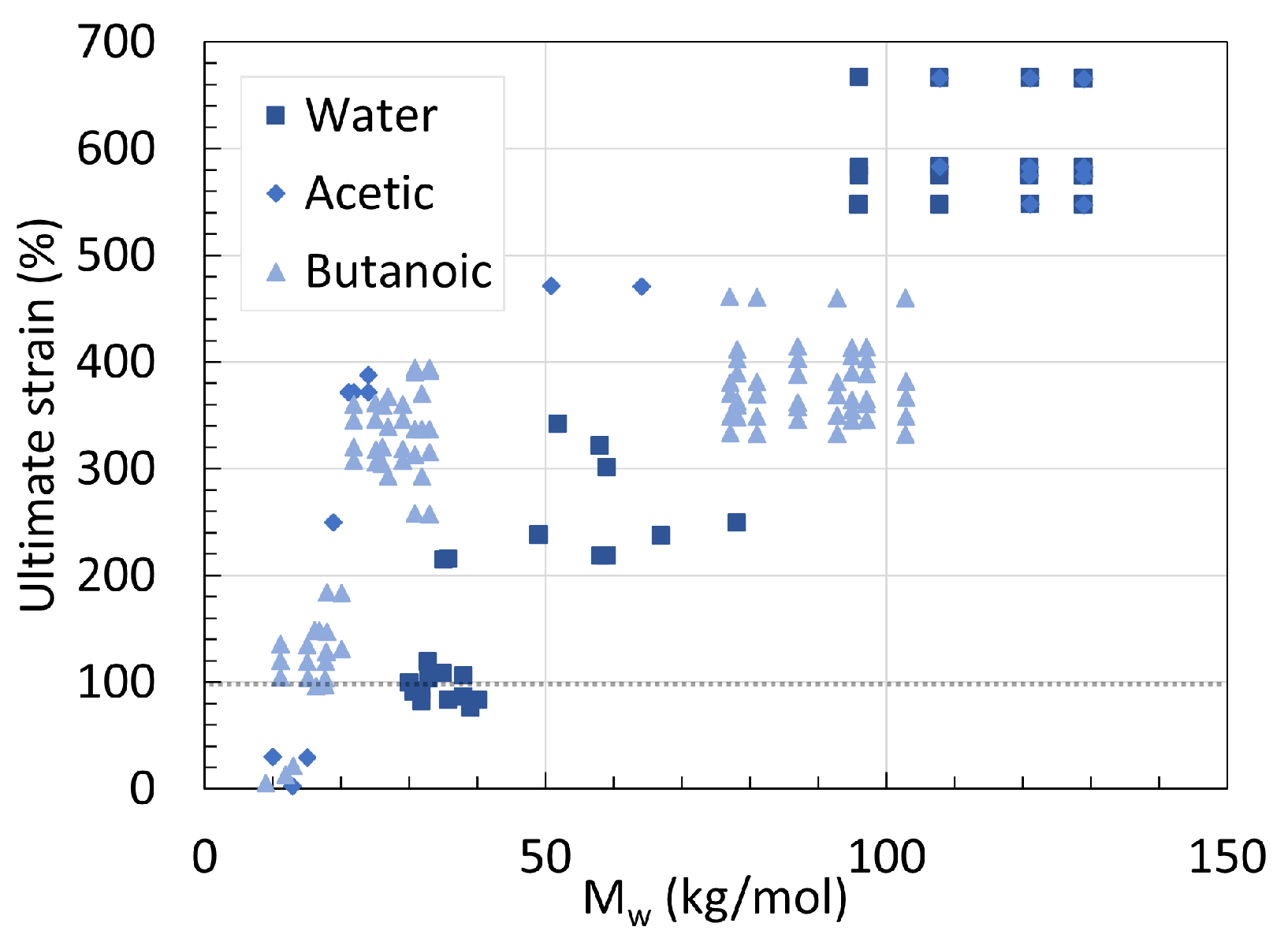
2. Molecular Weight
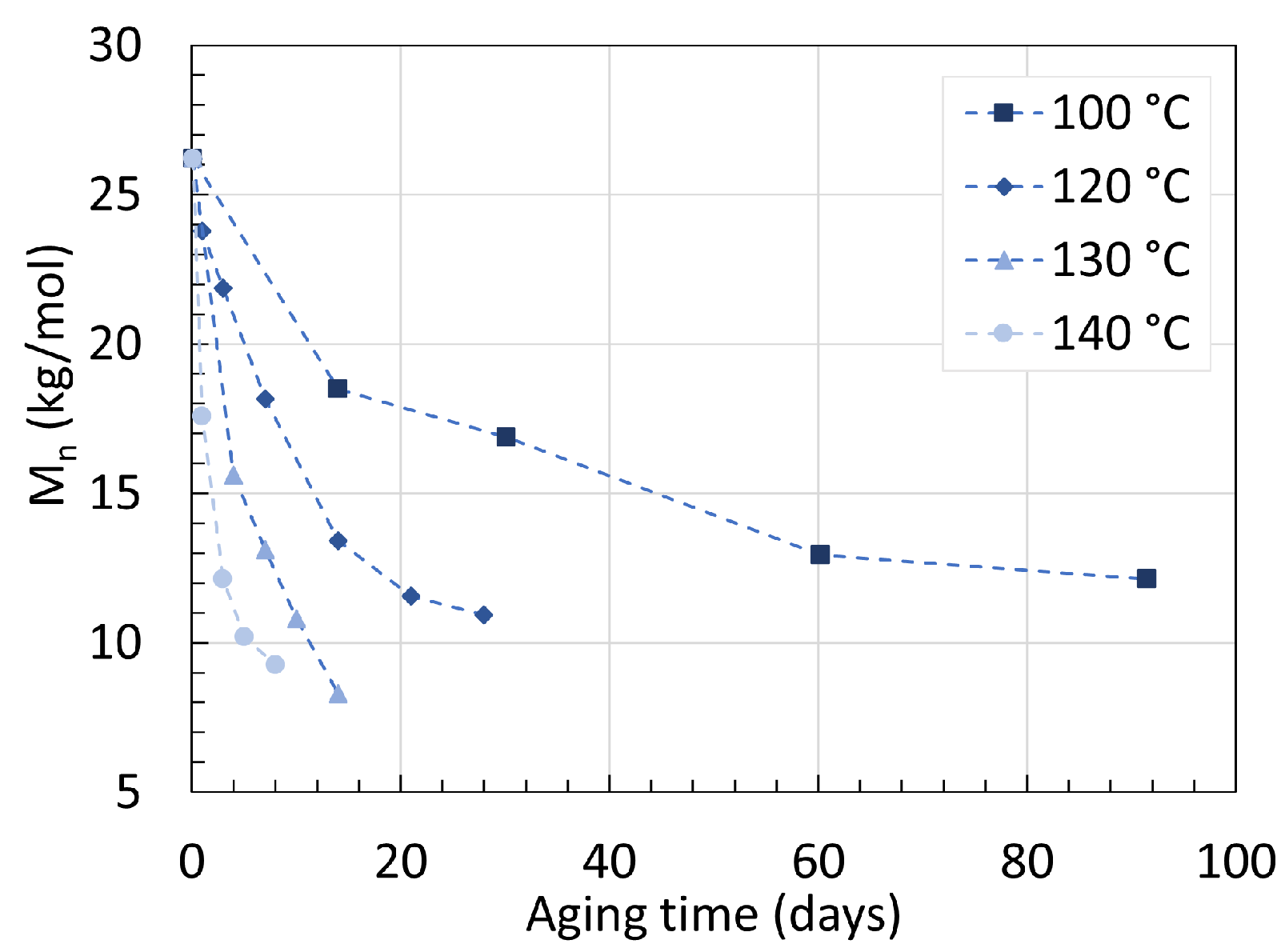
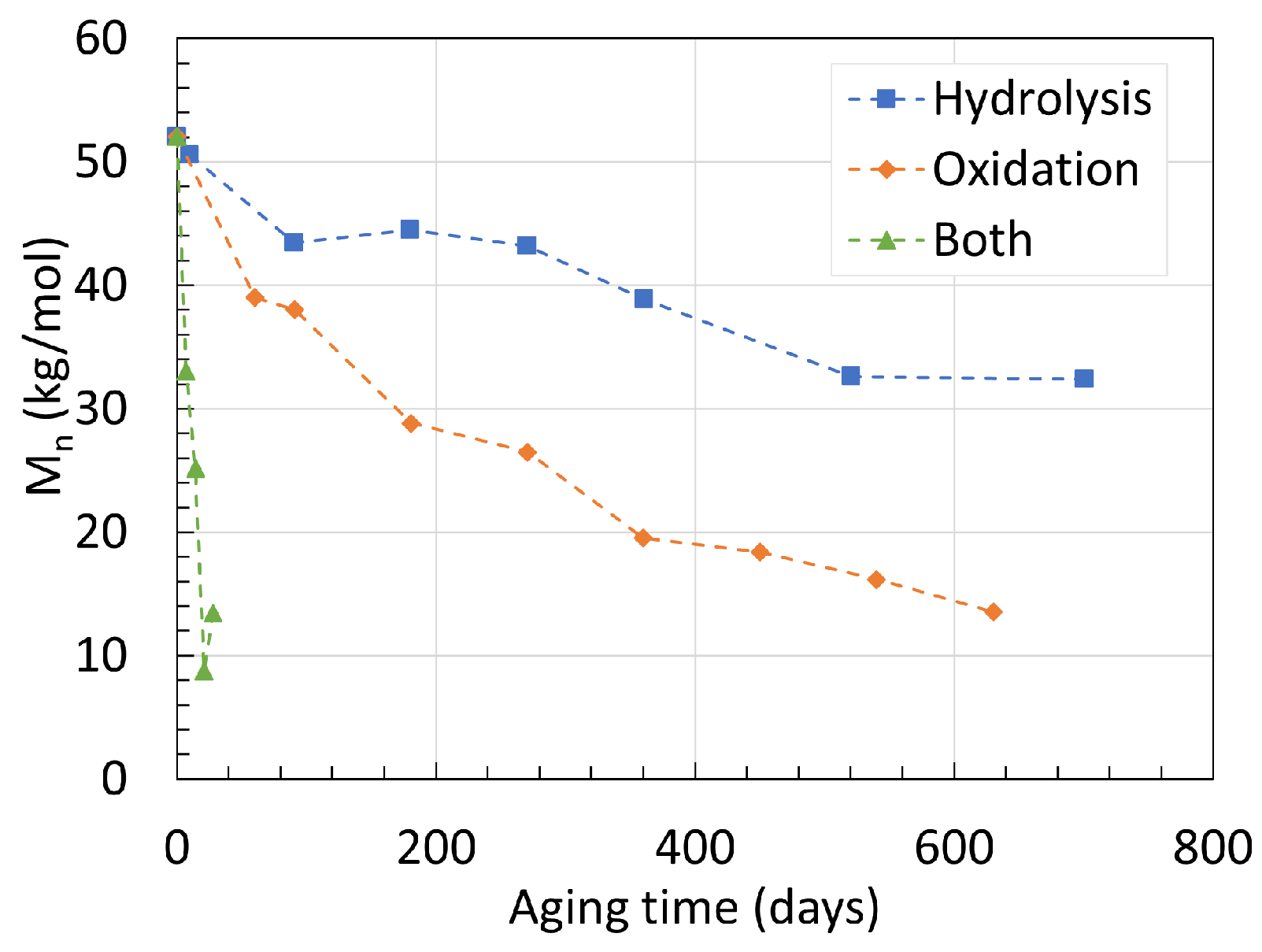
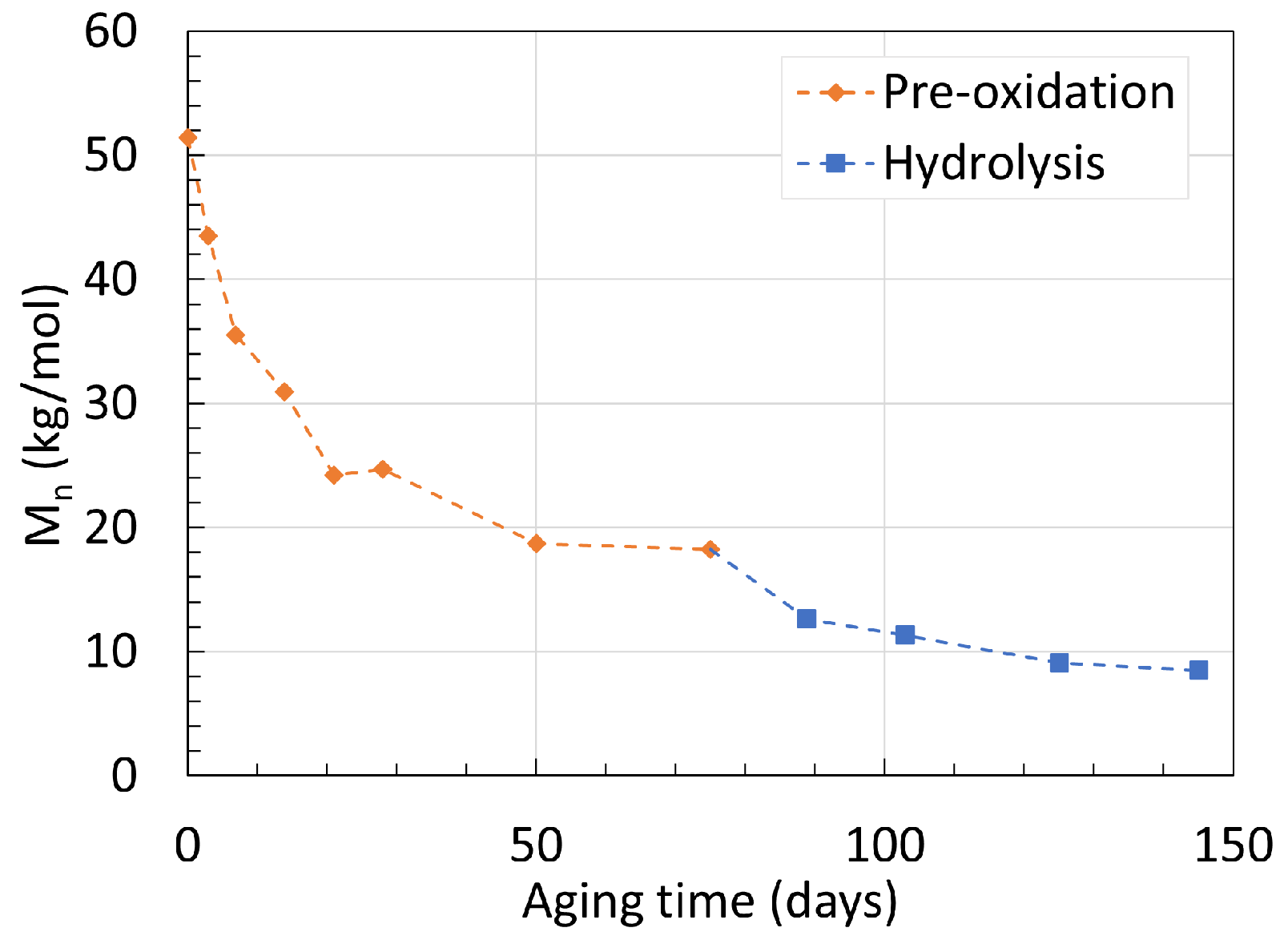
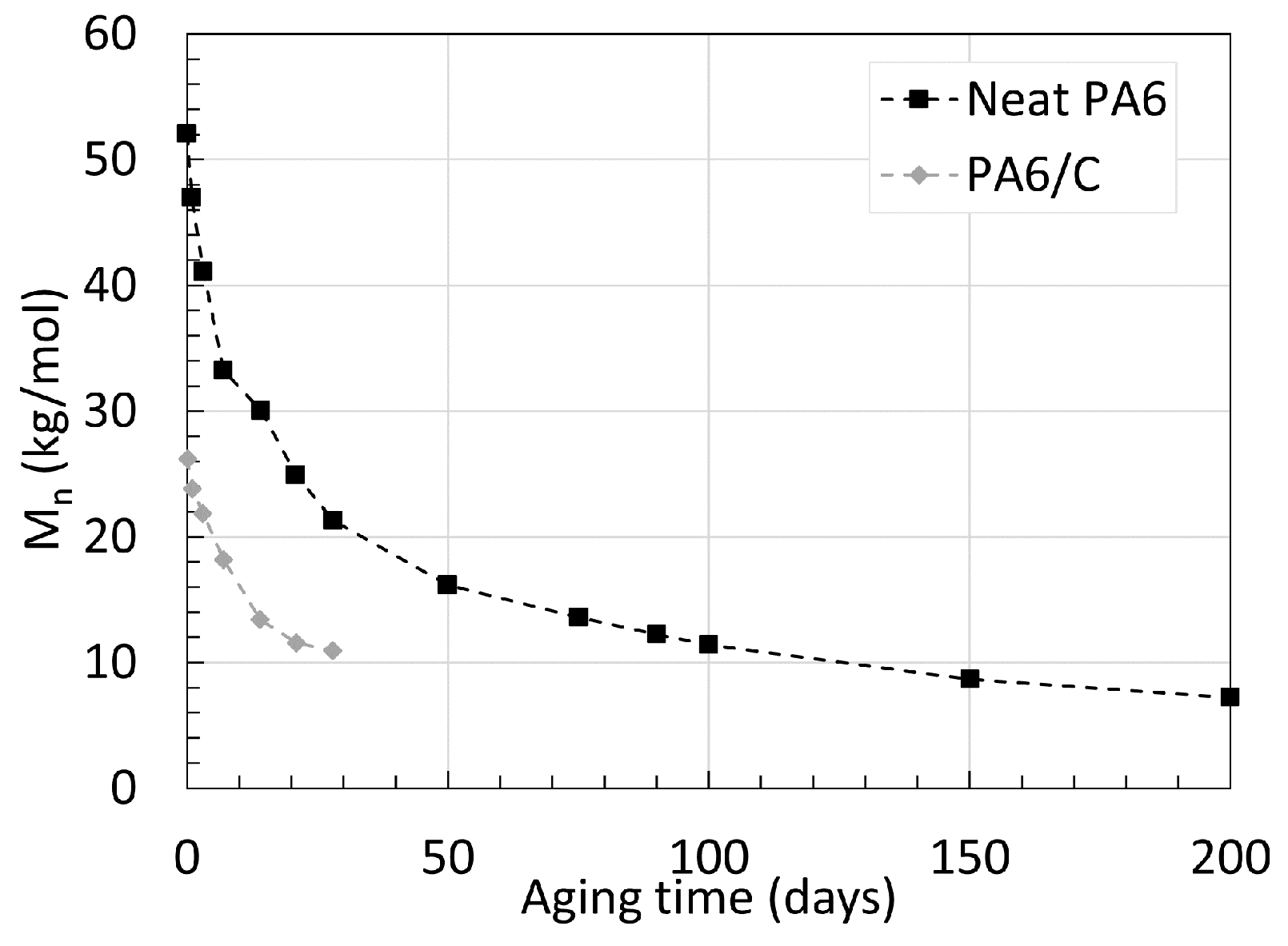
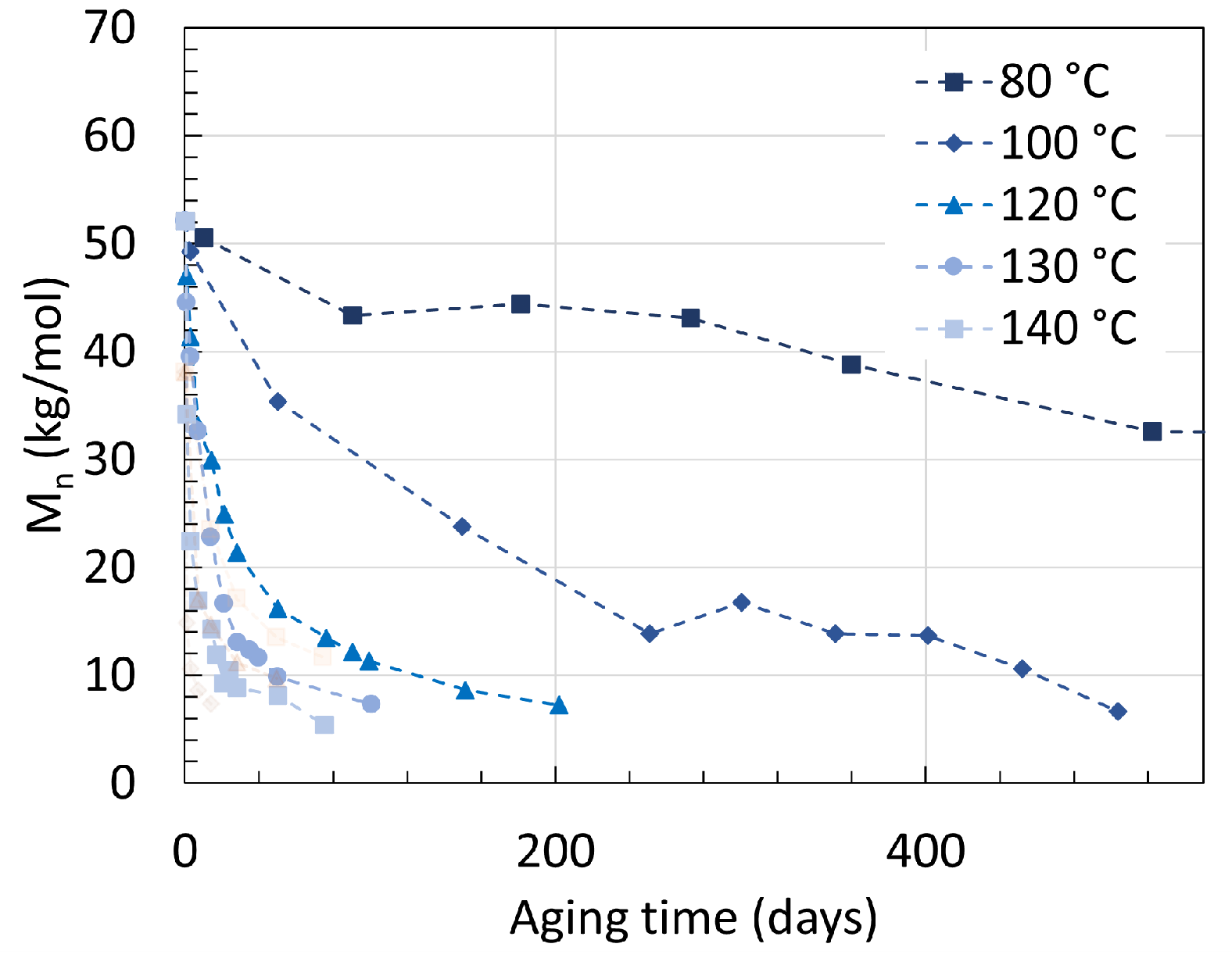
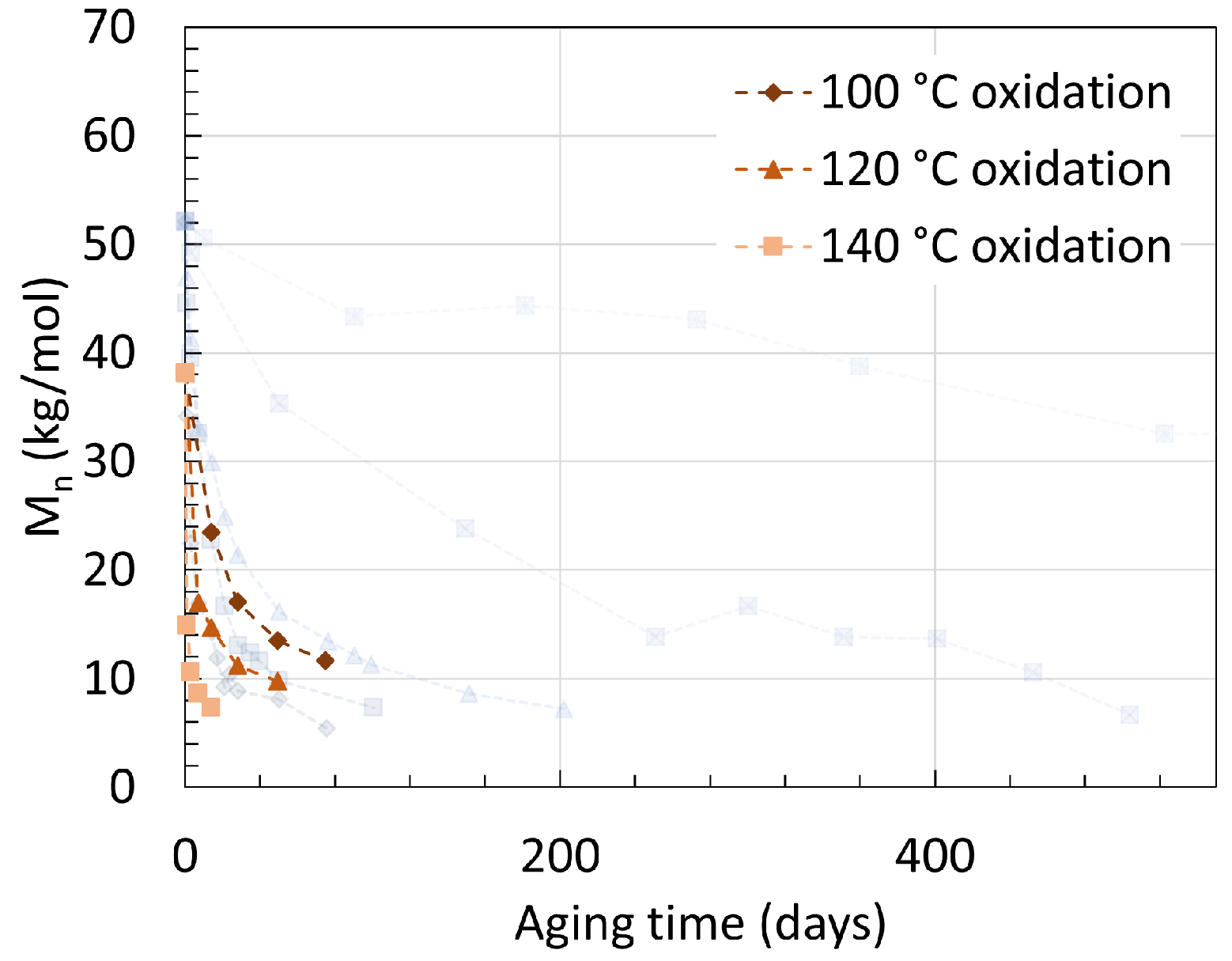
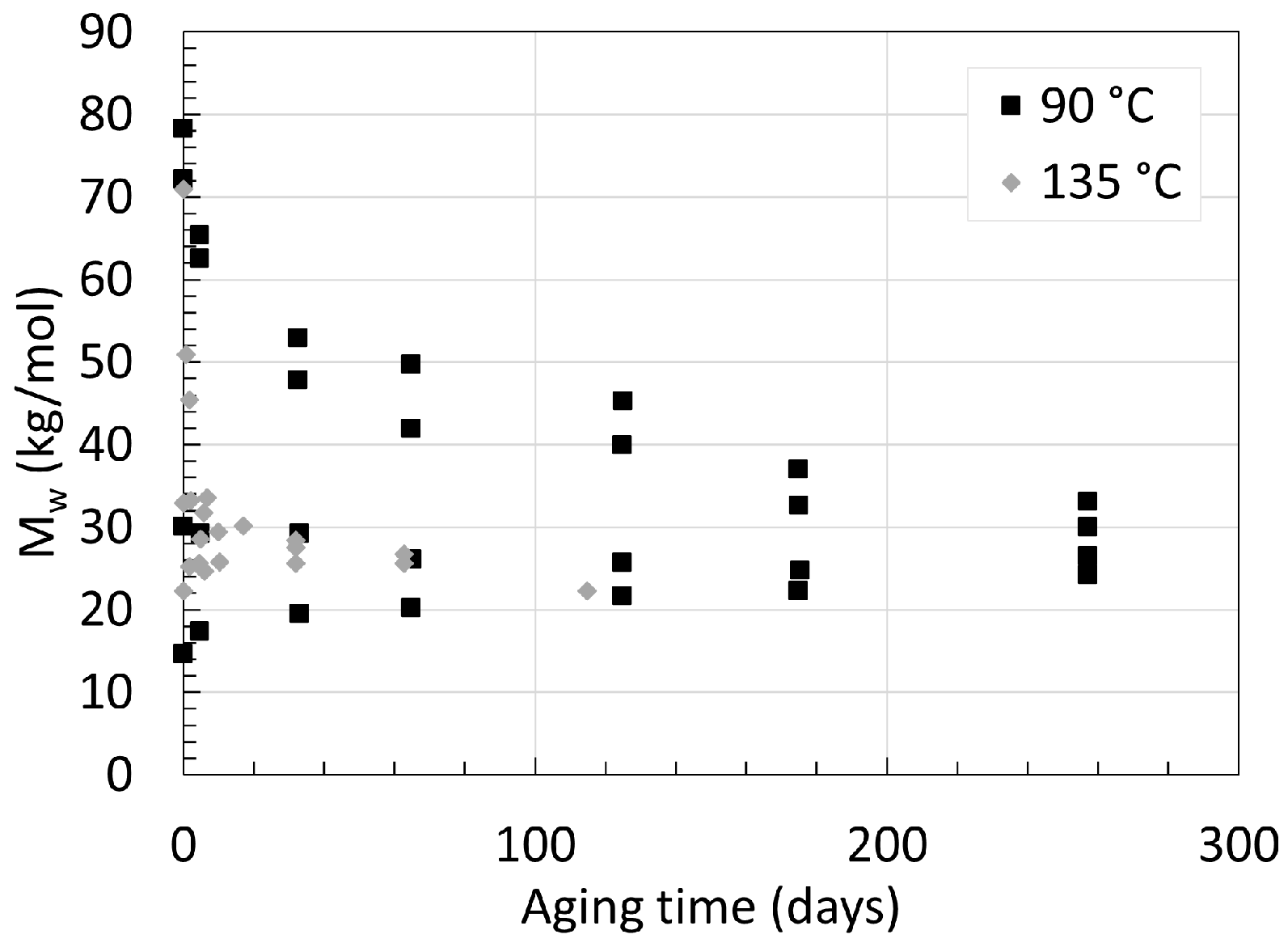
2.1. Polyamide 6
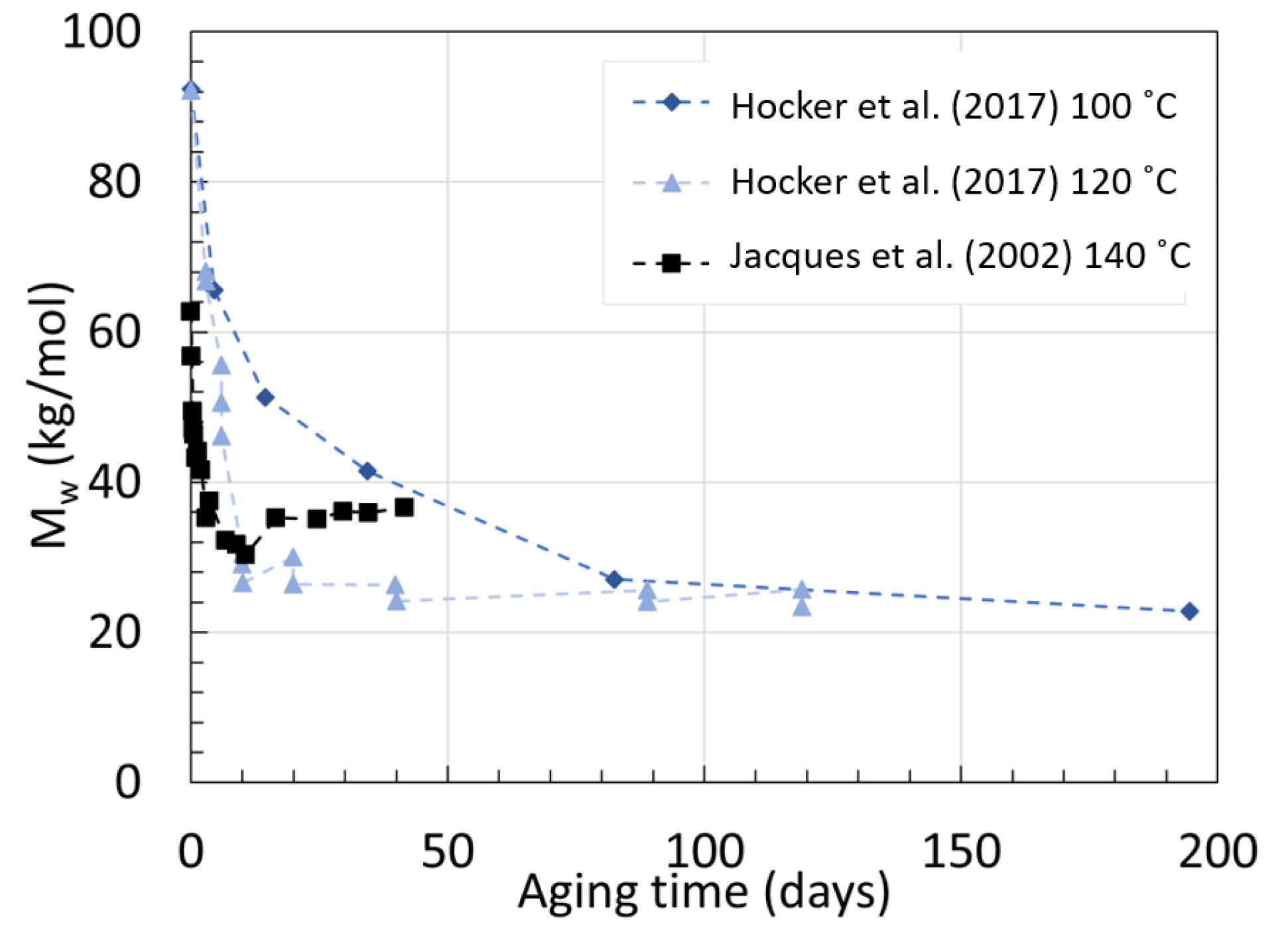
2.2. Polyamide 11
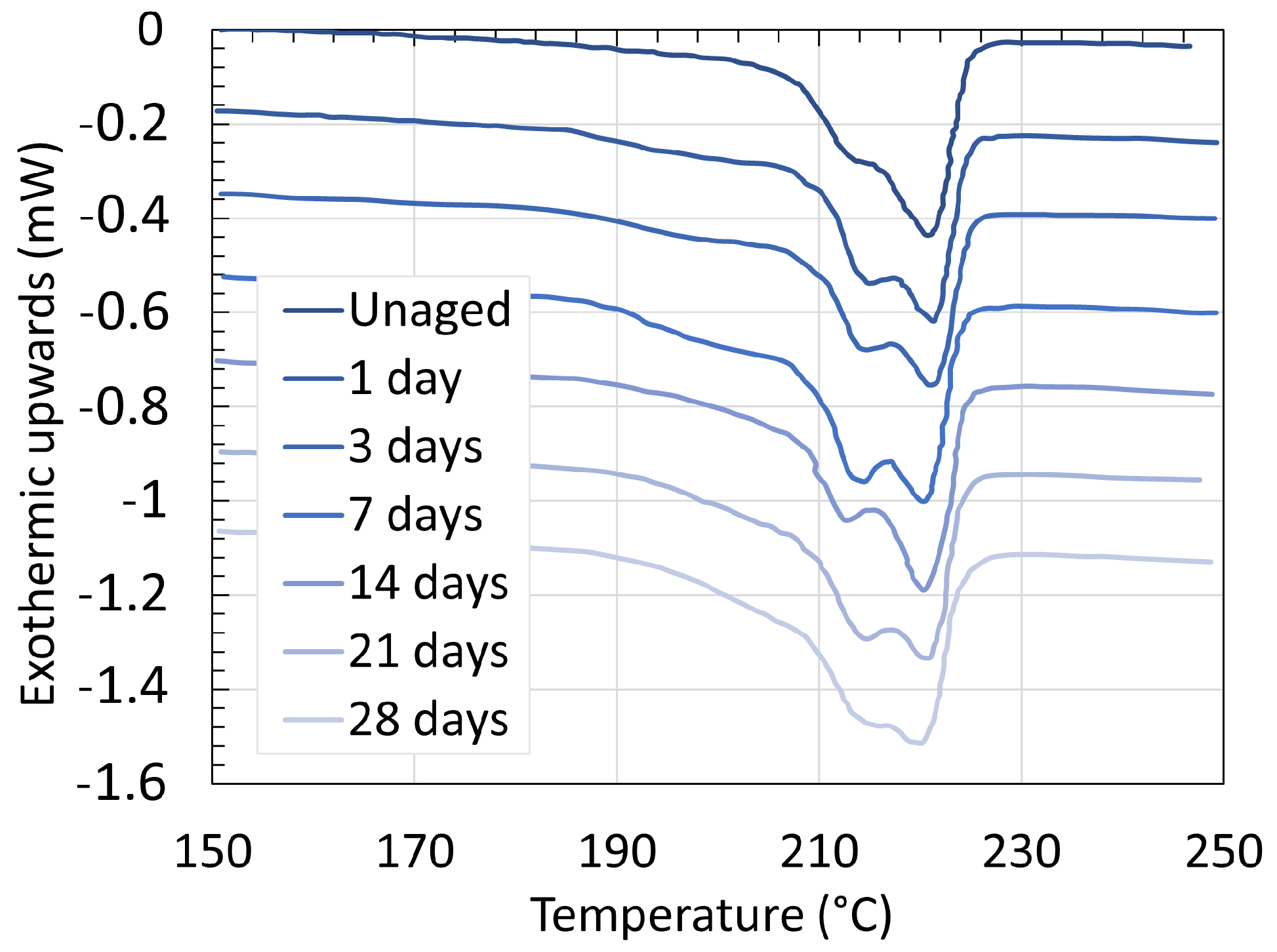
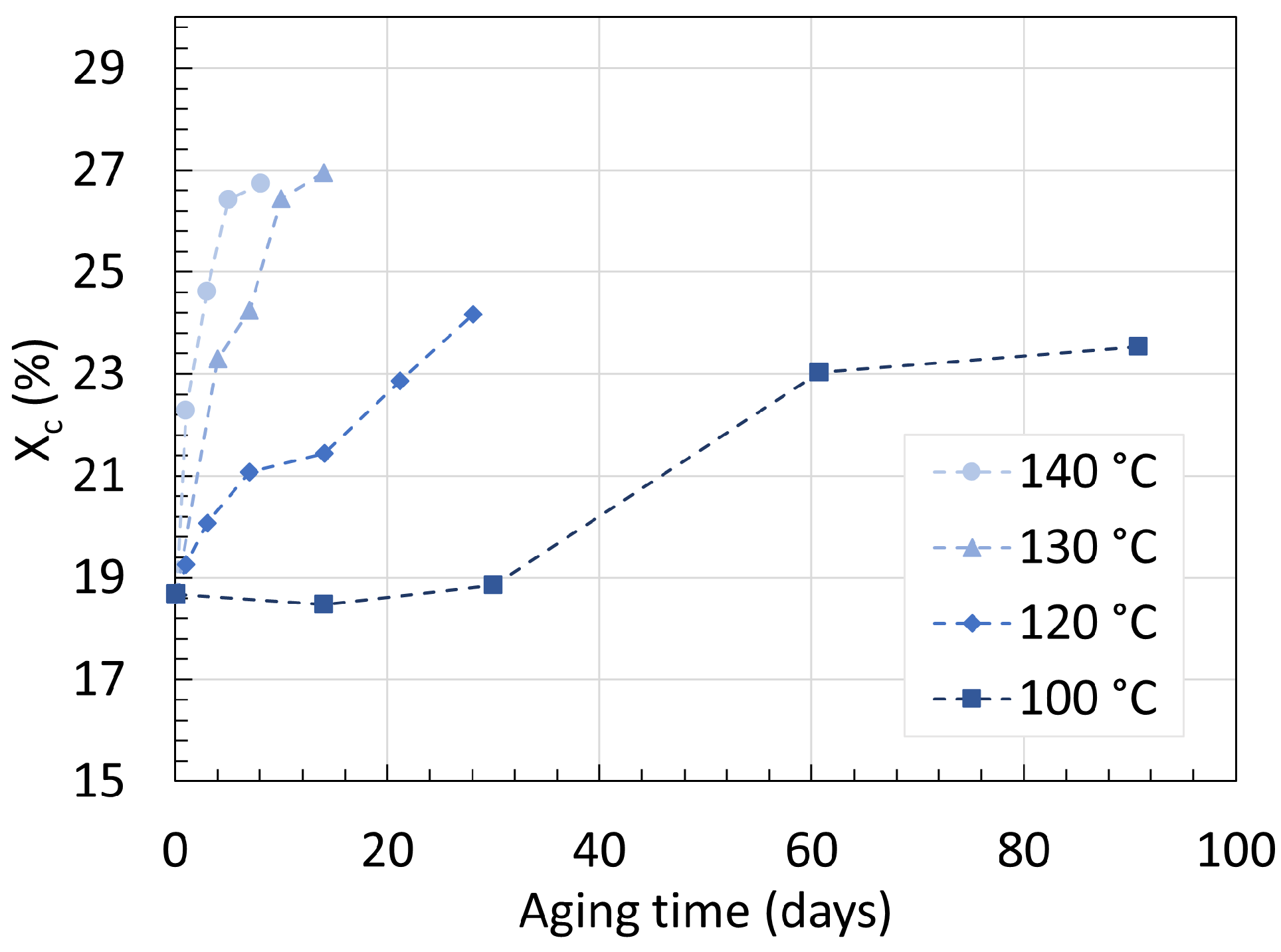
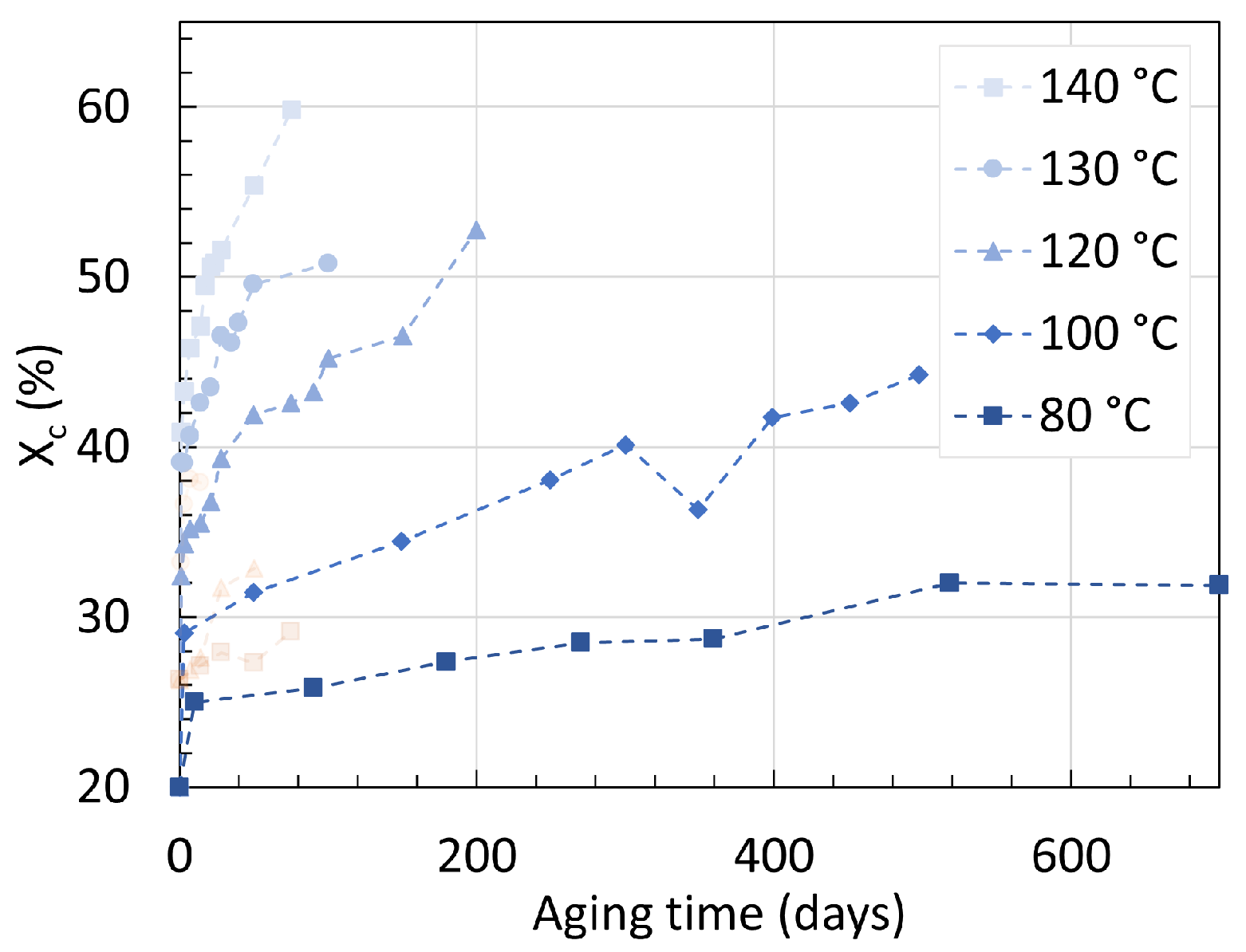
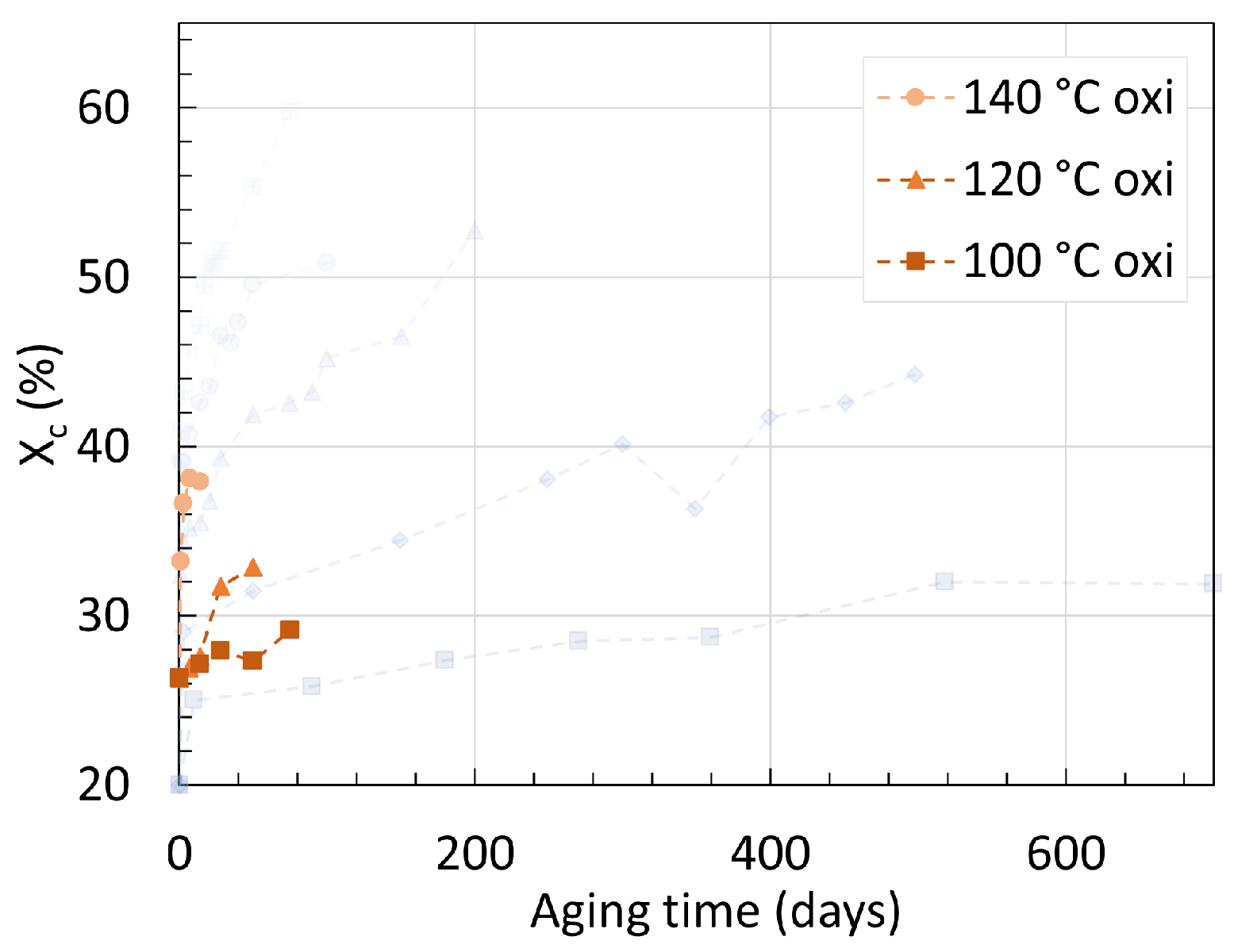
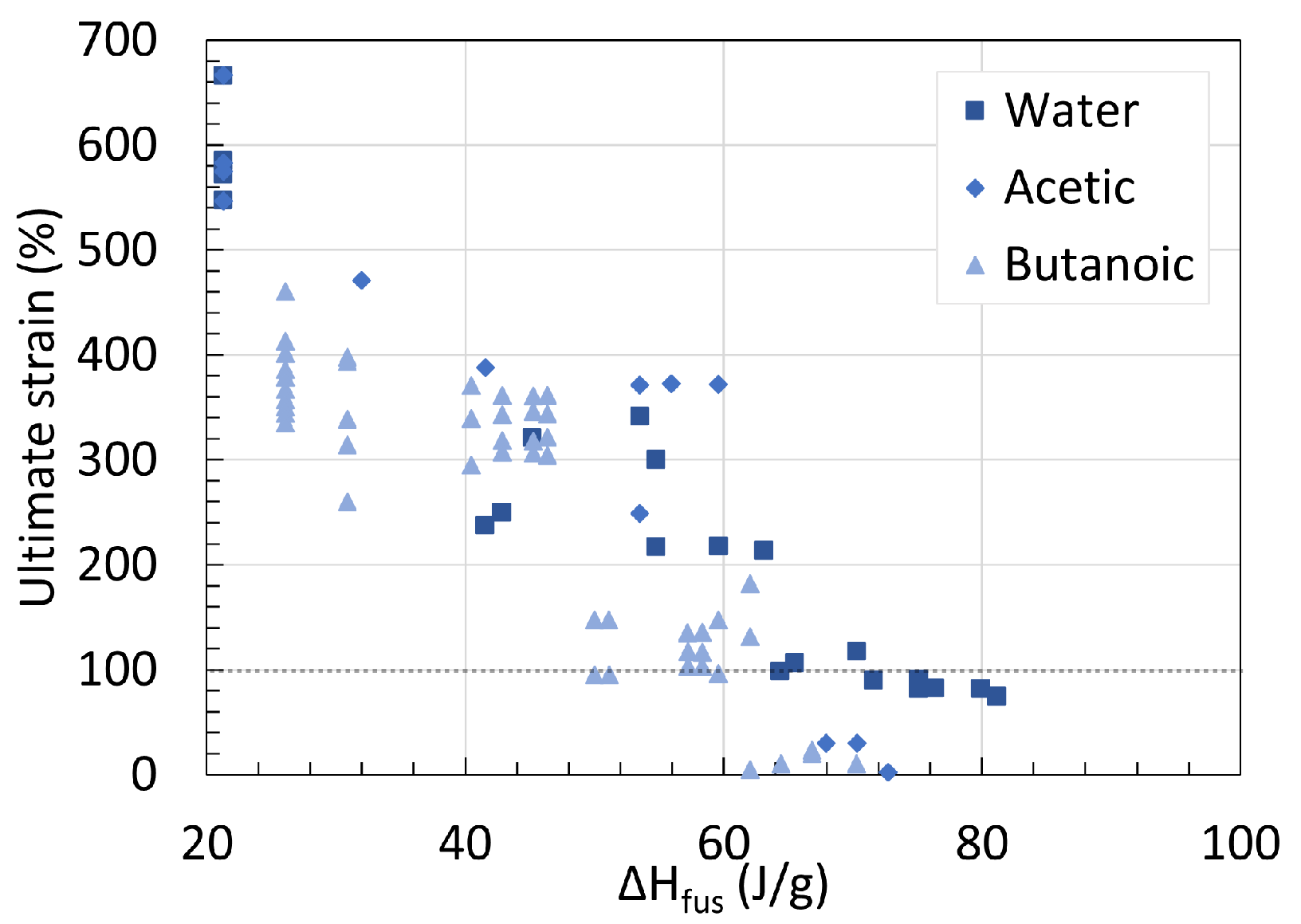
3. Crystallinity
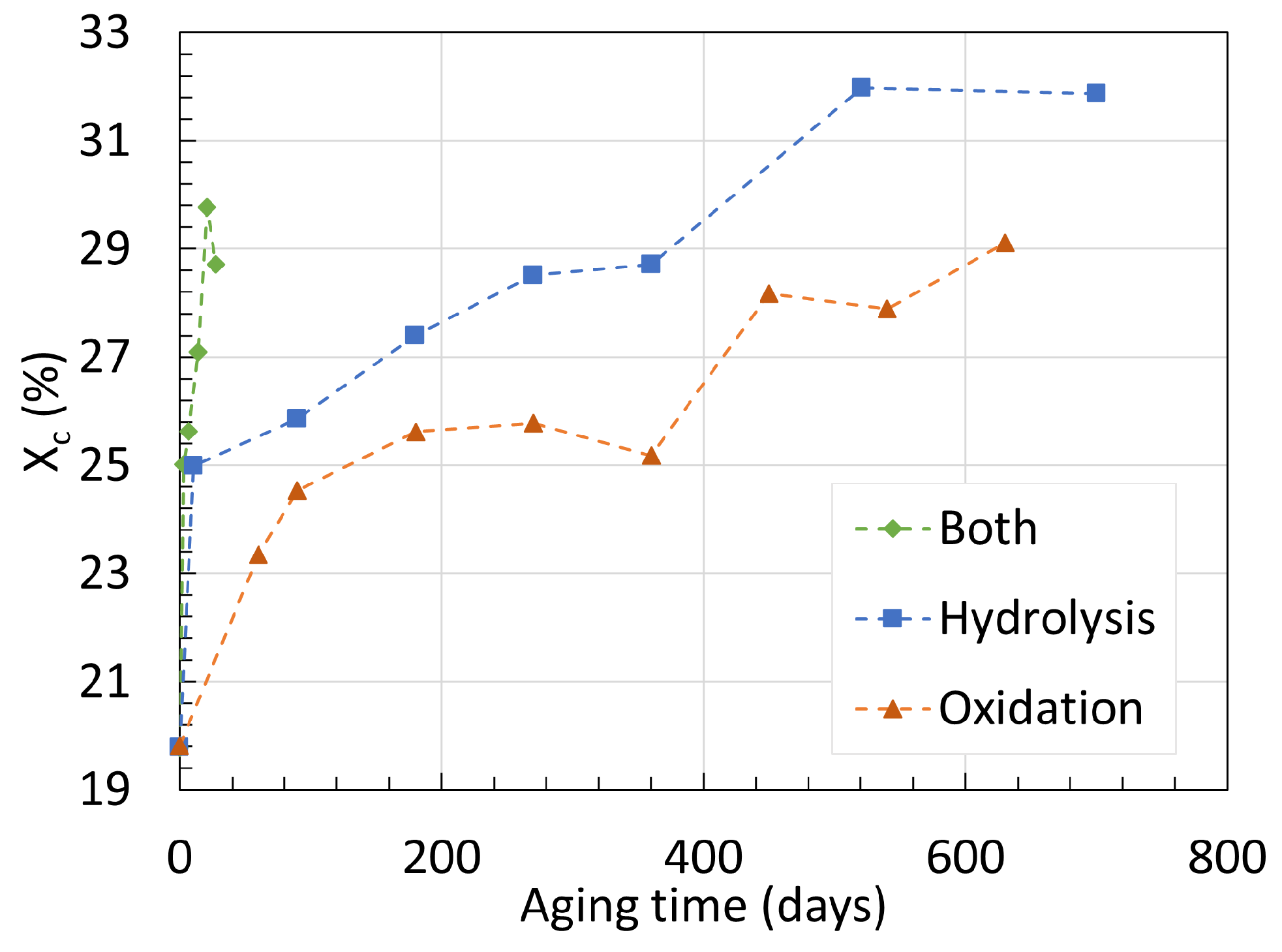
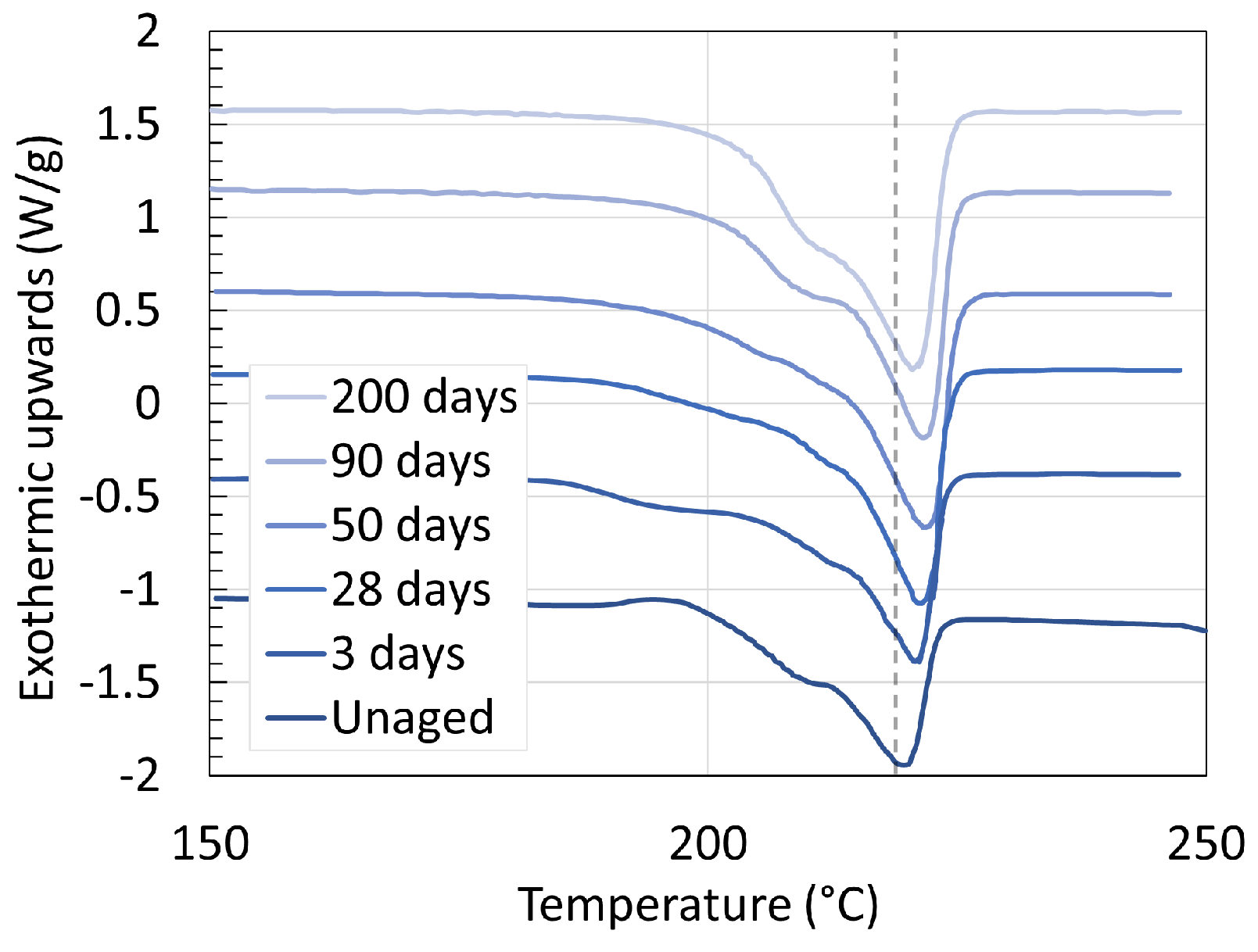
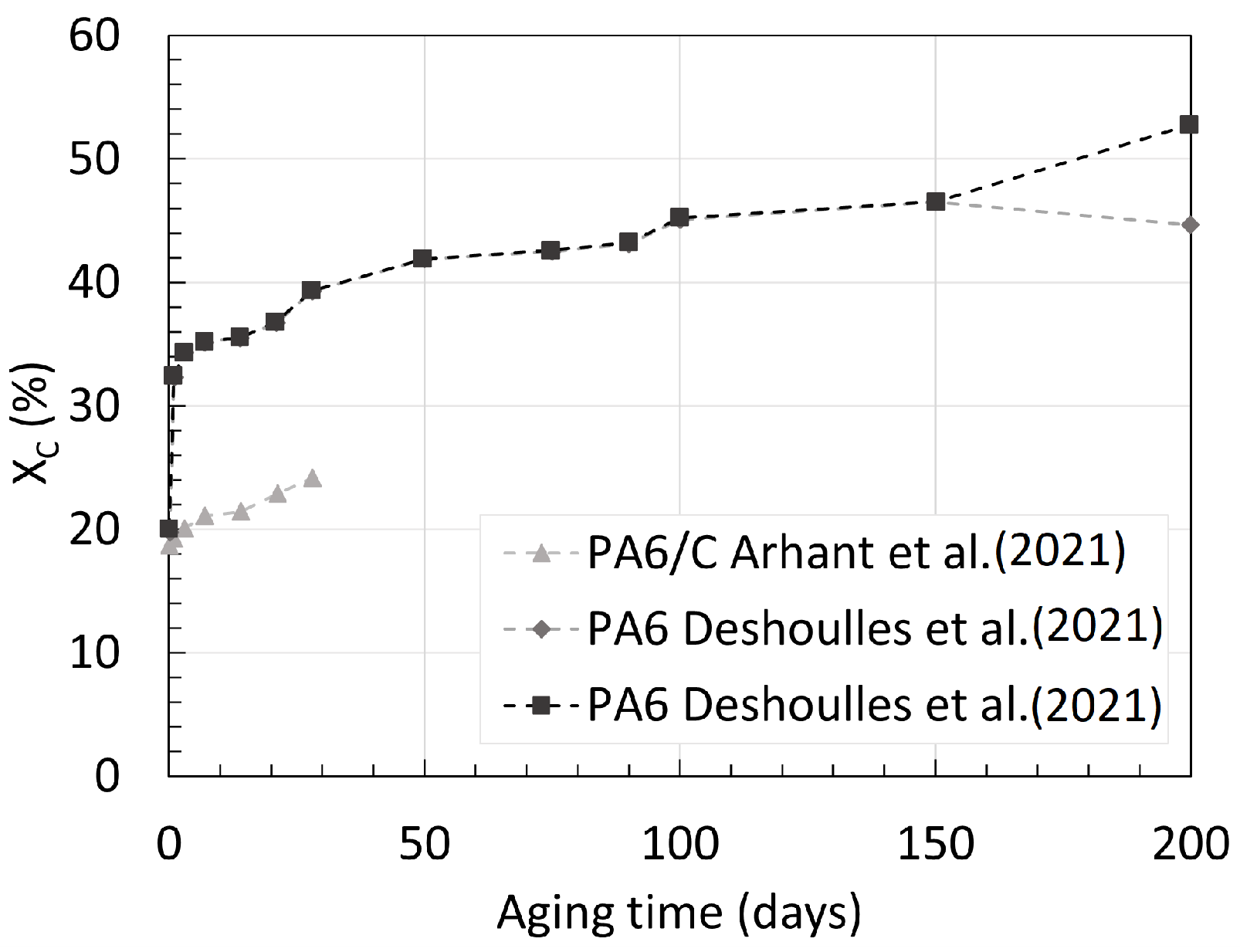
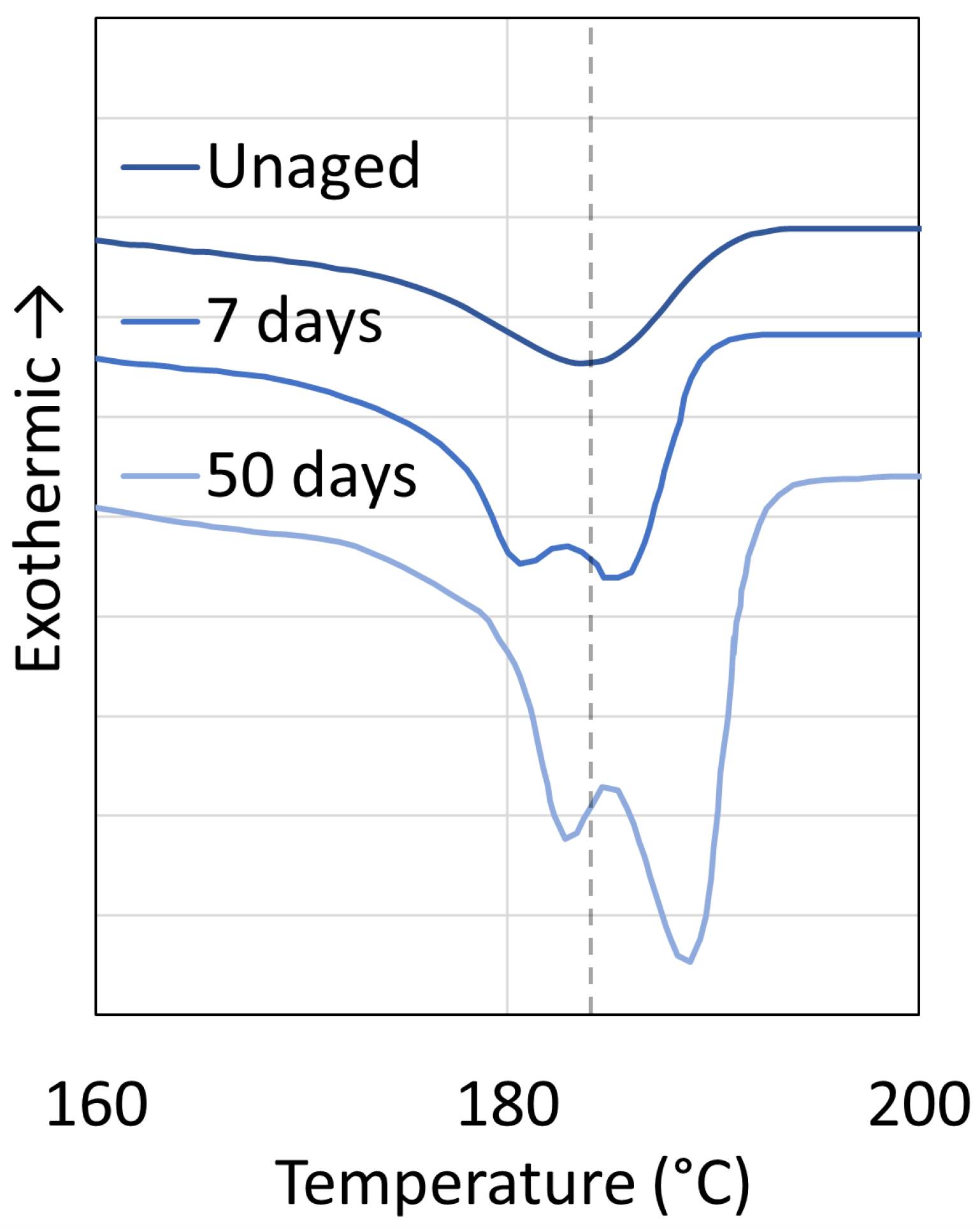
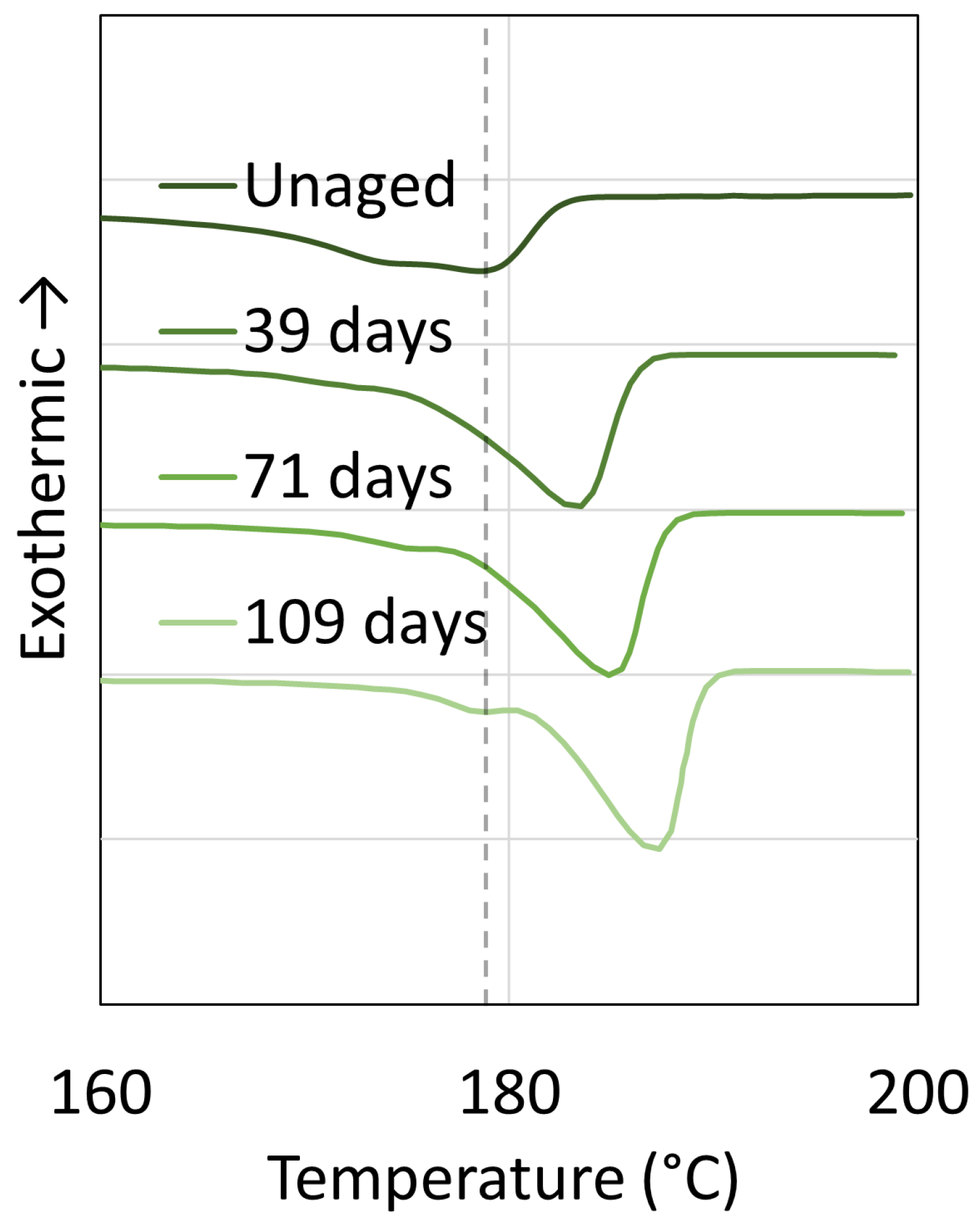
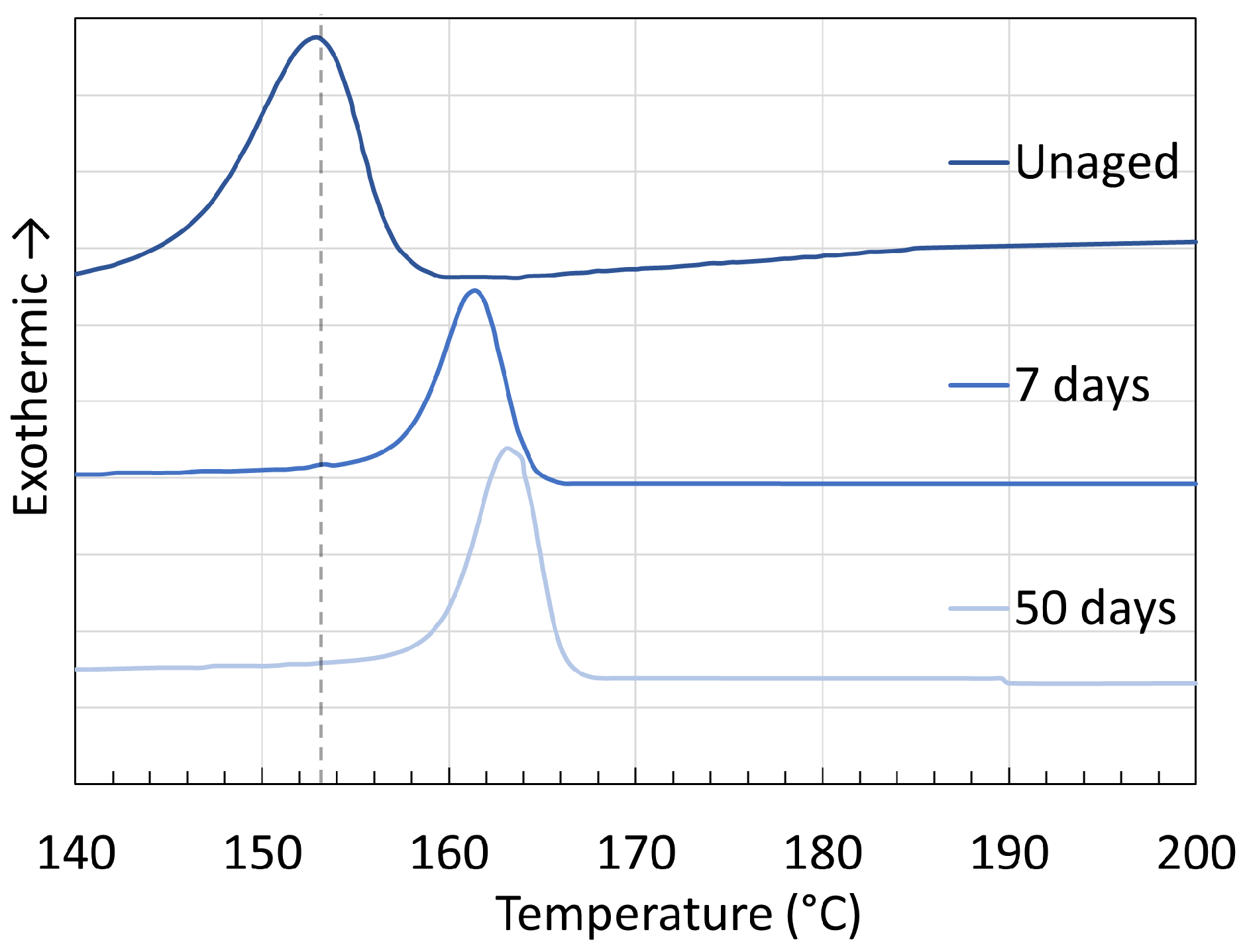
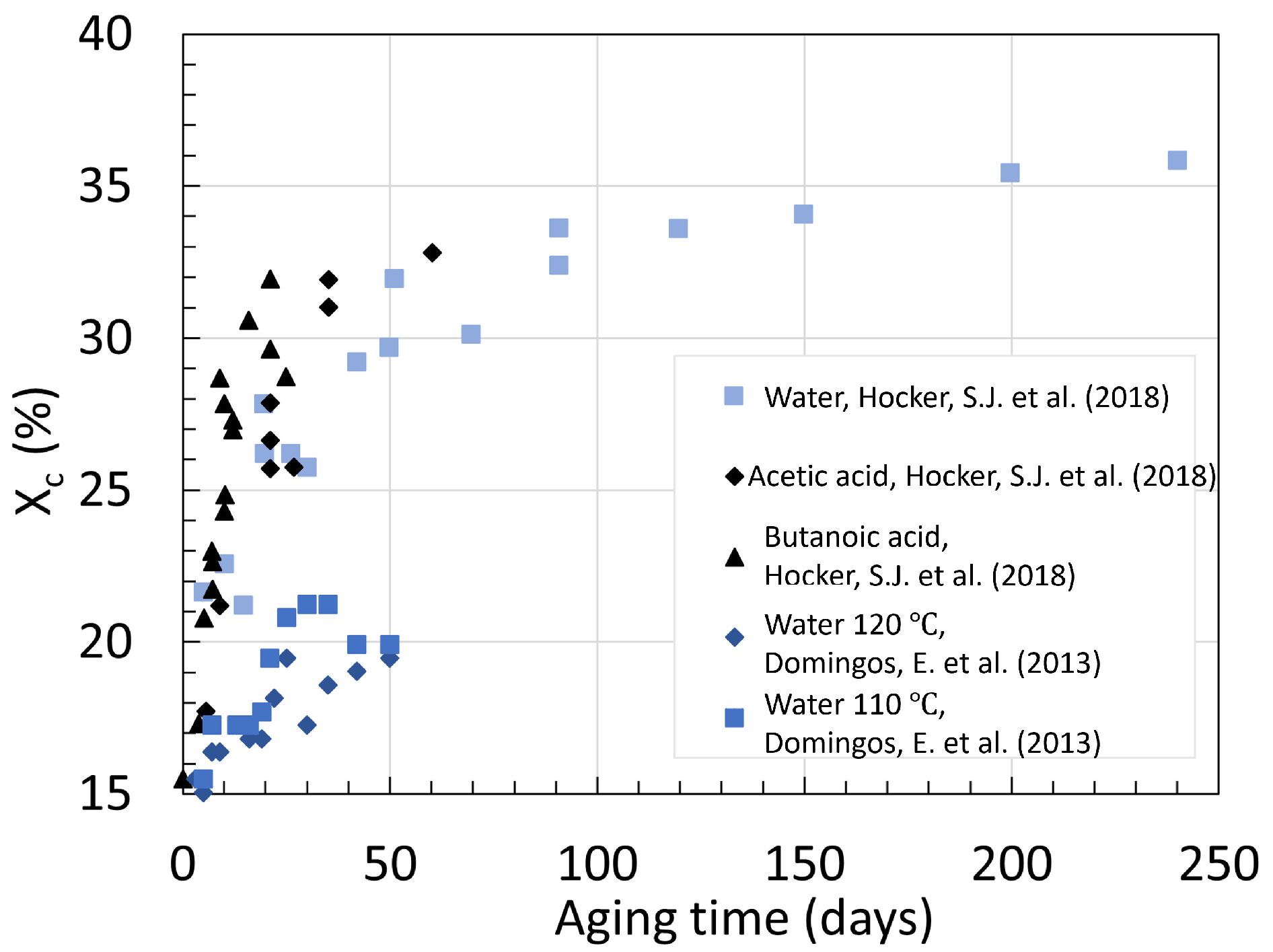
3.1. Polyamide 6
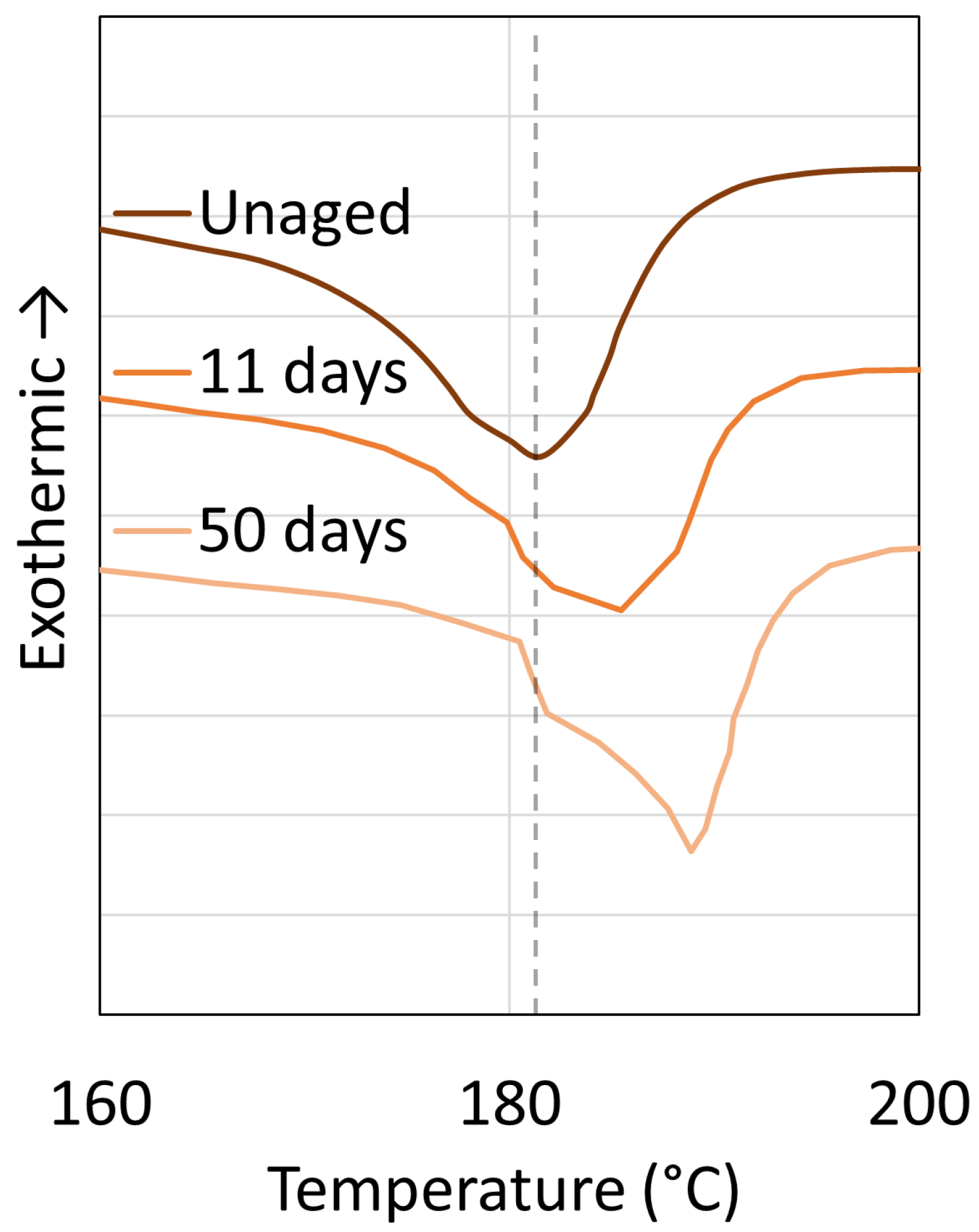
3.2. Polyamide 11
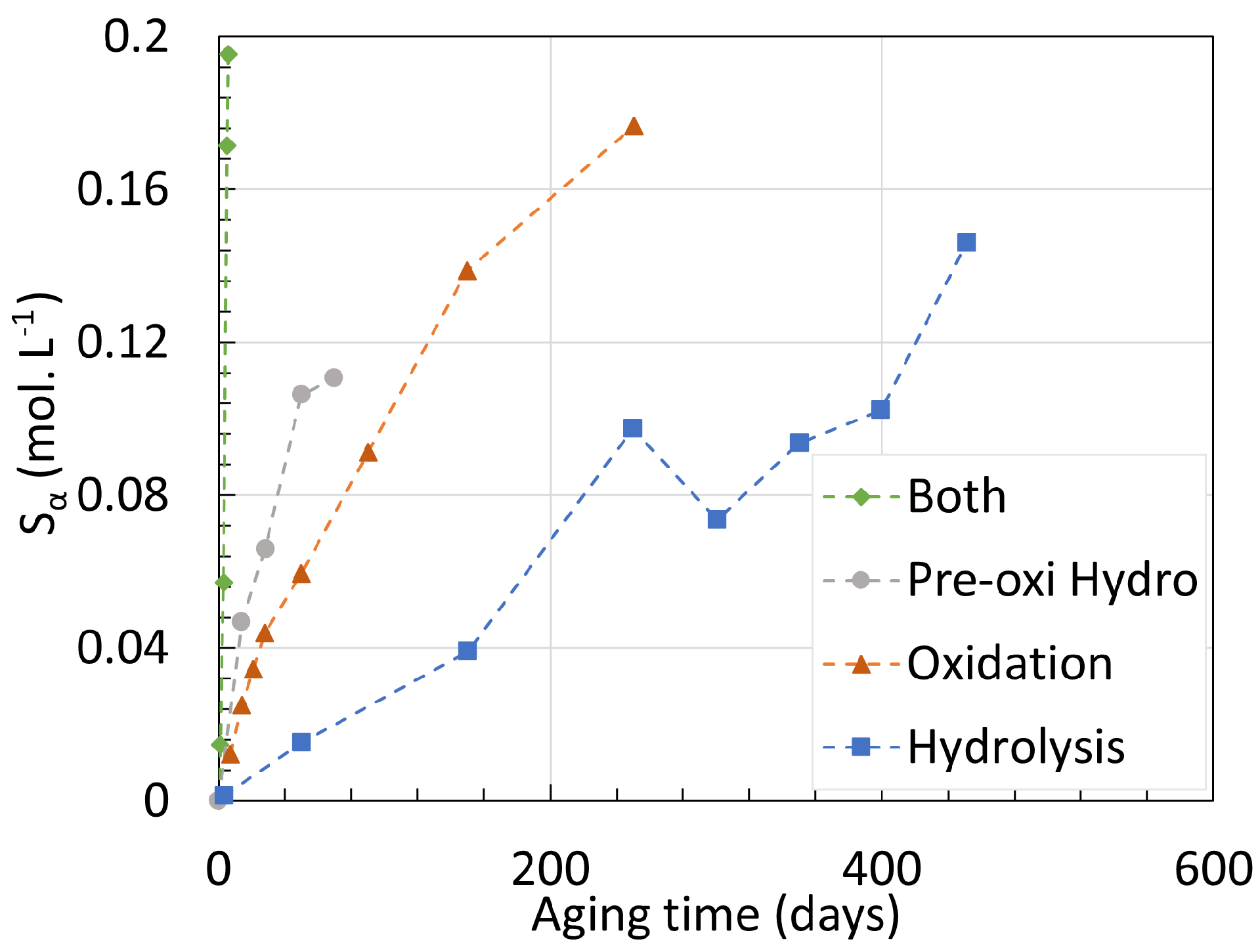
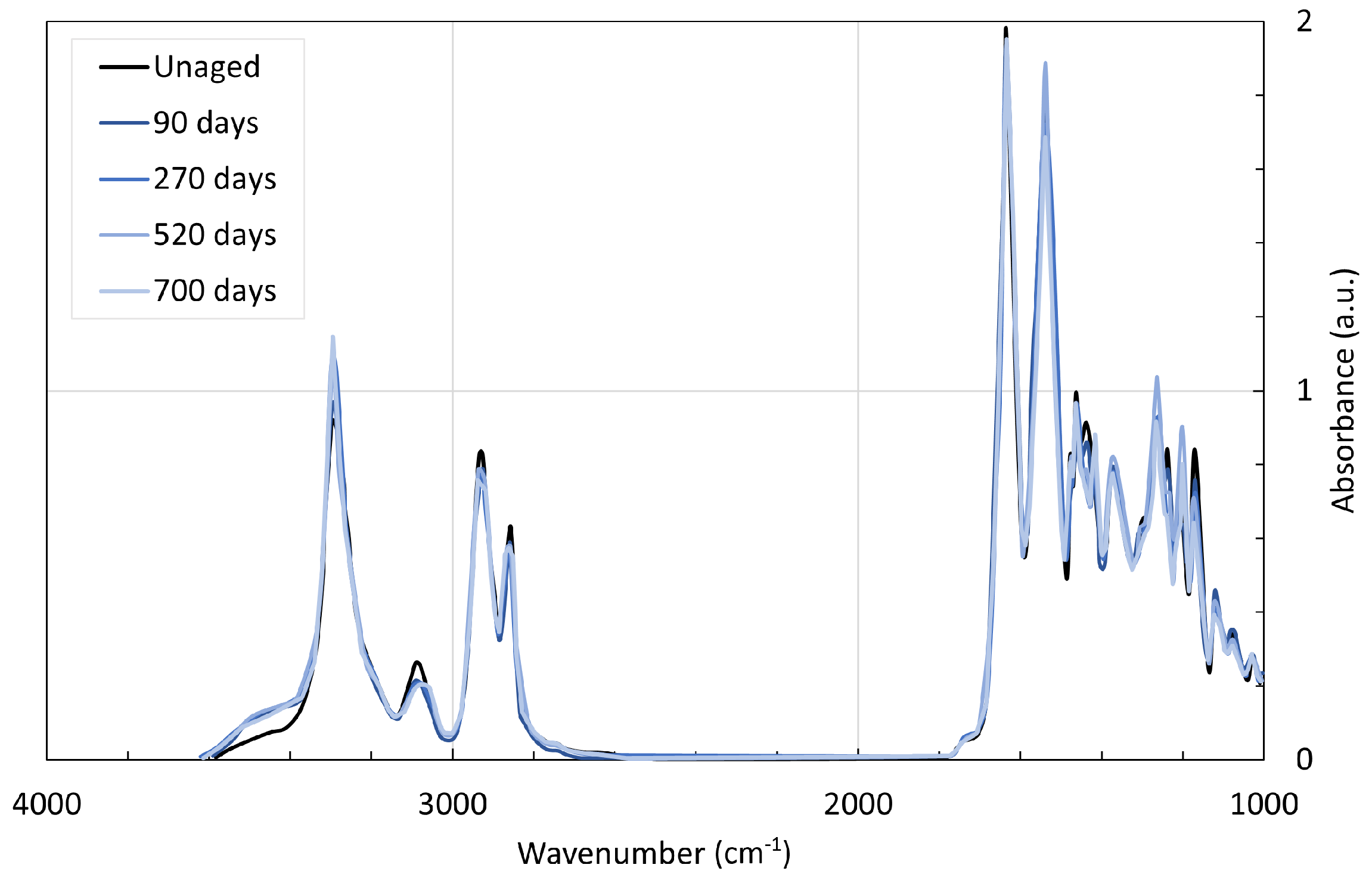
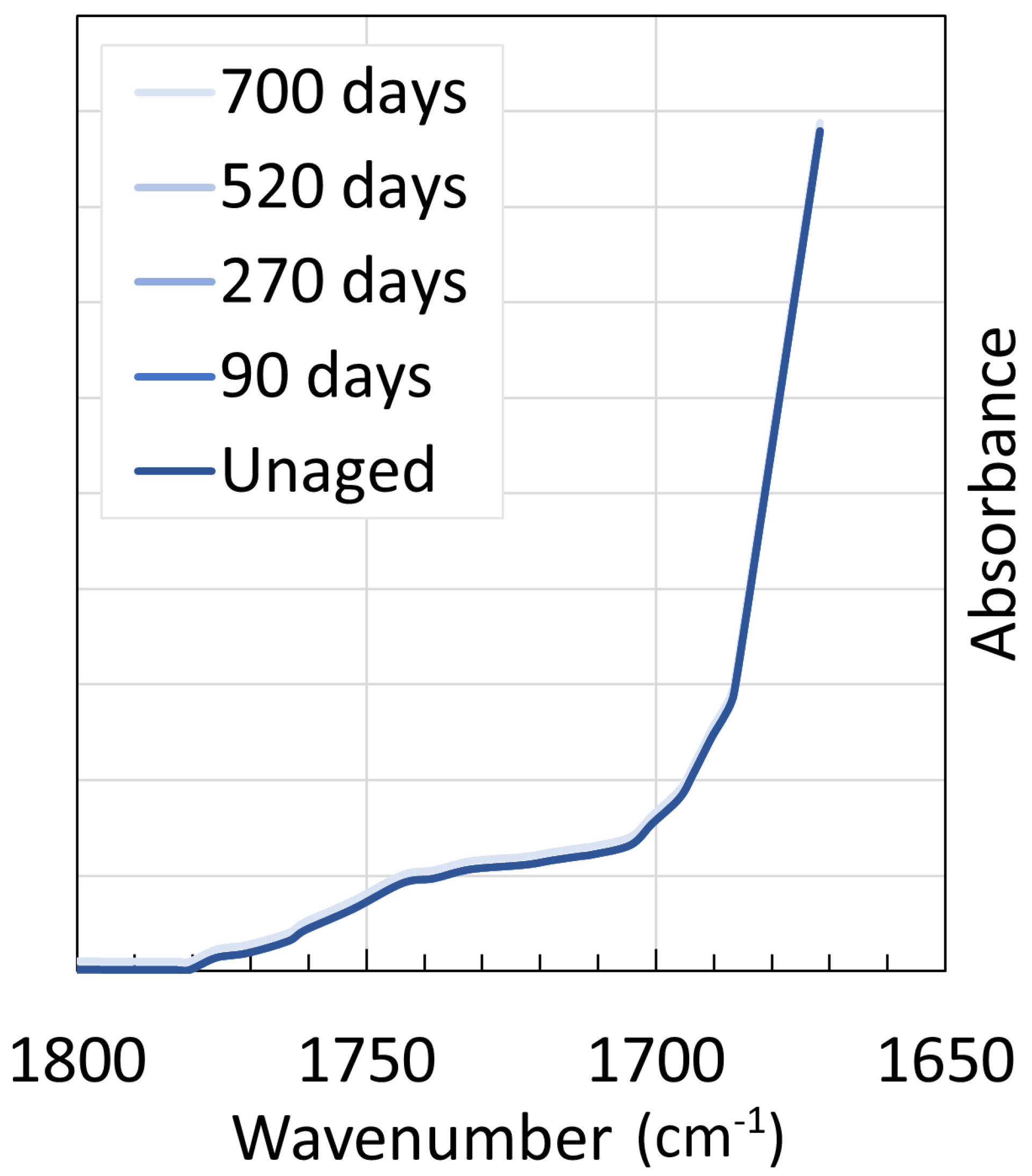
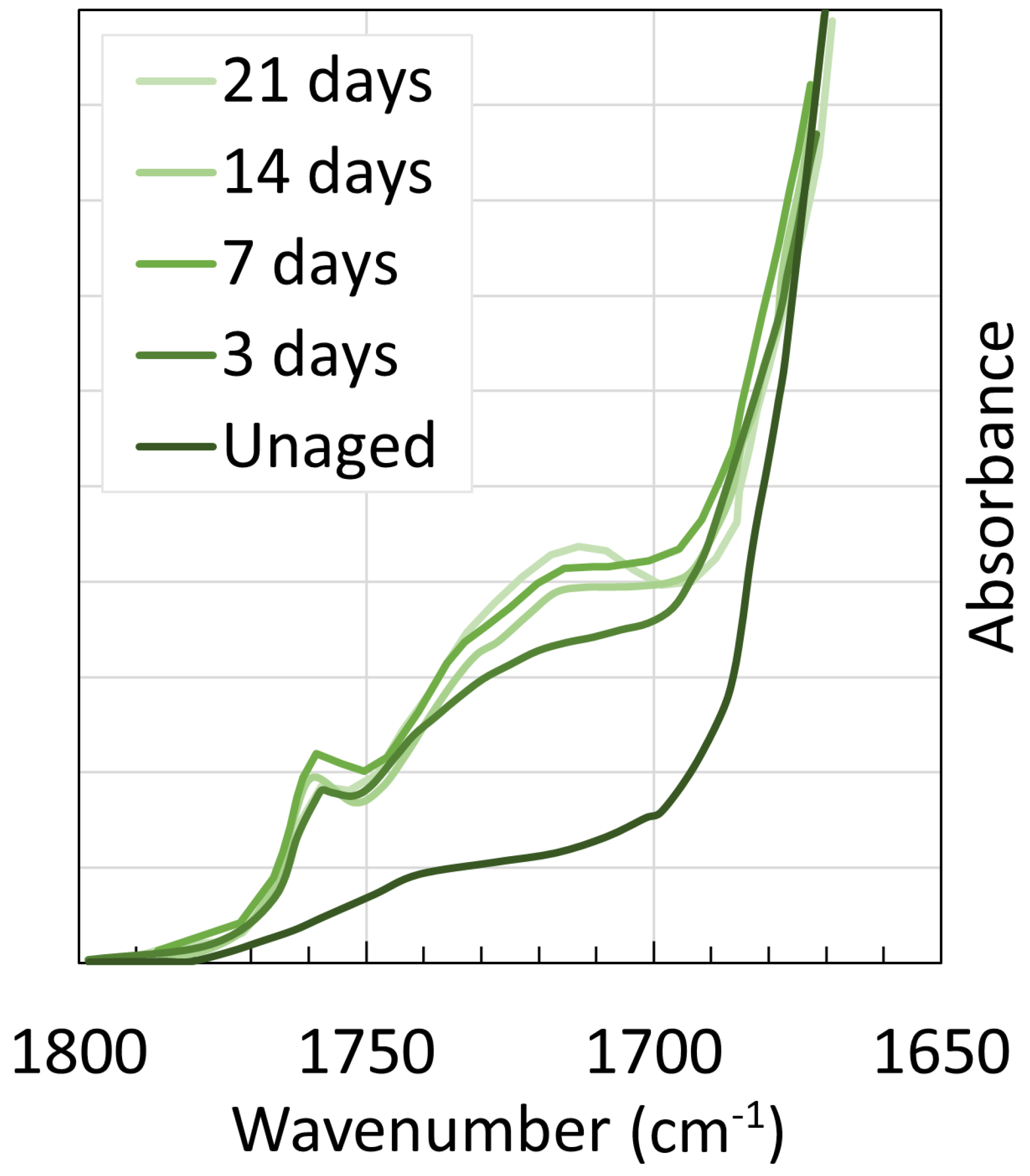
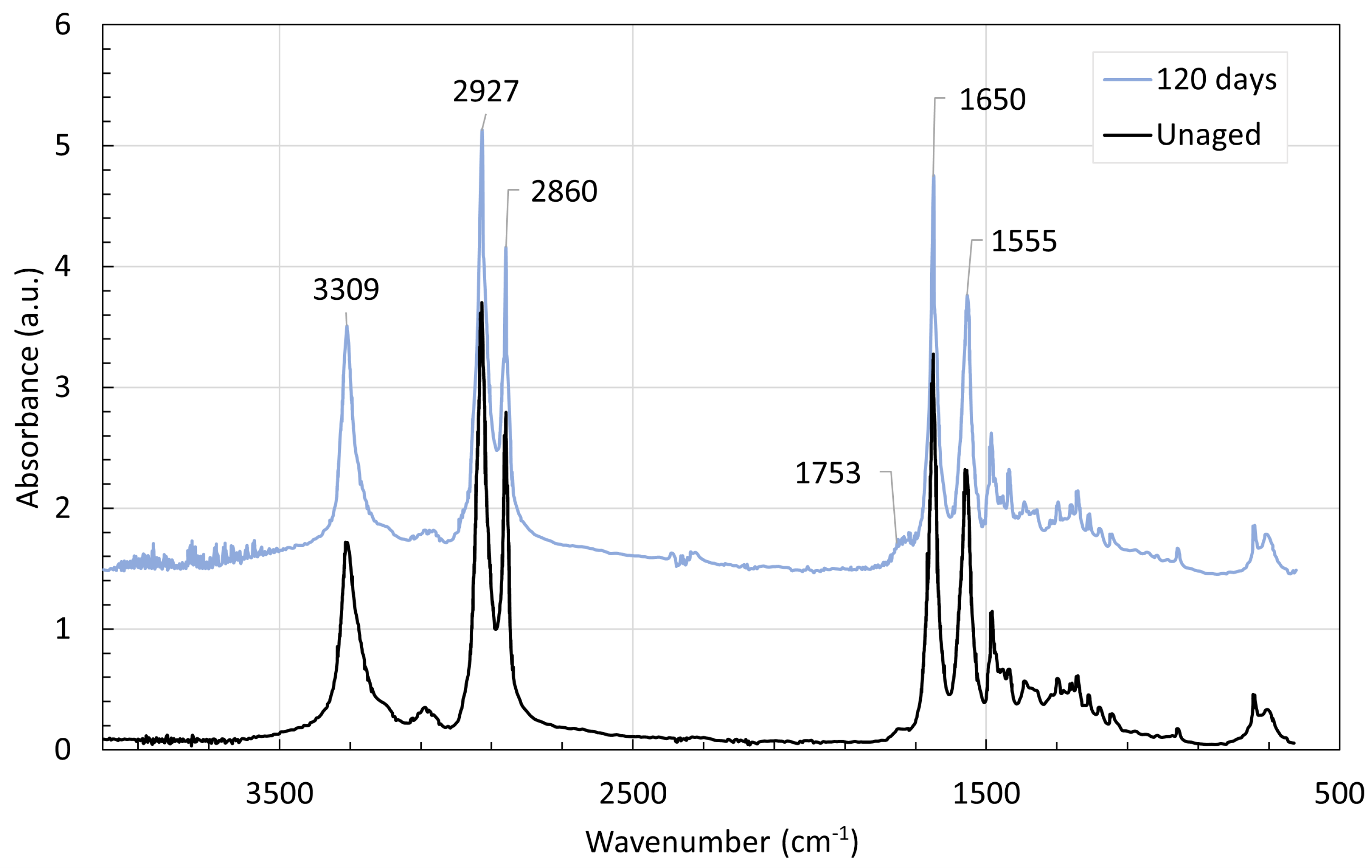
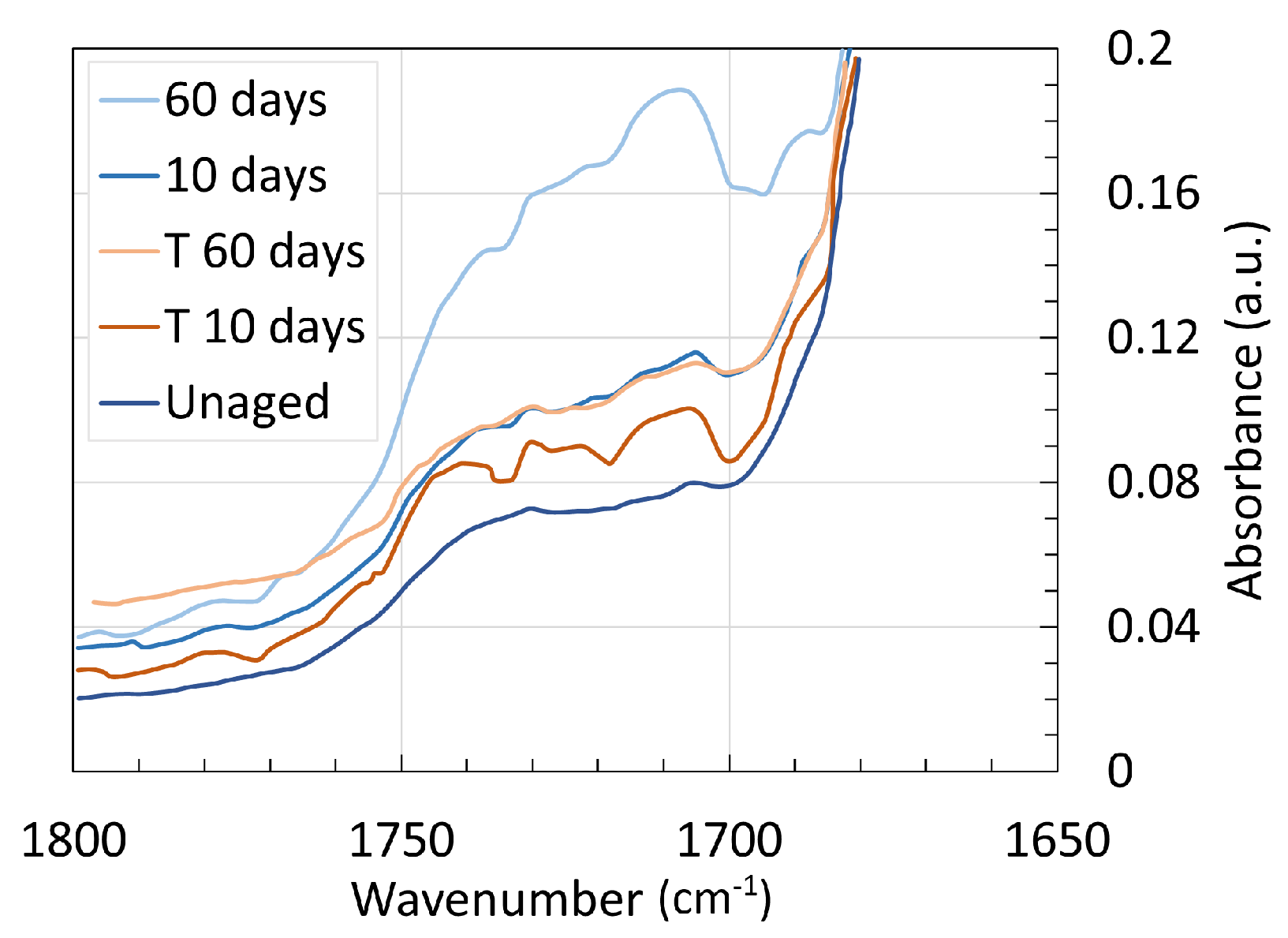
4. Chemical Analysis
4.1. Polyamide 6
4.2. Polyamide 11
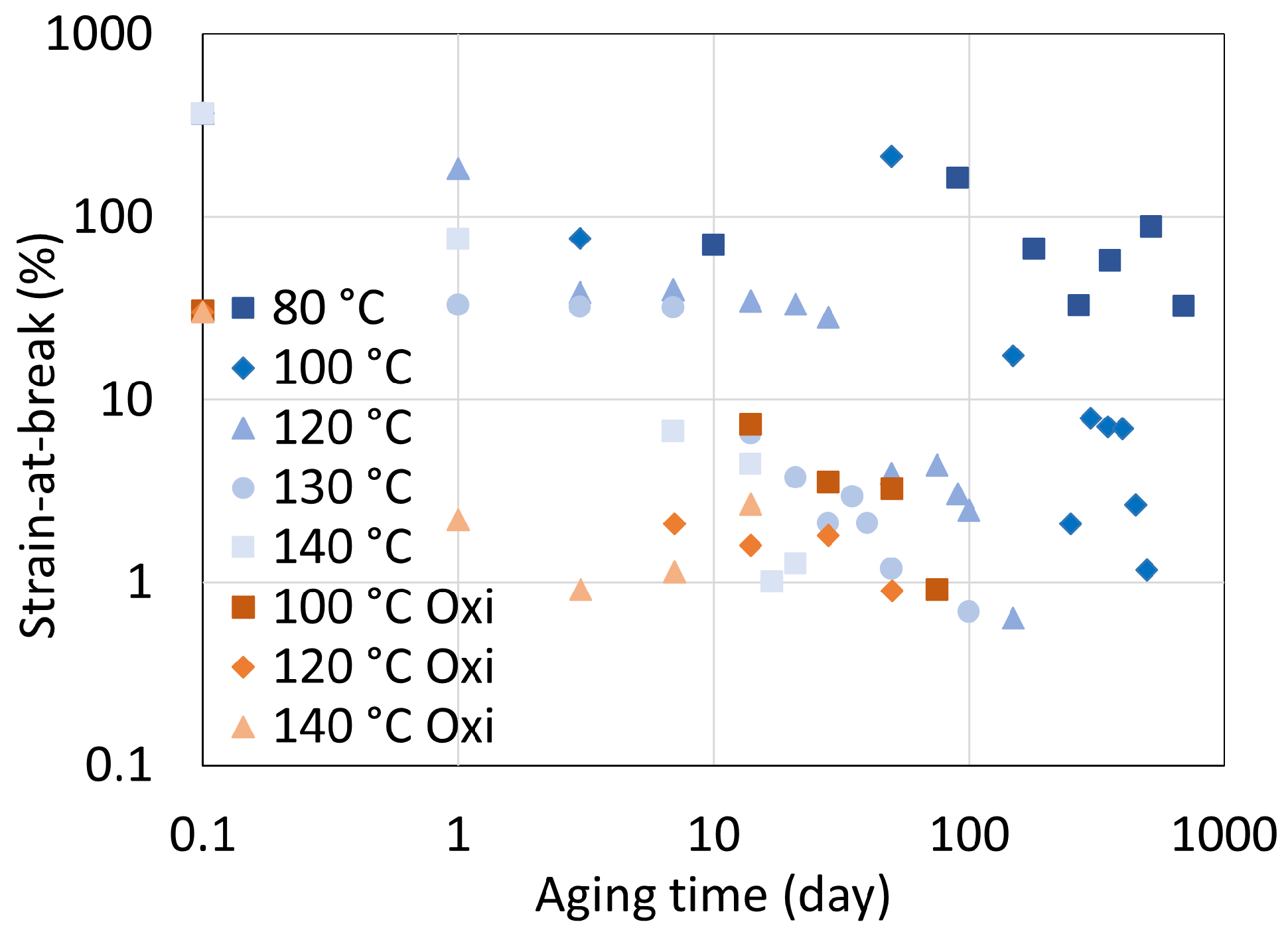
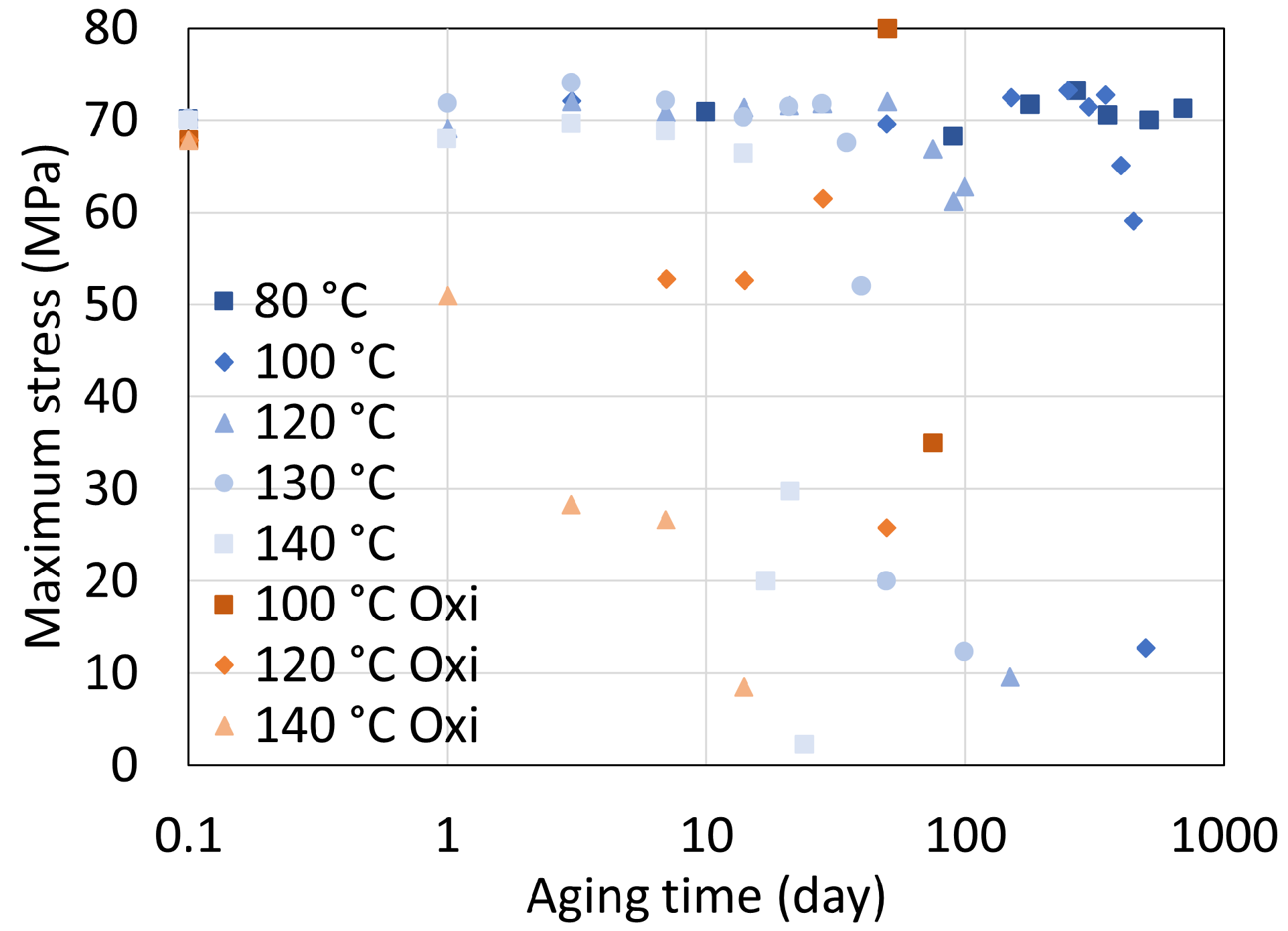
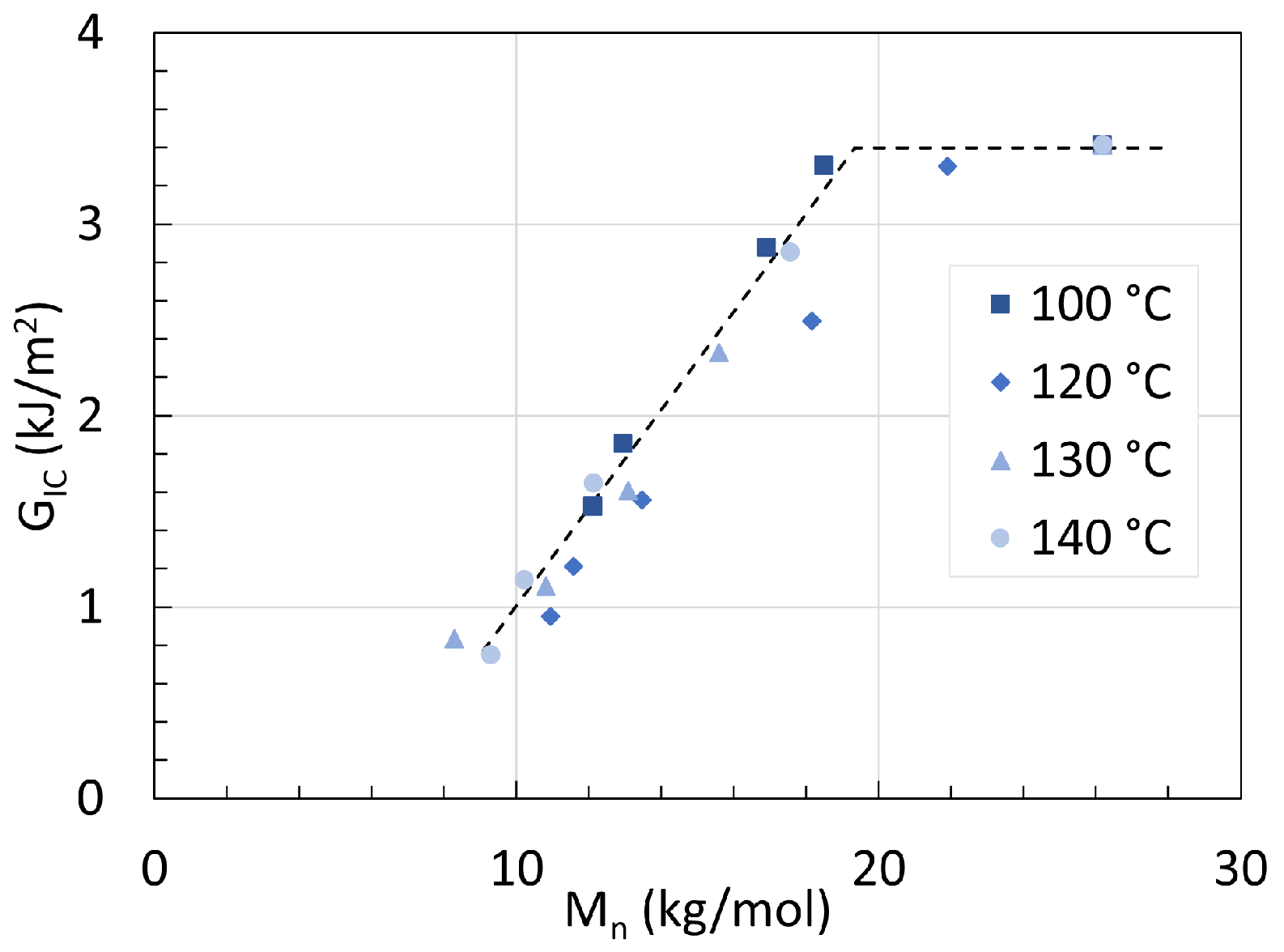
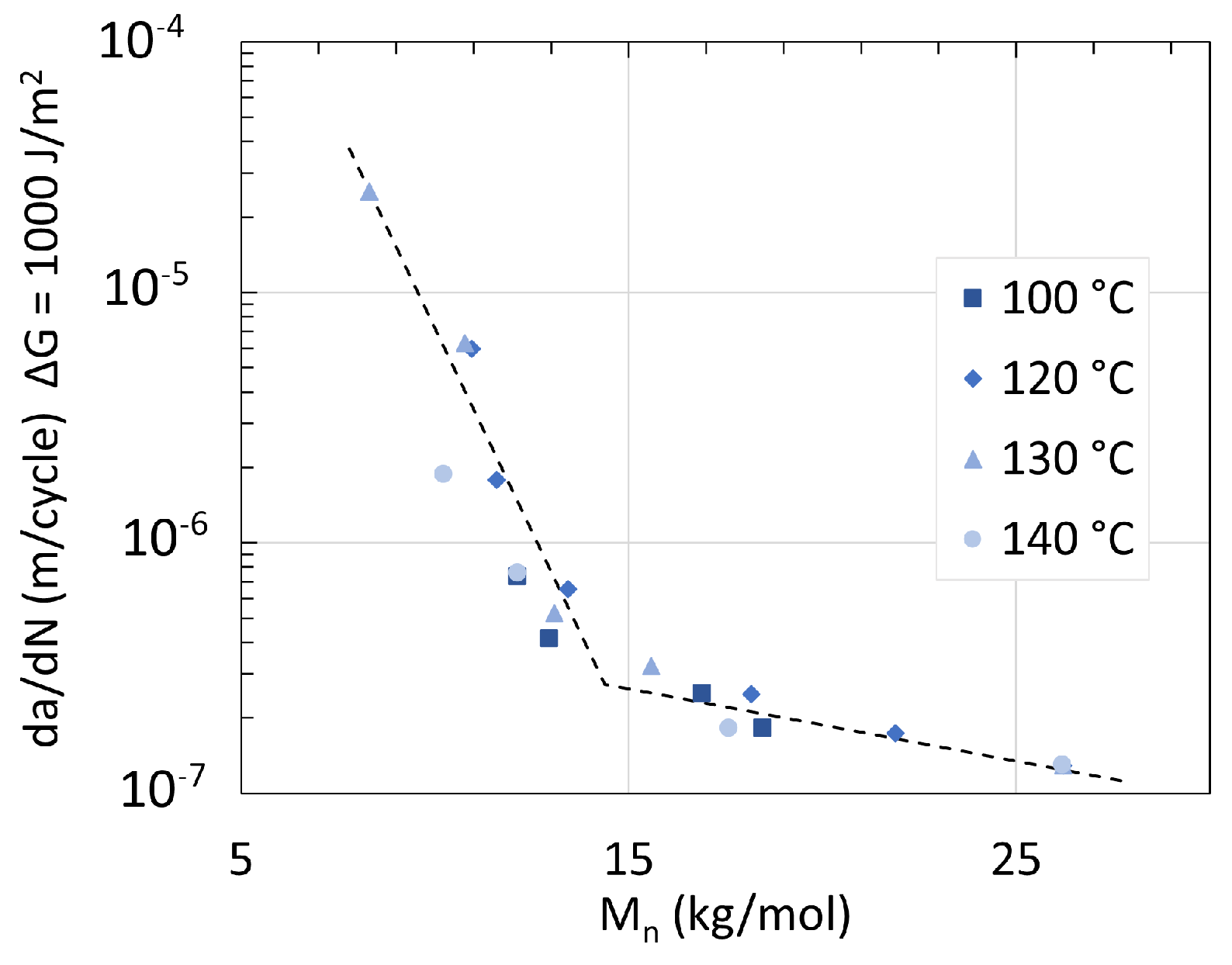
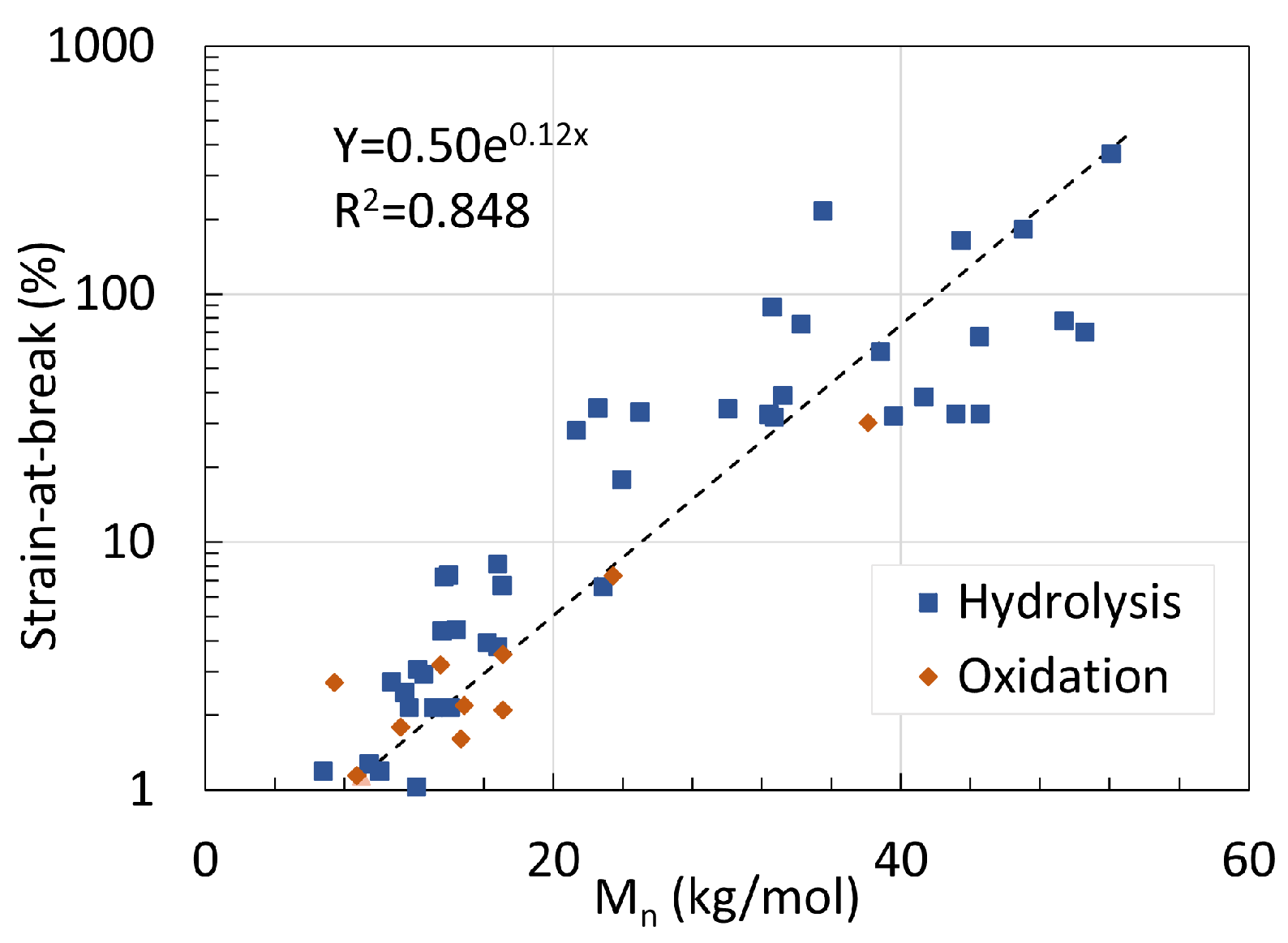
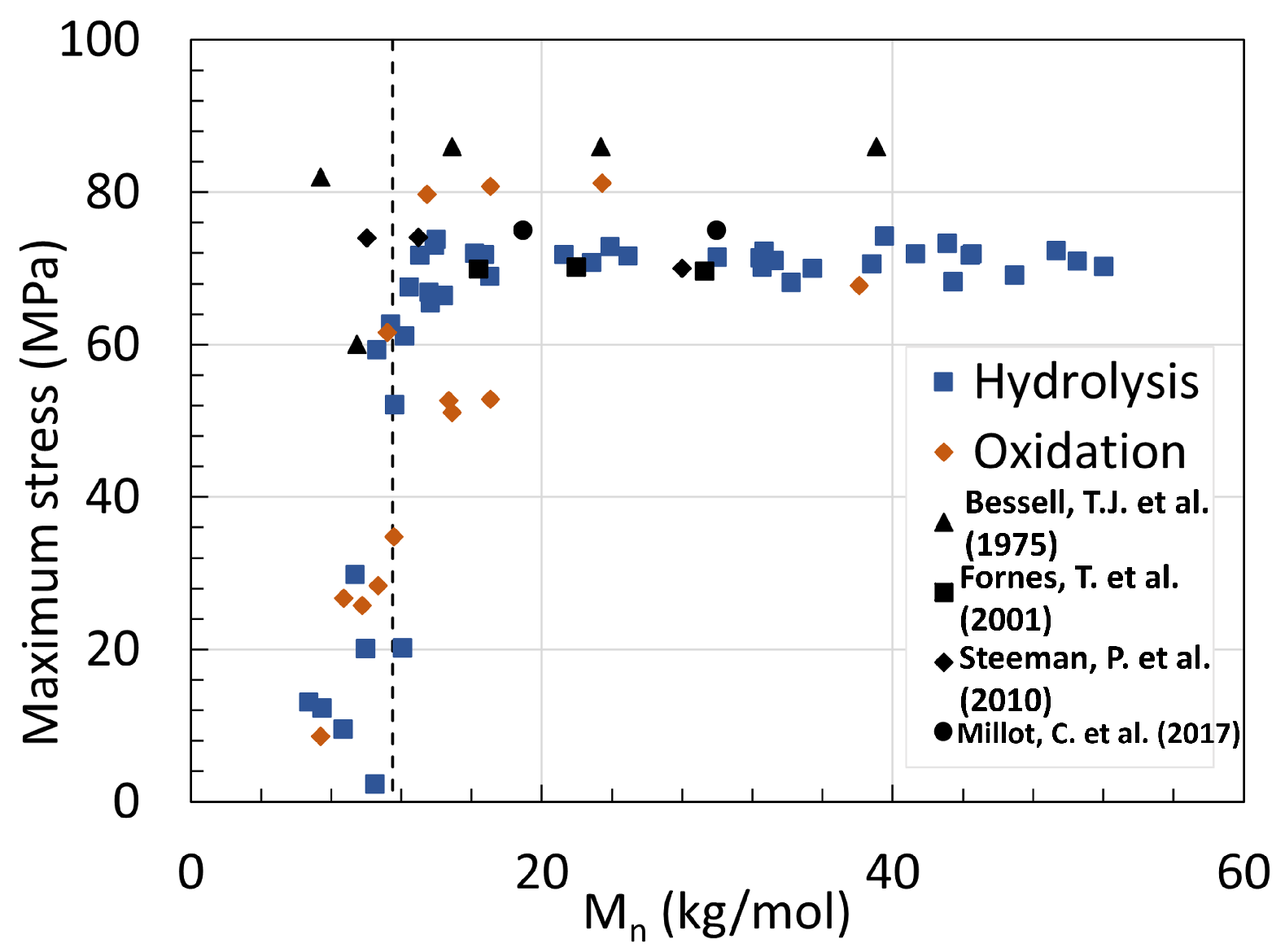
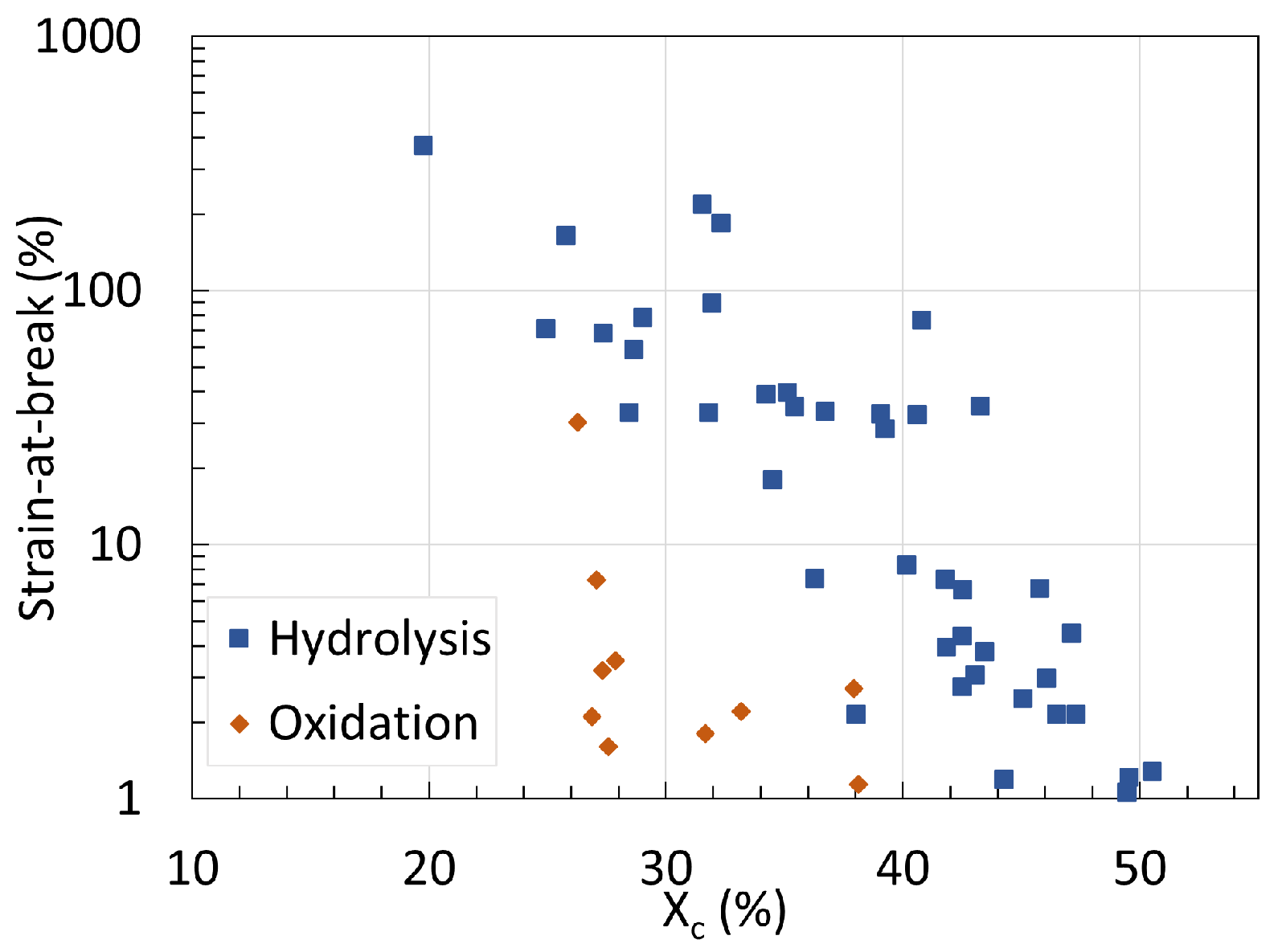
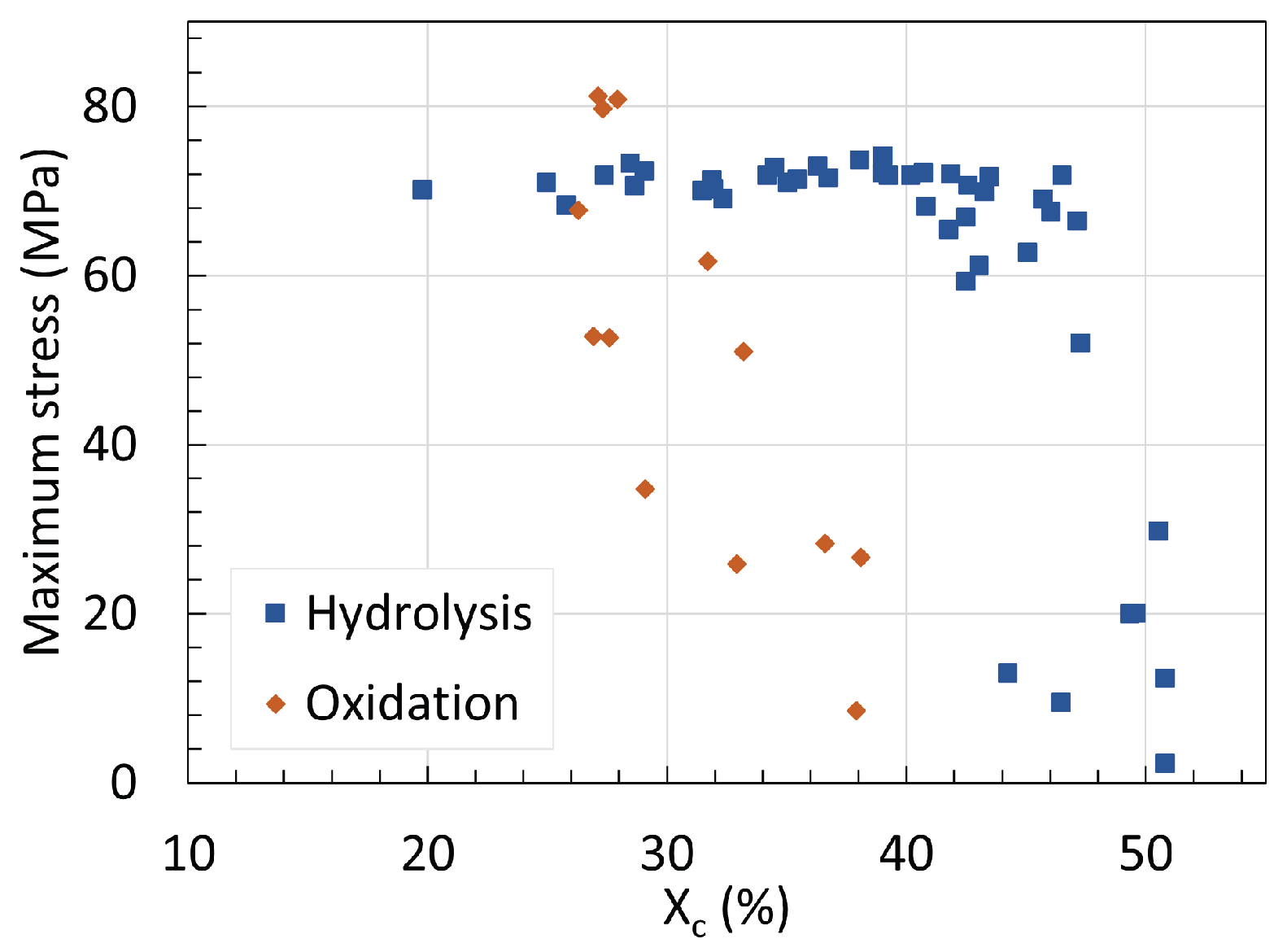
5. Mechanical Properties
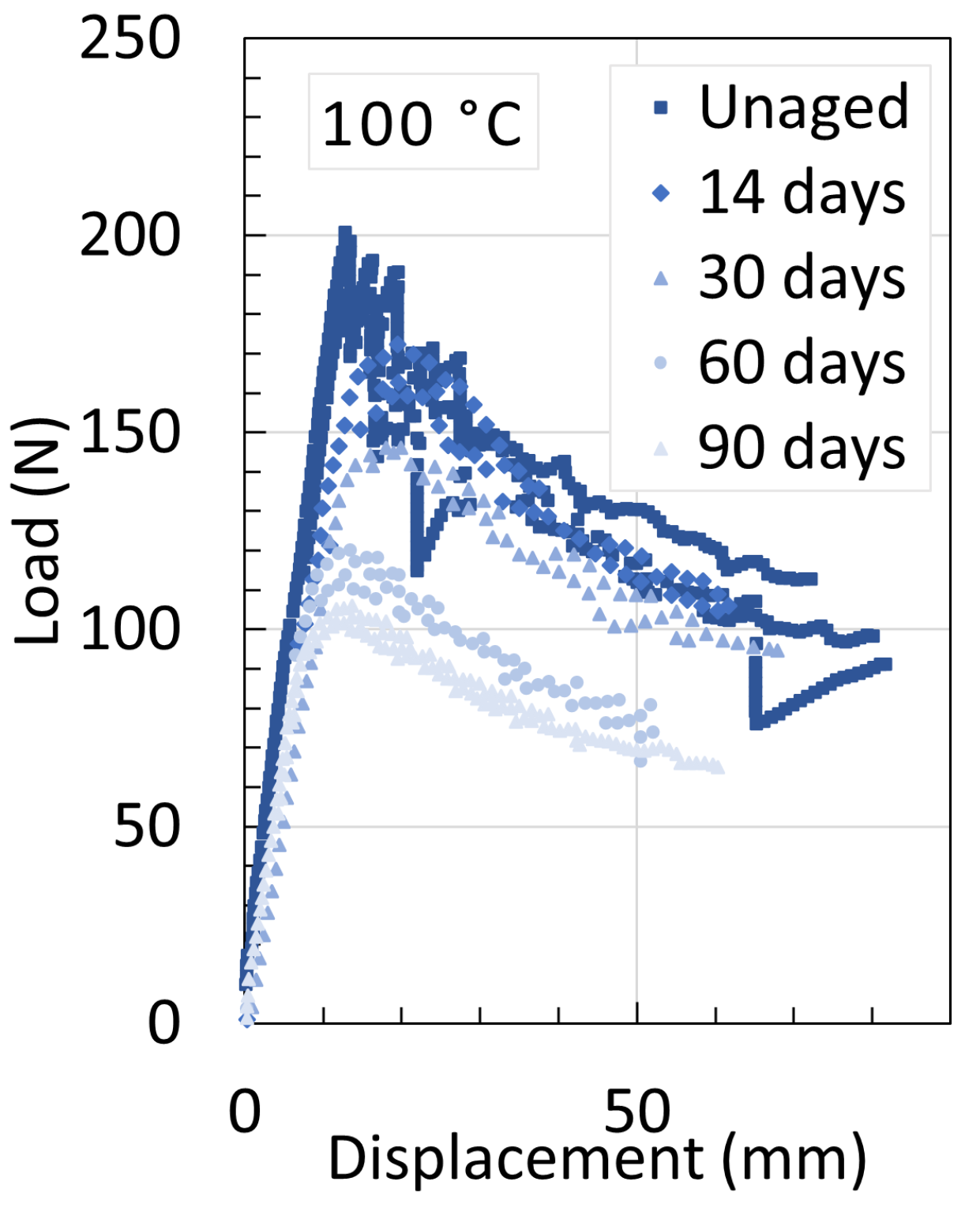
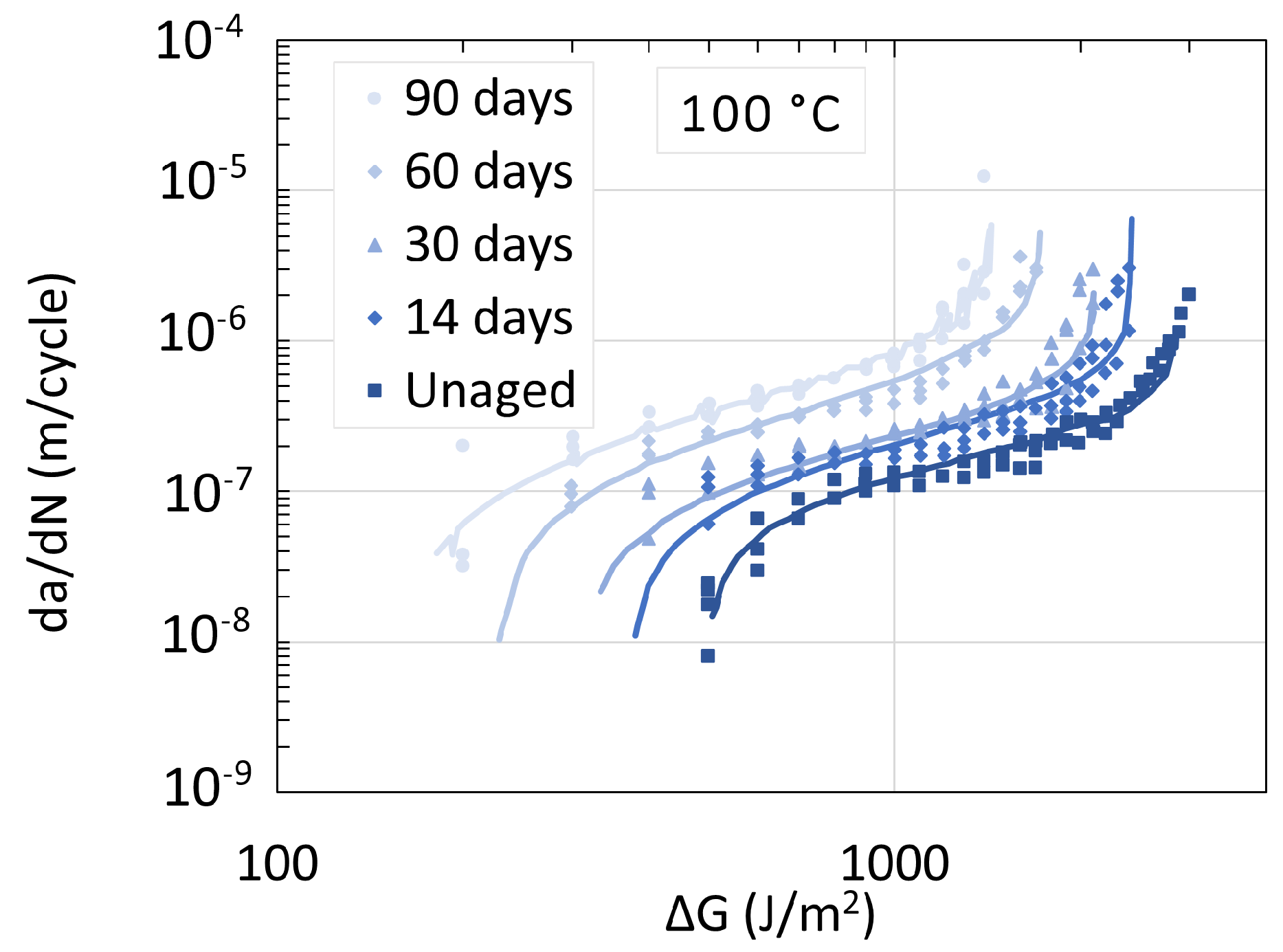
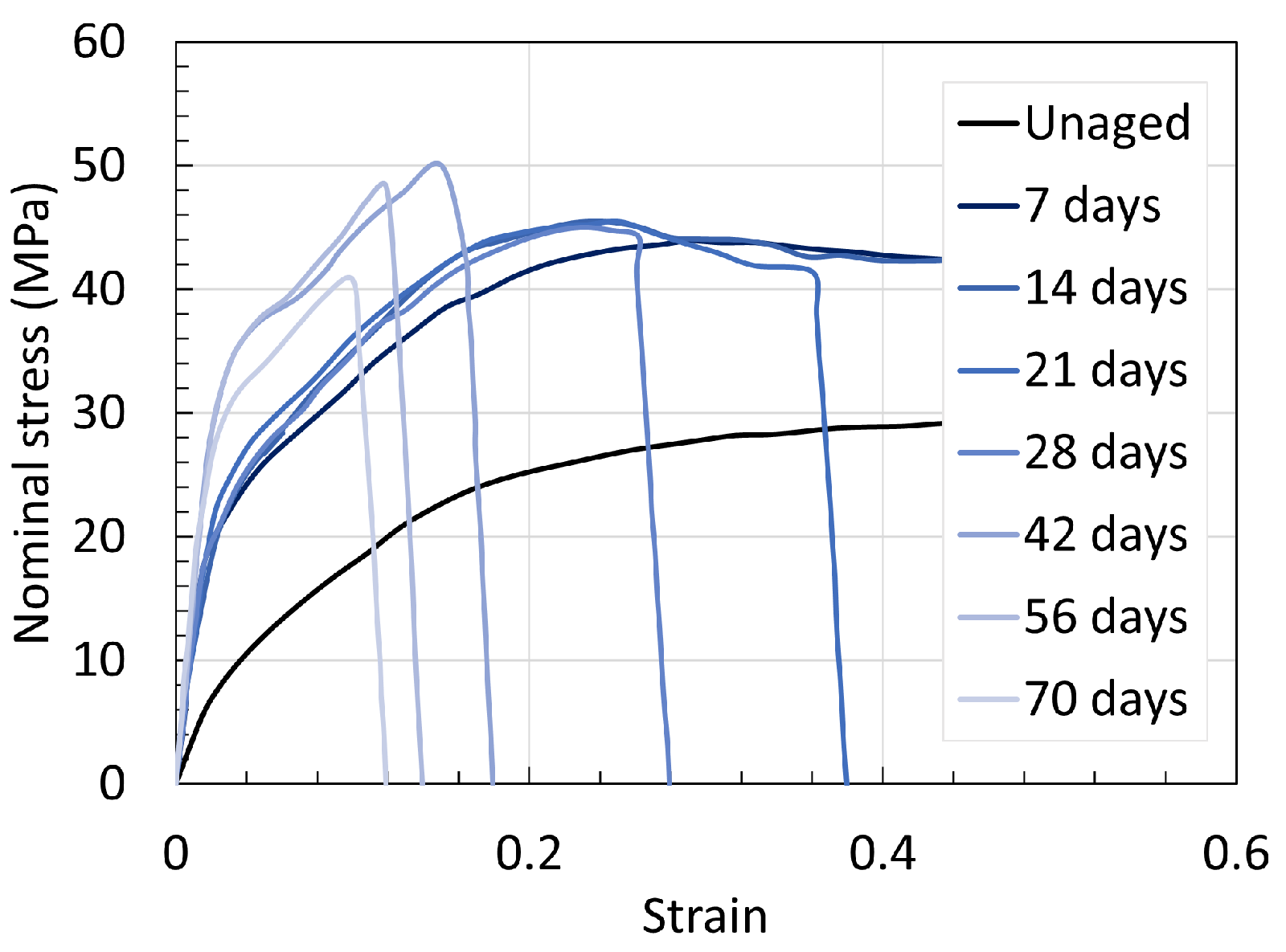
5.1. Polyamide 6
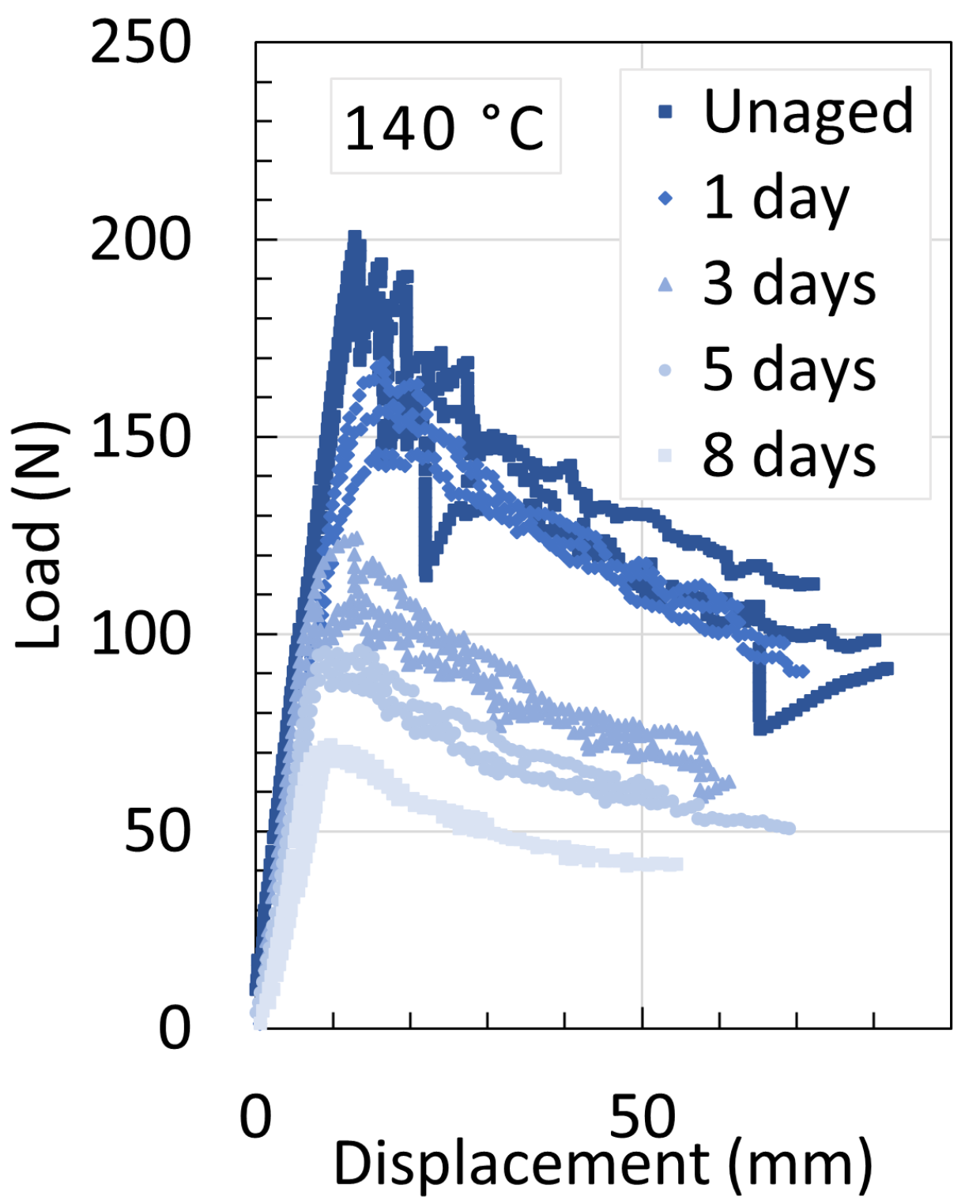
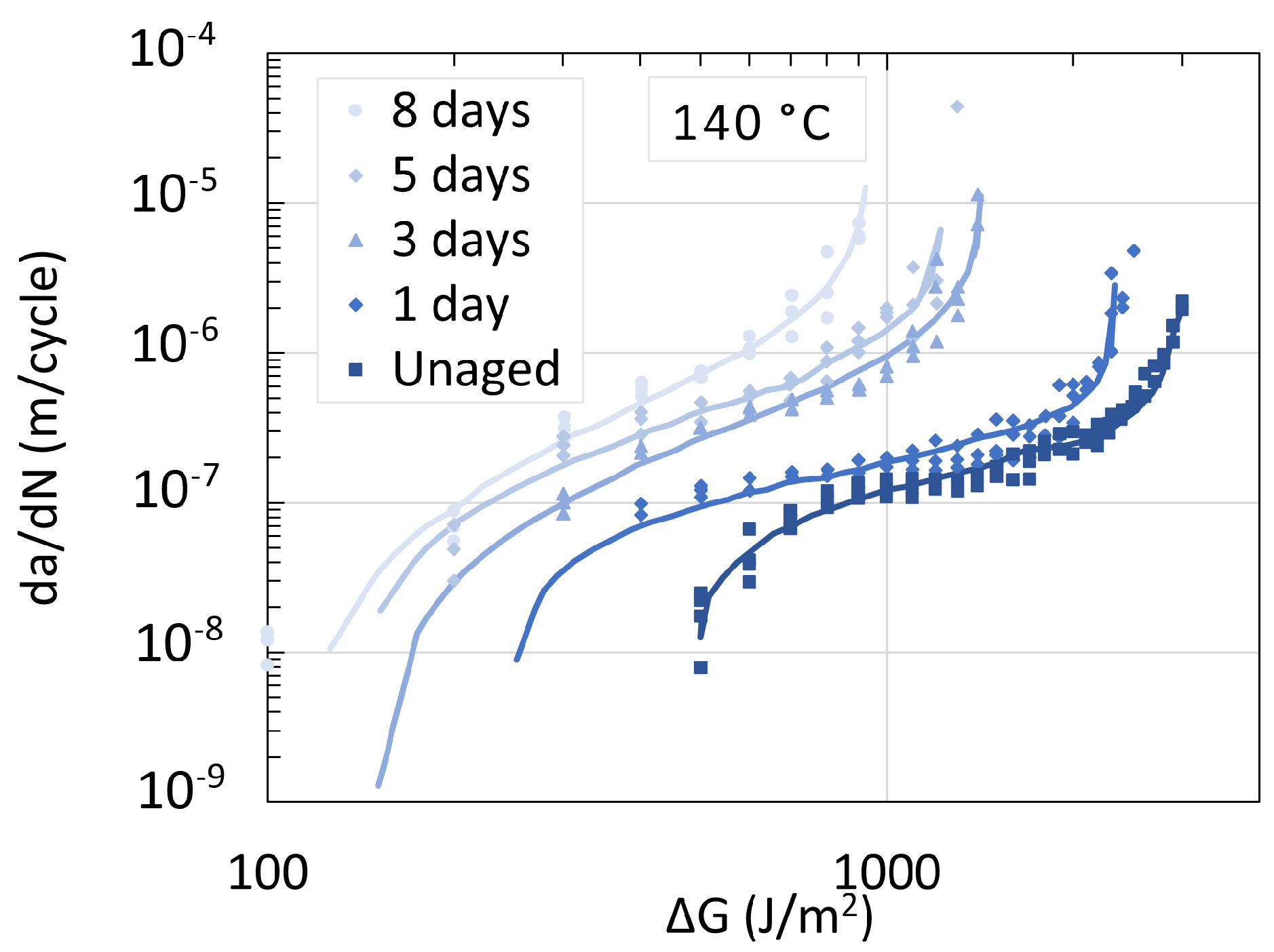
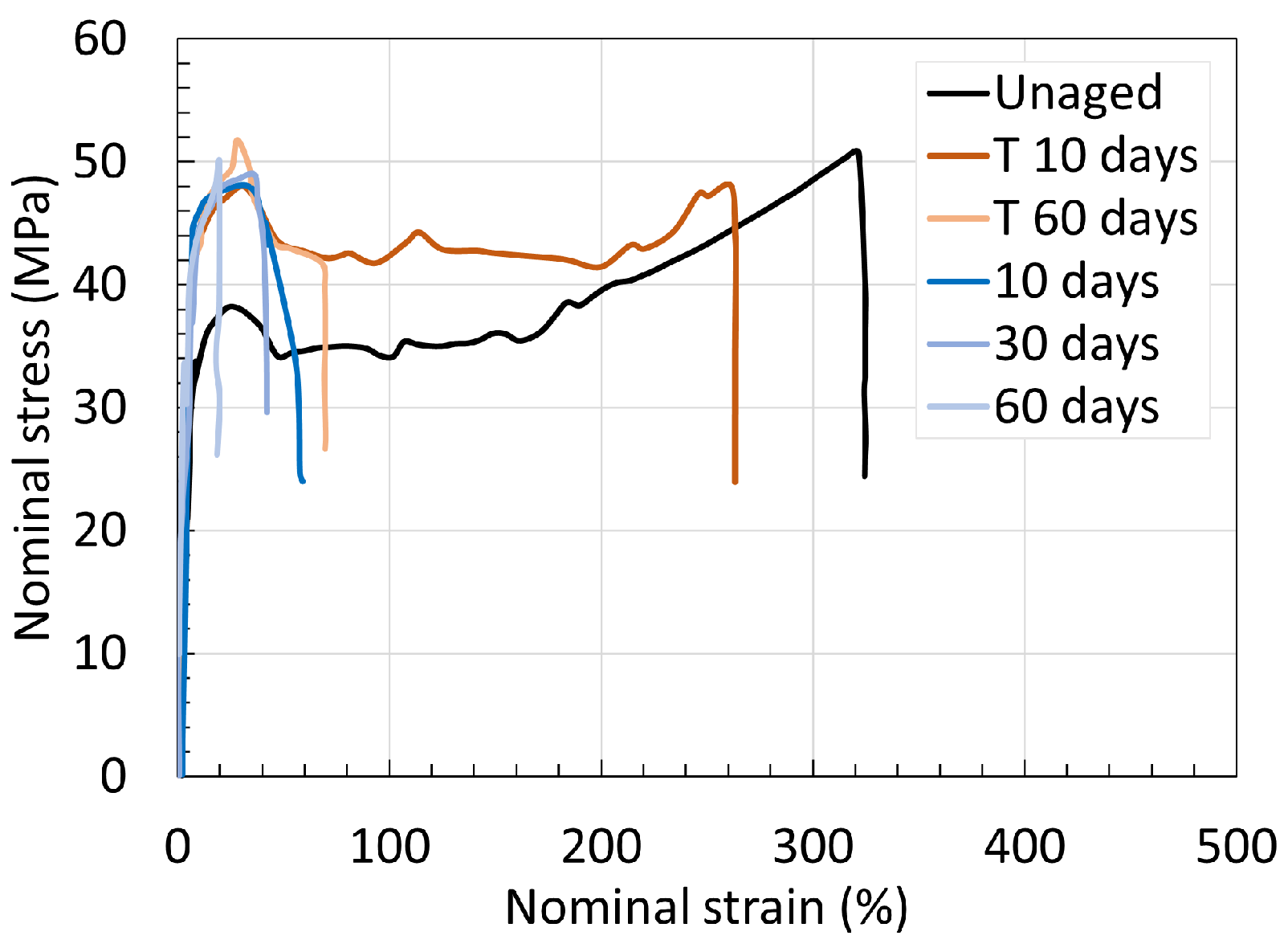
5.2. Polyamide 11
5.3. Determination of Embrittlement
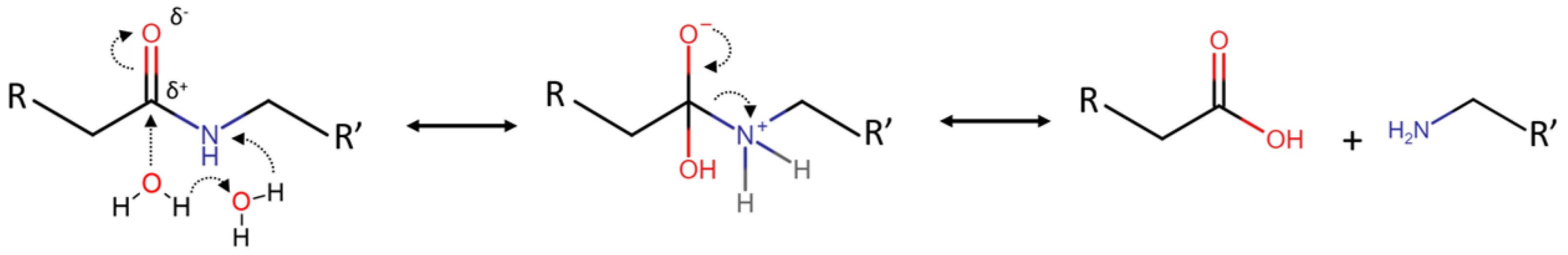
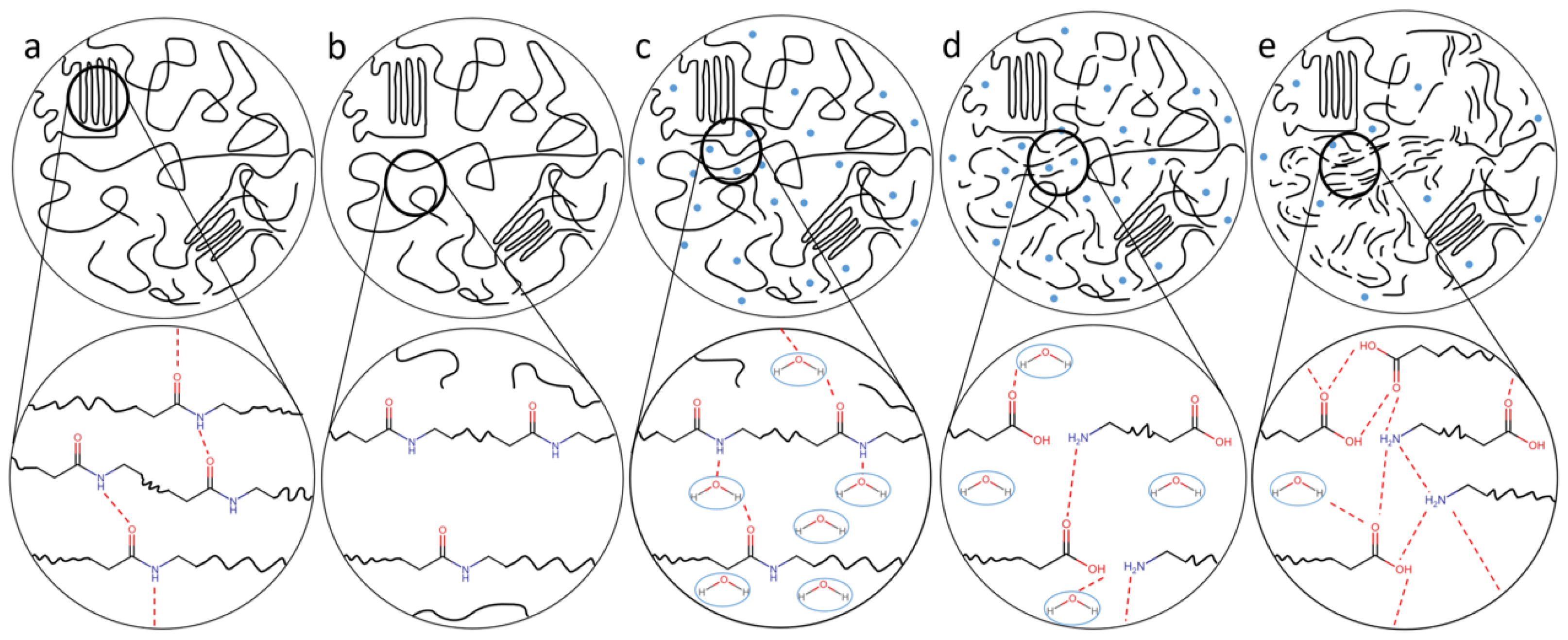
6. Mechanism Overview
7. Conclusions and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PA | Polyamide |
| GPC | Gel permeation chromatography |
| C | Carbon fiber |
| PDI | Polydispersity index |
| FTIR | Fourier-transform infrared |
| BBSA | N-butylbenzenesulfanomide |
| GO | Graphene oxide |
| DSC | Differential scanning calorimetry |
| XRD | X-ray diffraction |
| NMR | Nuclear magnetic resonance |
| SEM | Scanning electron microscopy |
| TGA | Thermogravimetric analysis |
| CIV | Correct inherent viscosity |
| WAXS | Wide-angle x-ray spectroscopy |
References
- Romão, W.; Castro, E.R.; Filho, E.A.S.; Guimarães, R.; Silva, A.L.N.; Teixeira, S.; Paoli, M.; Sena, G.L. Ageing of polyamide 11 used in the manufacture of flexible piping. J. Appl. Polym. Sci. 2009, 114, 1777–1783. [Google Scholar] [CrossRef]
- El-Mazry, C.; Correc, O.; Colin, X. A new kinetic model for predicting polyamide 6-6 hydrolysis and its mechanical embrittlement. Polym. Degrad. Stab. 2012, 97, 1049–1059. [Google Scholar] [CrossRef]
- Shakiba, M.; Rezvani Ghomi, E.; Khosravi, F.; Jouybar, S.; Bigham, A.; Zare, M.; Abdouss, M.; Moaref, R.; Ramakrishna, S. Nylon—A material introduction and overview for biomedical applications. Polym. Adv. Technol. 2021, 32, 3368–3383. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Yi, Y.; Wang, W.; Yang, J. Synthesis and Properties of Polyamide 6 Random Copolymers Containing an Aromatic Imide Structure. Polymers 2023, 15, 2812. [Google Scholar] [CrossRef] [PubMed]
- Shamey, R.; Sinha, K. A review of degradation of nylon 6. 6 as a result of exposure to environmental conditions. Color. Technol. 2003, 33, 93–107. [Google Scholar] [CrossRef]
- Varghese, M.; Grinstaff, M.W. Beyond nylon 6: Polyamides via ring opening polymerization of designer lactam monomers for biomedical applications. Chem. Soc. Rev. 2022, 51, 8258–8275. [Google Scholar] [CrossRef]
- Grigg, M.N. Thermo-Oxidative Degradation of Polyamide 6. Ph.D. Thesis, School of Physical and Chemical Sciences Queensland University of Technology, Brisbane, Australia, 2006. [Google Scholar]
- Gonçalves, E.S.; Poulsen, L.; Ogilby, P.R. Mechanism of the temperature-dependent degradation of polyamide 66 films exposed to water. Polym. Degrad. Stab. 2007, 92, 1977–1985. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. Thermal degradation of nylon polymers. Polym. Int. 2000, 49, 943–948. [Google Scholar] [CrossRef]
- Ullah, I.; Gul, T.; Ali, M.; Khan, I.; Khan, W.; Asghar, H.; Saeed, K. Preparation, Analysis and UV-Accelerated Photocatalytic Degradation of Pesticide Over Mg Doped ZnO/Nylon 6,6/PMMA Ternary Blend. J. Inorg. Organomet. Polym. Mater. 2023, 33, 3441–3453. [Google Scholar] [CrossRef]
- Gröning, M.; Hakkarainen, M. Headspace solid-phase microextraction–a tool for new insights into the long-term thermo-oxidation mechanism of polyamide 6.6. J. Chromatogr. A 2001, 932, 1–11. [Google Scholar] [CrossRef]
- Vasanthan, N.; Murthy, N.; Bray, R. Investigation of Brill transition in nylon 6 and nylon 6,6 by infrared spectroscopy. Macromolecules 1998, 31, 8433–8435. [Google Scholar] [CrossRef]
- Okamba-Diogo, O.; Richaud, E.; Verdu, J.; Fernagut, F.; Guilment, J.; Fayolle, B. Molecular and macromolecular structure changes in polyamide 11 during thermal oxidation-Kinetic modeling. Polym. Degrad. Stab. 2014, 108, 123–132. [Google Scholar] [CrossRef]
- Krejsa, M.; Udipi, K.; Middleton, J. NMR Analysis of UV- and Heat-Aged Nylon-6,6. Macromolecules 1997, 30, 4695–4703. [Google Scholar] [CrossRef]
- Sanders, B.; Cant, E.; Amel, H.; Jenkins, M. The Effect of Physical Aging and Degradation on the Re-Use of Polyamide 12 in Powder Bed Fusion. Polymers 2022, 14, 2682. [Google Scholar] [CrossRef] [PubMed]
- Moore, R. The photochemical degradation of polyamides and related model N-alkylamides. Polymer 1963, 4, 493–513. [Google Scholar] [CrossRef]
- Levchik, S.; Weil, E.; Lewin, M. Thermal decomposition of aliphatic nylons. Polym. Int. 1999, 48, 532–557. [Google Scholar] [CrossRef]
- Mazry, C.E.; Hassine, M.B.; Correc, O.; Colin, X. Thermal oxidation kinetics of additive free polyamide 6-6. Polym. Degrad. Stab. 2013, 98, 22–36. [Google Scholar] [CrossRef]
- Forsström, D.; Terselius, B. Thermo oxidative stability of polyamide 6 films. I. Mechanical and chemical characterisation. Polym. Degrad. Stab. 2000, 67, 69–78. [Google Scholar] [CrossRef]
- Thanki, P.N.; Singh, R.P. Progress in the Area of Degradation and Stabilization of Nylon 66. J. Macromol. Sci.-Rev. in Macromol. Chem. Phys. 1998, 38, 595–614. [Google Scholar] [CrossRef]
- Venoor, V.; Park, J.H.; Kazmer, D.; Sobkowicz, M.J. Understanding the Effect of Water in Polyamides: A Review. Polym. Rev. 2021, 61, 598–645. [Google Scholar] [CrossRef]
- Lottier, S.; Tencé-Girault, S.; Gervat, L.; Saintier, N.; Miquelard-Garnier, G.; Fayolle, B. Effect of chemical ageing on fatigue life of short glass fiber-reinforced Polyamide 6,6. Polym. Degrad. Stab. 2023, 208, 110244. [Google Scholar] [CrossRef]
- He, X.; Xiao, B.; Cai, G.; Takahashi, J. Morphological, physicochemical, and flexural characterization of carbon fiber paper–reinforced polyamide 6 for long-term application in aqueous environments. J. Polym. Res. 2021, 28, 362. [Google Scholar] [CrossRef]
- Bäckström, E.; Odelius, K.; Hakkarainen, M. Microwave Assisted Selective Hydrolysis of Polyamides from Multicomponent Carpet Waste. Glob. Challenges 2021, 5, 2000119. [Google Scholar] [CrossRef]
- Harrass, K.; Mauer, S.; Tsekov, R. Resistance of glass fiber reinforced polyamide 6.6 materials to automotive cooling fluids: An analytical method for lifetime prediction. Polym. Int. 2021, 71, 724–733. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, C.; Zhao, H.; Hu, W.; Liu, G.; Zhao, Y.; Dong, X.; Wang, K.; Zhang, J.; Li, X.; et al. Effect of nanoparticle and glass fiber on the hydrothermal aging of polyamide 6. J. Appl. Polym. Sci. 2020, 137, 49585. [Google Scholar] [CrossRef]
- Češarek, U.; Pahovnik, D.; Žagar, E. Chemical Recycling of Aliphatic Polyamides by Microwave-Assisted Hydrolysis for Efficient Monomer Recovery. ACS Sustain. Chem. Eng. 2020, 8, 16274–16282. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, K.J. MEG Effects on Hydrolysis of Polyamide 66/Glass Fiber Composites and Mechanical Property Changes. Molecules 2019, 24, 755. [Google Scholar] [CrossRef]
- Sang, L.; Wang, C.; Wang, Y.; Hou, W. Effects of hydrothermal aging on moisture absorption and property prediction of short carbon fiber reinforced polyamide 6 composites. Compos. Part B Eng. 2018, 153, 306–314. [Google Scholar] [CrossRef]
- Li, R.; Ye, L.; Li, G. Long-Term Hydrothermal Aging Behavior and Aging Mechanism of Glass Fibre Reinforced Polyamide 6 Composites. J. Macromol. Sci. Part B 2018, 57, 67–82. [Google Scholar] [CrossRef]
- Ksouri, I.; Haddar, N. Long term ageing of polyamide 6 and polyamide 6 reinforced with 30% of glass fibers: Temperature effect. J. Polym. Res. 2018, 25, 153. [Google Scholar] [CrossRef]
- Geretschläger, K.J.; Wallner, G. Aging characteristics of glass fiber-reinforced polyamide in hot water and air. Polym. Compos. 2018, 39, 997–1005. [Google Scholar] [CrossRef]
- Wang, W.; Meng, L.; Leng, K.; Huang, Y. Hydrolysis of waste monomer casting nylon catalyzed by solid acids. Polym. Degrad. Stab. 2017, 136, 112–120. [Google Scholar] [CrossRef]
- Ksouri, I.; De Almeida, O.; Haddar, N. Long term ageing of polyamide 6 and polyamide 6 reinforced with 30% of glass fibers: Physicochemical, mechanical and morphological characterization. J. Polym. Res. 2017, 24, 133. [Google Scholar] [CrossRef]
- Arash, B.; Thijsse, B.J.; Pecenko, A.; Simone, A. Effect of water content on the thermal degradation of amorphous polyamide 6,6: A collective variable-driven hyperdynamics study. Polym. Degrad. Stab. 2017, 146, 260–266. [Google Scholar] [CrossRef]
- Gao, L.; Ye, L.; Li, G. Long-Term Hydrothermal Aging Behavior and Life-Time Prediction of Polyamide 6. J. Macromol. Sci. Part B 2015, 54, 239–252. [Google Scholar] [CrossRef]
- Cruz, C.; Belmonte, E.; Lux, A.; Monte, M.D.; Quaresimin, M. Multi-scale analysis of the ageing of a reinforced polyamide 66 in ethanol-based fuels. In Proceedings of the 20th International Conference on Composite Materials, Copenhagen, Denmark, 19–24 July 2015. [Google Scholar]
- Wang, W.; Meng, L.; Huang, Y. Hydrolytic degradation of monomer casting nylon in subcritical water. Polym. Degrad. Stab. 2014, 110, 312–317. [Google Scholar] [CrossRef]
- Haddar, N.; Ksouri, I.; Kallel, T.; Mnif, N. Effect of hygrothermal ageing on the monotonic and cyclic loading of glass fiber reinforced polyamide. Polym. Compos. 2014, 35, 501–508. [Google Scholar] [CrossRef]
- Launay, A.; Marco, Y.; Maitournam, H.; Raoult, I. Modelling the influence of temperature and relative humidity on the time-dependent mechanical behaviour of a short glass fibre reinforced polyamide. Mech. Mater. 2013, 56, 1–10. [Google Scholar] [CrossRef]
- Thomason, J.; Ali, J.; Anderson, J. The thermo-mechanical performance of glass-fibre reinforced polyamide 66 during glycol–water hydrolysis conditioning. Compos. Part Appl. Sci. Manuf. 2010, 41, 820–826. [Google Scholar] [CrossRef]
- Chen, J.; Liu, G.; Jin, L.; Ni, P.; Li, Z.; He, H.; Xu, Y.; Zhang, J.; Dong, J. Catalytic hydrolysis of waste nylon 6 to produce ϵ-caprolactam in sub-critical water. J. Anal. Appl. Pyrolysis 2010, 87, 50–55. [Google Scholar] [CrossRef]
- Bernstein, R.; Gillen, K.T. Nylon 6.6 accelerating aging studies: II. Long-term thermal-oxidative and hydrolysis results. Polym. Degrad. Stab. 2010, 95, 1471–1479. [Google Scholar] [CrossRef]
- Cribbs, D.; Ogale, A. Hydrolytic Degradation of Nylon 66 Pile Carpet Fibers. Text. Res. J. 2003, 95, 1471–1479. [Google Scholar] [CrossRef]
- Ishak, Z.A.M.; Berry, J.P. Hygrothermal aging studies of short carbon fiber reinforced nylon 6.6. J. Appl. Polym. Sci. 1994, 51, 2145–2155. [Google Scholar] [CrossRef]
- Hunt, D.G.; Darlington, M.W. Creep of nylon-6,6 during concurrent moisture changes. Polymer 1980, 21, 502–508. [Google Scholar] [CrossRef]
- Gac, P.Y.L.; Fayolle, B. Impact of fillers (short glass fibers and rubber) on the hydrolysis-induced embrittlement of polyamide 6.6. Compos. Part B-Eng. 2018, 153, 256–263. [Google Scholar] [CrossRef]
- Deshoulles, Q.; Le Gall, M.; Dreanno, C.; Arhant, M.; Priour, D.; Le Gac, P. Chemical coupling between oxidation and hydrolysis in polyamide 6-A key aspect in the understanding of microplastic formation. Polym. Degrad. Stab. 2022, 197, 109851. [Google Scholar] [CrossRef]
- Darzi, R.; Dubowski, Y.; Posmanik, R. Hydrothermal processing of polyethylene-terephthalate and nylon-6 mixture as a plastic waste upcycling treatment: A comprehensive multi-phase analysis. Waste Manag. 2022, 143, 223–231. [Google Scholar] [CrossRef]
- Da Cruz, B.d.S.M.; Tienne, L.G.P.; Gondim, F.F.; Candido, L.d.S.; Chaves, E.G.; Marques, M.d.F.V.; da Luz, F.S.; Monteiro, S.N. Graphene nanoplatelets reinforced Polyamide-11 nanocomposites thermal stability and aging for application in flexible pipelines. J. Mater. Res. Technol. 2022, 18, 1842–1854. [Google Scholar] [CrossRef]
- Costa, D.R.; Costa, M.F.; Grytten, F. Morphological changes of polyamide 11 through the corrected inherent viscosity plateau. J. Appl. Polym. Sci. 2022, 139, 52223. [Google Scholar] [CrossRef]
- Arhant, M.; Lolive, E.; Bonnemains, T.; Davies, P. A study of pure hydrolysis of carbon fibre reinforced polyamide 6 composites tested under mode I loading. Compos. Part A Appl. Sci. Manuf. 2021, 152, 106719. [Google Scholar] [CrossRef]
- Arhant, M.; Lolive, E.; Bonnemains, T.; Davies, P. Effect of aging on the fatigue crack growth properties of carbon-polyamide 6 thermoplastic composites using the multi ΔG-control method. Compos. Part A Appl. Sci. Manuf. 2022, 161, 107105. [Google Scholar] [CrossRef]
- Olufsen, S.N.; Nygård, P.; Teixeira Pais, C.I.; Perillo, G.; Hopperstad, O.S.; Clausen, A.H. Influence of loading conditions on the tensile response of degraded polyamide 11. Polymer 2021, 229, 123966. [Google Scholar] [CrossRef]
- Deshoulles, Q.; Gall, M.M.L.; Dreanno, C.; Arhant, M.; Stoclet, G.; Priour, D.; Gac, P.L. Origin of embrittlement in Polyamide 6 induced by chemical degradations: Mechanisms and governing factors. Polym. Degrad. Stab. 2021, 191, 109657. [Google Scholar] [CrossRef]
- Deshoulles, Q.; Gall, M.M.L.; Dreanno, C.; Arhant, M.; Priour, D.; Gac, P.Y.L. Modelling pure polyamide 6 hydrolysis: Influence of water content in the amorphous phase. Polym. Degrad. Stab. 2021, 183, 109435. [Google Scholar] [CrossRef]
- Maïza, S.; Lefebvre, X.; Brusselle-Dupend, N.; Klopffer, M.H.; Cangémi, L.; Castagnet, S.; Grandidier, J.C. Physicochemical and mechanical degradation of polyamide 11 induced by hydrolysis and thermal aging. J. Appl. Polym. Sci. 2019, 136, 47628. [Google Scholar] [CrossRef]
- Hocker, S.J.; Kim, W.T.; Schniepp, H.C.; Kranbuehl, D.E. Polymer crystallinity and the ductile to brittle transition. Polymer 2018, 158, 72–76. [Google Scholar] [CrossRef]
- Hocker, S.J.; Hudson-Smith, N.V.; Smith, P.T.; Komatsu, C.H.; Dickinson, L.R.; Schniepp, H.C.; Kranbuehl, D.E. Graphene oxide reduces the hydrolytic degradation in polyamide-11. Polymer 2017, 126, 248–258. [Google Scholar] [CrossRef]
- Mazan, T.; Berggren, R.; Jørgensen, J.K.; Echtermeyer, A. Aging of polyamide 11. Part 1: Evaluating degradation by thermal, mechanical, and viscometric analysis. J. Appl. Polym. Sci. 2015, 132, 41971. [Google Scholar] [CrossRef]
- Hocker, S.; Rhudy, A.K.; Ginsburg, G.; Kranbuehl, D.E. Polyamide hydrolysis accelerated by small weak organic acids. Polymer 2014, 55, 5057–5064. [Google Scholar] [CrossRef]
- Domingos, E.; Pereira, T.; Filgueiras, P.; Bueno, M.; Castro, E.R.; Guimarães, R.; Sena, G.L.; Rocha, W.F.; Romão, W. Monitoring the polyamide 11 degradation by thermal properties and X-ray fluorescence spectrometry allied to chemometric methods. X-ray Spectrom. 2013, 42, 79–86. [Google Scholar] [CrossRef]
- Bergeret, A.; Ferry, L.; Ienny, P. Influence of the fibre/matrix interface on ageing mechanisms of glass fibre reinforced thermoplastic composites (PA-6,6, PET, PBT) in a hygrothermal environment. Polym. Degrad. Stab. 2009, 94, 1315–1324. [Google Scholar] [CrossRef]
- Iwaya, T.; Sasaki, M.; Goto, M. Kinetic analysis for hydrothermal depolymerization of nylon 6. Polym. Degrad. Stab. 2006, 91, 1989–1995. [Google Scholar] [CrossRef]
- Bernstein, R.; Derzon, D.K.; Gillen, K.T. Nylon 6.6 accelerated aging studies: Thermal–oxidative degradation and its interaction with hydrolysis. Polym. Degrad. Stab. 2005, 88, 480–488. [Google Scholar] [CrossRef]
- Merdas, I.; Thominette, F.; Verdu, J. Hydrolytic ageing of polyamide 11—effect of carbon dioxide on polyamide 11 hydrolysis. Polym. Degrad. Stab. 2003, 79, 419–425. [Google Scholar] [CrossRef]
- Alam, T.M. Solution 17O NMR study of thermal hydrolysis in nylon 6,6. Polymer 2003, 44, 6531–6536. [Google Scholar] [CrossRef]
- Meyer, A.; Jones, N.; Lin, Y.; Kranbuehl, D.E. Characterizing and Modeling the Hydrolysis of Polyamide-11 in a pH 7 Water Environment. Macromolecules 2002, 44, 6531–6536. [Google Scholar] [CrossRef]
- Jacques, B.; Werth, M.; Merdas, I.; Thominette, F.; Verdu, J. Hydrolytic ageing of polyamide 11. 1. Hydrolysis kinetics in water. Polymer 2002, 43, 6439–6447. [Google Scholar] [CrossRef]
- Esmizadeh, E.; Vahidifar, A.; Shojaie, S.; Naderi, G.; Kalaei, M.R.; Mekonnen, T.H. Tailoring the properties of PA6 into high-performance thermoplastic elastomer: Simultaneous reinforcement and impact property modification. Mater. Today Commun. 2021, 26, 102027. [Google Scholar] [CrossRef]
- Latko-Durałek, P.; Kolbuk, D.; Kozera, R.; Boczkowska, A. Microstructural Characterization and Mechanical Properties of PA11 Nanocomposite Fibers. J. Mater. Eng. Perform. 2015, 25, 68–75. [Google Scholar] [CrossRef]
- Chaupart, N.; Serpe, G.; Verdu, J. Molecular weight distribution and mass changes during polyamide hydrolysis. Polymer 1998, 39, 1375–1380. [Google Scholar] [CrossRef]
- Abastari; Sakai, T.; Sembokuya, H.; Kubouchi, M.; Tsuda, K. Study on permeation behavior and chemical degradation of PA66 in acid solution. Polym. Degrad. Stab. 2007, 39, 1375–1380. [Google Scholar] [CrossRef]
- Liang, J.; Xu, Y.; Wei, Z.; Song, P.; Chen, G.; Zhang, W. Mechanical properties, crystallization and melting behaviors of carbon fiber-reinforced PA6 composites. J. Therm. Anal. Calorim. 2014, 115, 209–218. [Google Scholar] [CrossRef]
- Gianchandani, J.; Spruiell, J.E.; Clark, E.S. Polymorphism and orientation development in melt spinning, drawing, and annealing of nylon-6 filaments. J. Appl. Polym. Sci. 1982, 27, 3527–3551. [Google Scholar] [CrossRef]
- Li, L.; Li, C.Y.; Ni, C.; Rong, L.; Hsiao, B. Structure and crystallization behavior of Nylon 66/multi-walled carbon nanotube nanocomposites at low carbon nanotube contents. Polymer 2007, 48, 3452–3460. [Google Scholar] [CrossRef]
- Gurato, G.; Fichera, A.; Grandi, F.Z.; Zannetti, R.; Canal, P. Crystallinity and polymorphism of 6-polyamide. Die Makromol. Chem. 1974, 175, 953–975. [Google Scholar] [CrossRef]
- Inoue, M. Studies on crystallization of high polymers by differential thermal analysis. J. Polym. Sci. Part A Gen. Pap. 1963, 1, 2697–2709. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Frone, A.N.; Nicolae, C. Micro- and nano-mechanical characterization of polyamide 11 and its composites containing cellulose nanofibers. Eur. Polym. J. 2013, 49, 3857–3866. [Google Scholar] [CrossRef]
- Roguet, E.; Tencé-Girault, S.; Castagnet, S.; Grandidier, J.; Hochstetter, G. Micromechanisms involved in the atypical tensile behavior observed in polyamide 11 at high temperature. J. Polym. Sci. Part Polym. Phys. 2007, 45, 3046–3059. [Google Scholar] [CrossRef]
- Pepin, J.; Miri, V.; Lefebvre, J.M. New Insights into the Brill Transition in Polyamide 11 and Polyamide 6. Macromolecules 2016, 49, 564–573. [Google Scholar] [CrossRef]
- Zhang, Q.; Mo, Z.; Liu, S.; Zhang, H. Influence of Annealing on Structure of Nylon 11. Macromolecules 2000, 33, 5999–6005. [Google Scholar] [CrossRef]
- Ram, F.; Radhakrishnan, S.; Ambone, T.; Shanmuganathan, K. Highly Flexible Mechanical Energy Harvester Based on Nylon 11 Ferroelectric Nanocomposites. ACS Appl. Polym. Mater. 2019, 1, 1998–2005. [Google Scholar] [CrossRef]
- Dhanalakshmi, M.; Jog, J.P. Preparation and characterization of electrospun fibers of Nylon 11. Express Polym. Lett. 2008, 2, 540–545. [Google Scholar] [CrossRef]
- Fernandez, J.O.; Swallowe, G.M.; Lee, S.F. Crystallization of Nylon 11 under compressive high strain rates. J. Appl. Polym. Sci. 2001, 80, 2031–2038. [Google Scholar] [CrossRef]
- Benobeidallah, B.; Benhamida, A.; Dorigato, A.; Sola, A.; Messori, M.; Pegoretti, A. Structure and Properties of Polyamide 11 Nanocomposites Filled with Fibrous Palygorskite Clay. J. Renew. Mater. 2019, 7, 89–102. [Google Scholar] [CrossRef]
- Mago, G.; Kalyon, D.M.; Fisher, F.T. Nanocomposites of polyamide-11 and carbon nanostructures: Development of microstructure and ultimate properties following solution processing. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1311–1321. [Google Scholar] [CrossRef]
- Romão, W.; Franco, M.F.; Corilo, Y.E.; Eberlin, M.N.; Spinacé, M.A.; De Paoli, M.A. Poly (ethylene terephthalate) thermo-mechanical and thermo-oxidative degradation mechanisms. Polym. Degrad. Stab. 2009, 94, 1849–1859. [Google Scholar] [CrossRef]
- Romão, W.; Franco, M.F.; Bueno, M.I.M.; De Paoli, M.A. Distinguishing between virgin and post-consumption bottle-grade poly (ethylene terephthalate) using thermal properties. Polym. Test. 2010, 29, 879–885. [Google Scholar] [CrossRef]
- Wypych, G. 3-Mechanisms of UV Stabilization. In Handbook of UV Degradation and Stabilization, 2nd ed.; ChemTec Publishing: Toronto, ON, Canada, 2015; pp. 37–65. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Skrovanek, D.J.; Painter, P.C.; Coleman, M.M. Hydrogen bonding in polymers. 2. Infrared temperature studies of nylon 11. Macromolecules 1986, 19, 699–705. [Google Scholar] [CrossRef]
- Mazan, T.; Jørgensen, J.K.; Echtermeyer, A. Aging of polyamide 11. Part 2: General multiscale model of the hydrolytic degradation applied to predict the morphology evolution. J. Appl. Polym. Sci. 2015, 132, 42630. [Google Scholar] [CrossRef]
- Mazan, T.; Jørgensen, J.K.; Echtermeyer, A. Aging of polyamide 11. Part 3: Multiscale model predicting the mechanical properties after hydrolytic degradation. J. Appl. Polym. Sci. 2015, 132, 42792. [Google Scholar] [CrossRef]
- Andrady, A.L. The plastic in microplastics: A review. Mar. Pollut. Bull. 2017, 119, 12–22. [Google Scholar] [CrossRef]
- Greco, R.; Ragosta, G. Isotactic polypropylenes of different molecular characteristics: Influence of crystallization conditions and annealing on the fracture behaviour. J. Mater. Sci. 1988, 23, 4171–4180. [Google Scholar] [CrossRef]
- Bessell, T.J.; Hull, D.; Shortall, J.B. The effect of polymerization conditions and crystallinity on the mechanical properties and fracture of spherulitic nylon 6. J. Mater. Sci. 1975, 10, 1127–1136. [Google Scholar] [CrossRef]
- Fornes, T.; Yoon, P.; Keskkula, H.; Paul, D. Nylon 6 nanocomposites: The effect of matrix molecular weight. Polymer 2001, 42, 09929–09940. [Google Scholar] [CrossRef]
- Steeman, P.; Nijenhuis, A. The effect of random branching on the balance between flow and mechanical properties of polyamide-6. Polymer 2010, 51, 2700–2707. [Google Scholar] [CrossRef]
- Millot, C.; Séguéla, R.; Lame, O.; Fillot, L.A.; Rochas, C.; Sotta, P. Tensile Deformation of Bulk Polyamide 6 in the Preyield Strain Range. Micro–Macro Strain Relationships via in Situ SAXS and WAXS. Macromolecules 2017, 50, 1541–1553. [Google Scholar] [CrossRef]
- Huang, Y.L.; Brown, N. Dependence of slow crack growth in polyethylene on butyl branch density: Morphology and theory. J. Polym. Sci. Part B Polym. Phys. 1991, 29, 129–137. [Google Scholar] [CrossRef]
- Seguela, R. Critical review of the molecular topology of semicrystalline polymers: The origin and assessment of intercrystalline tie molecules and chain entanglements. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 1729–1748. [Google Scholar] [CrossRef]
- Arhant, M.; Gall, M.L.; Gac, P.Y.L.; Davies, P. Impact of hydrolytic degradation on mechanical properties of PET-Towards an understanding of microplastics formation. Polym. Degrad. Stab. 2019, 161, 175–182. [Google Scholar] [CrossRef]



















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | Strain Rate | Notch Radius | CIV | Plasticizer | |
|---|---|---|---|---|---|
| High E | Low | High | - | - | Low |
| High | - | High | High | Low | Low |
| Low H | - | High | High | - | - |
| Low | - | - | - | Low | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brette, M.M.; Holm, A.H.; Drozdov, A.D.; Christiansen, J.d.C. Pure Hydrolysis of Polyamides: A Comparative Study. Chemistry 2024, 6, 13-50. https://doi.org/10.3390/chemistry6010002
Brette MM, Holm AH, Drozdov AD, Christiansen JdC. Pure Hydrolysis of Polyamides: A Comparative Study. Chemistry. 2024; 6(1):13-50. https://doi.org/10.3390/chemistry6010002
Chicago/Turabian StyleBrette, Mathis Mortensen, Allan Hjarbæk Holm, Aleksey D. Drozdov, and Jesper de Claville Christiansen. 2024. "Pure Hydrolysis of Polyamides: A Comparative Study" Chemistry 6, no. 1: 13-50. https://doi.org/10.3390/chemistry6010002