
Recent Advances in C–H Functionalization of Pyrenes

Abstract
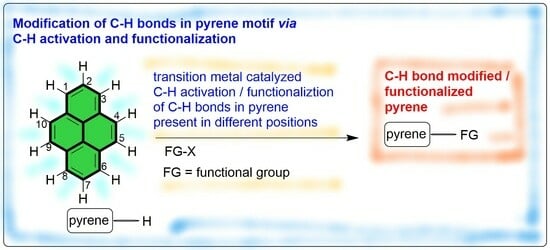
:
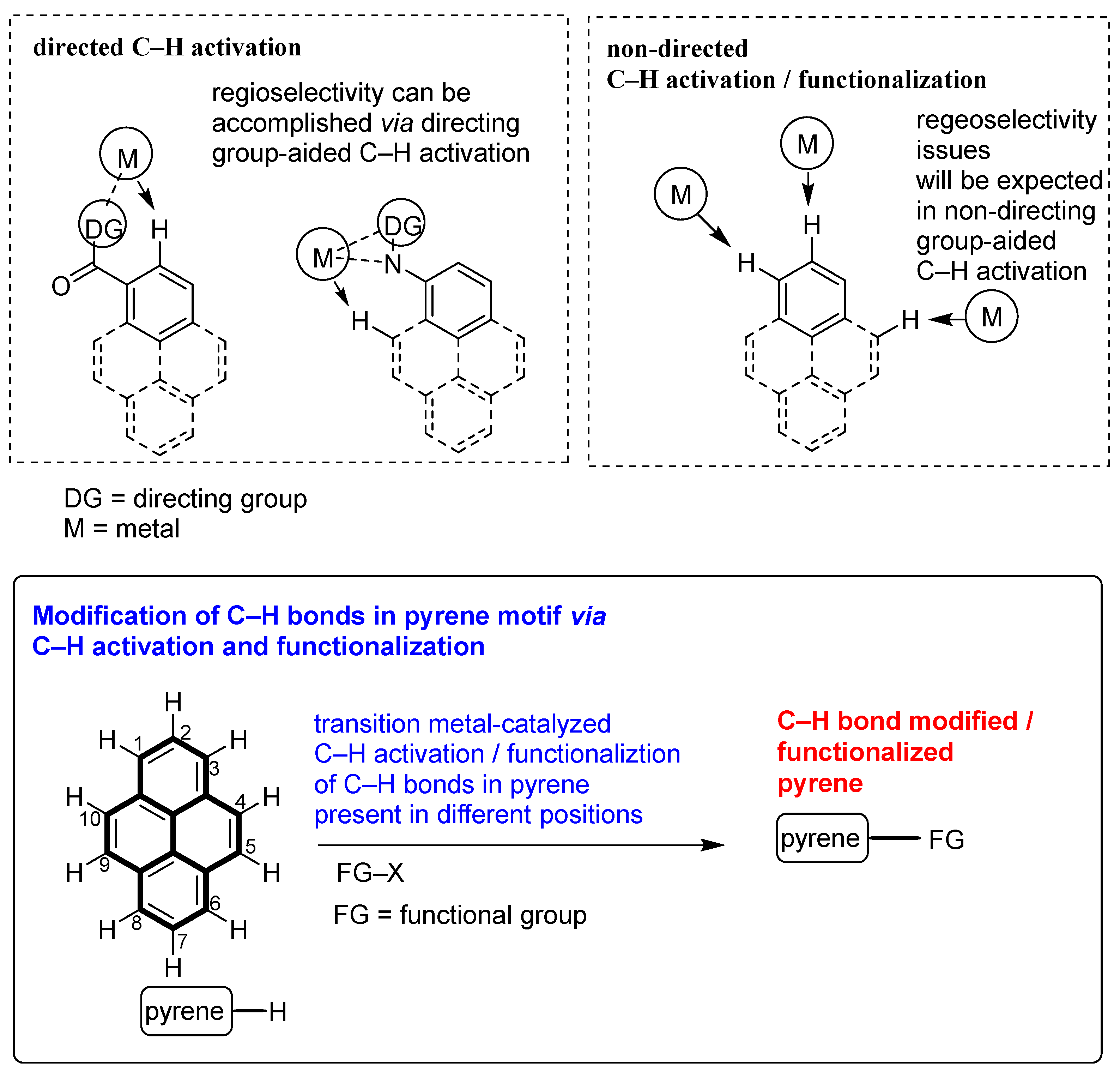
1. Introduction
2. Direct C–H Activation and Functionalization of Pyrene, Affording Functionalized Pyrenes
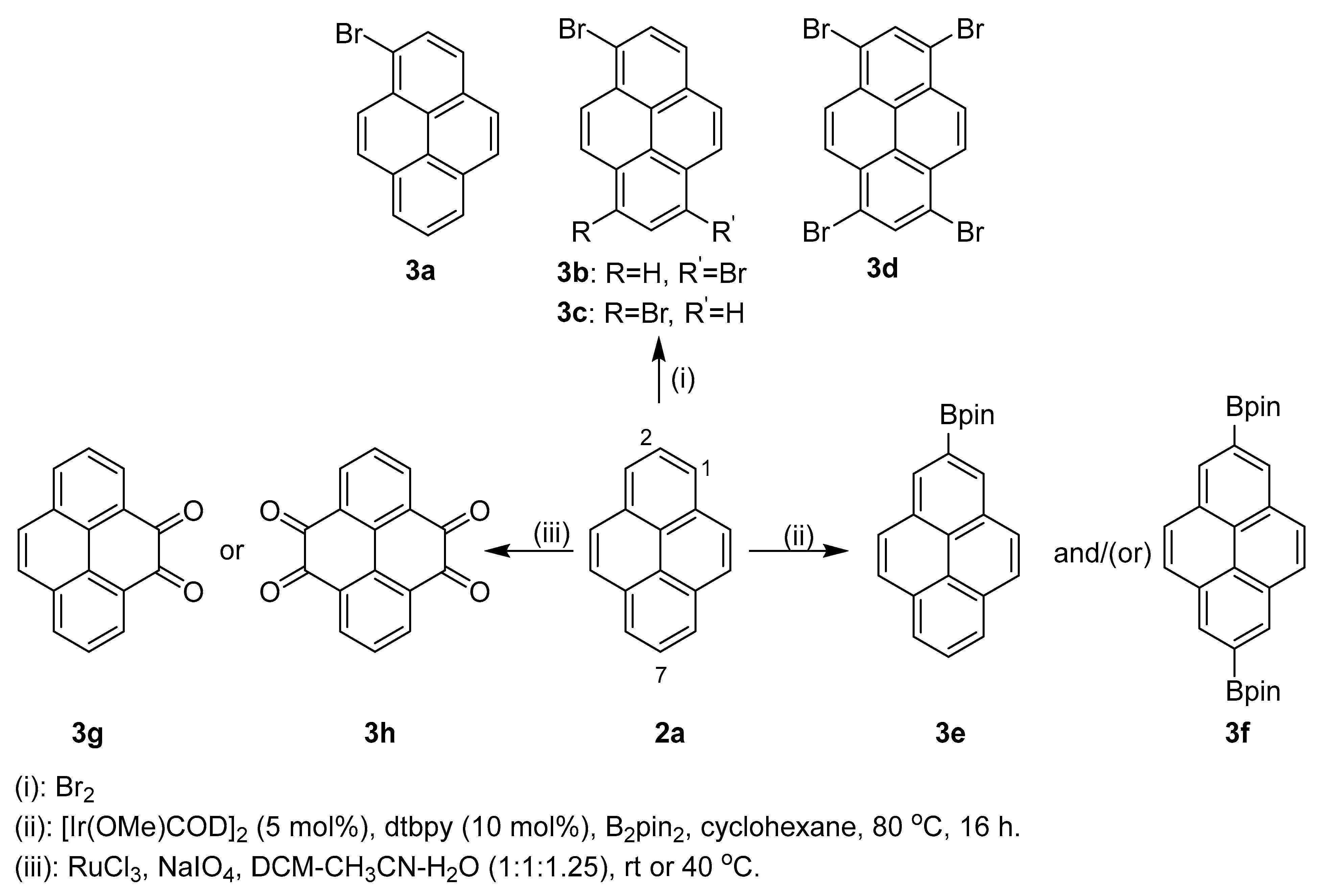
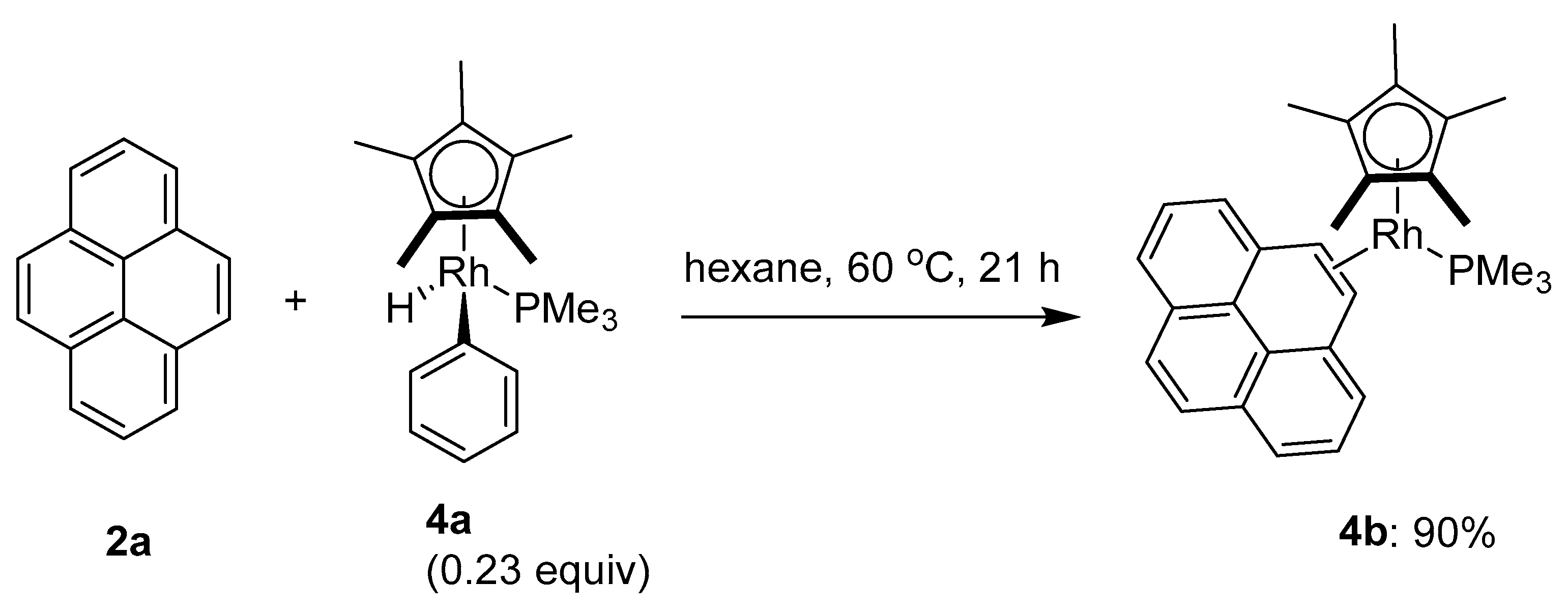
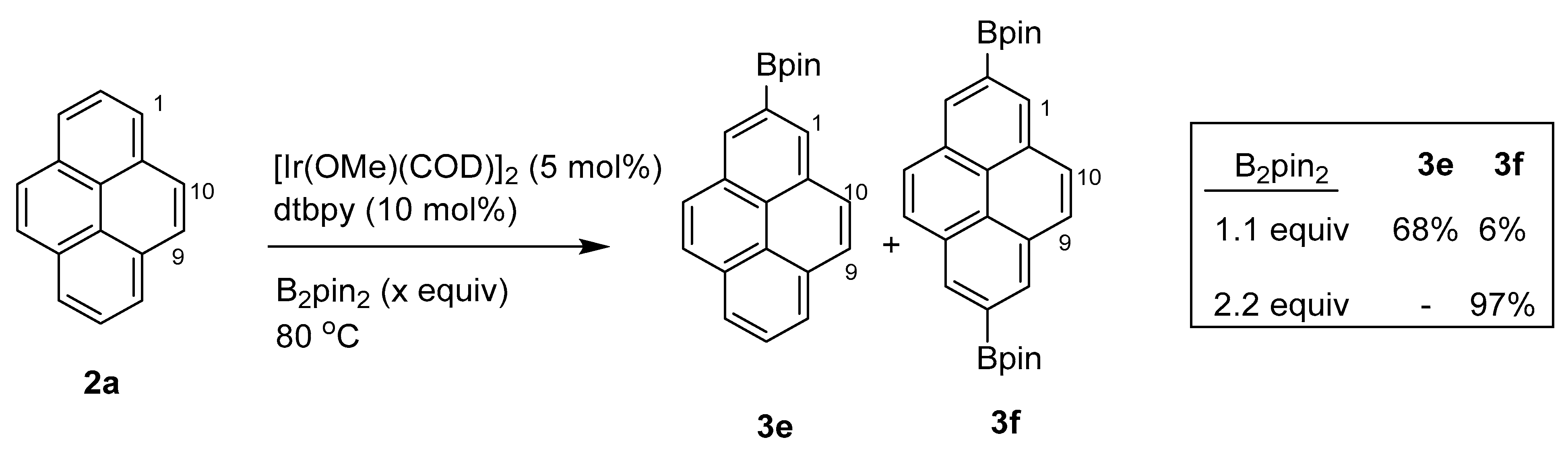
2.1. Direct Functionalization of the C2 and C7 Positions of Pyrenes
2.2. Direct Functionalization of the C4 Position of Pyrenes
2.3. Direct Functionalization of the C1 and C6 Positions of Pyrenes
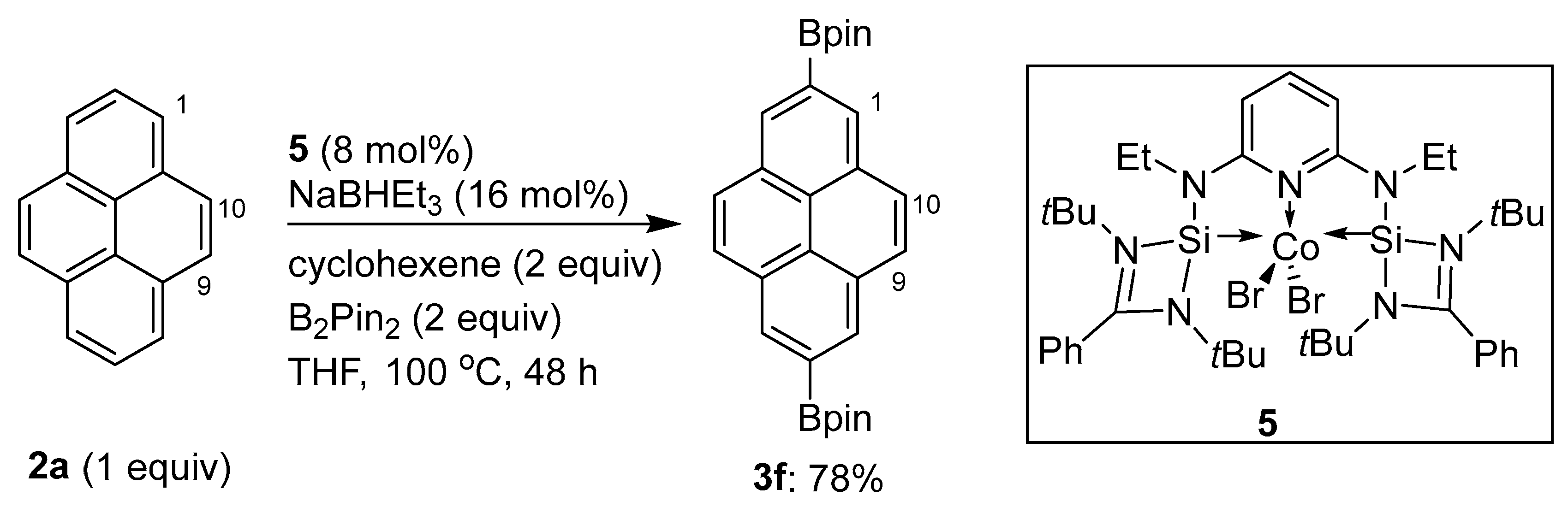
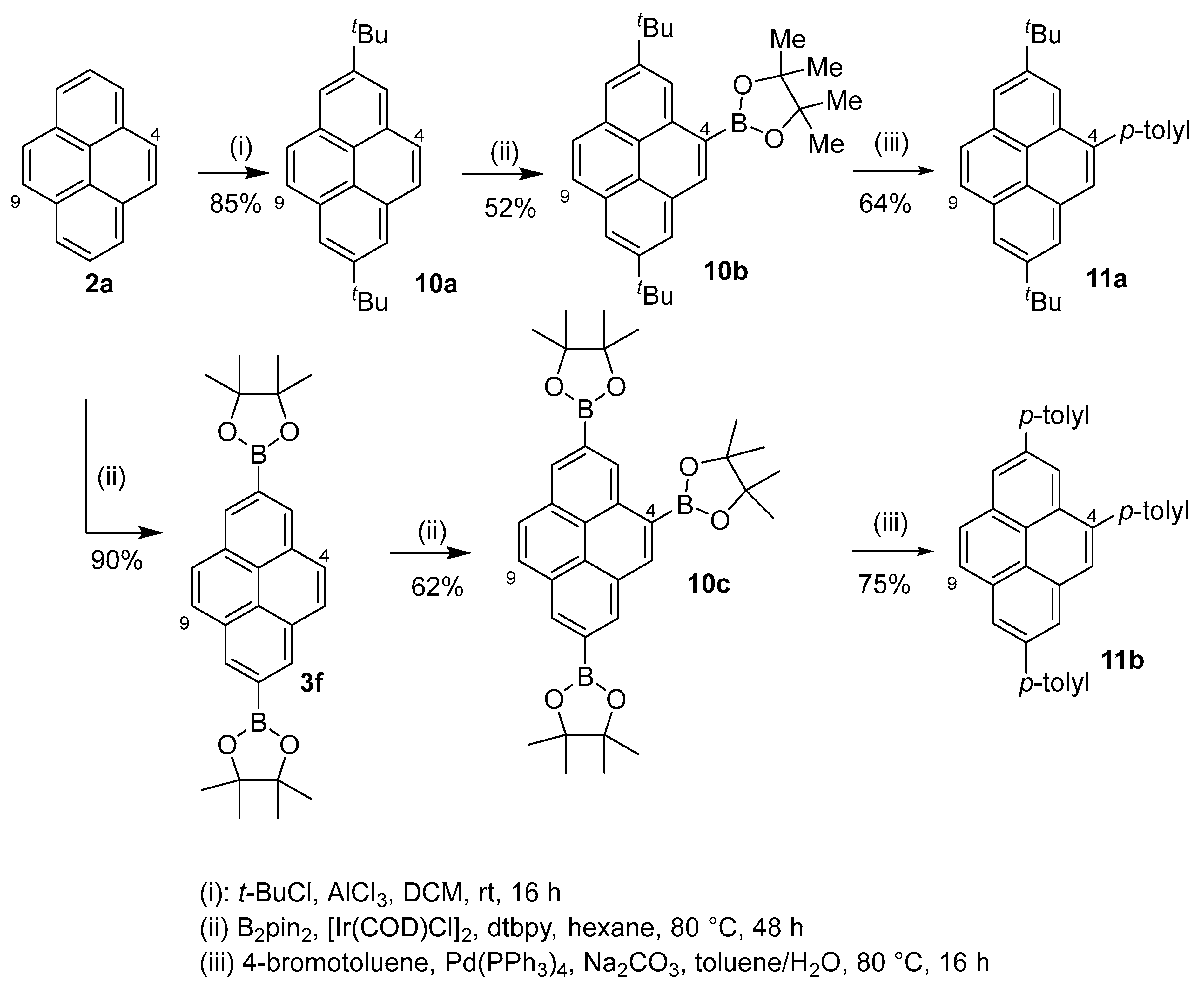
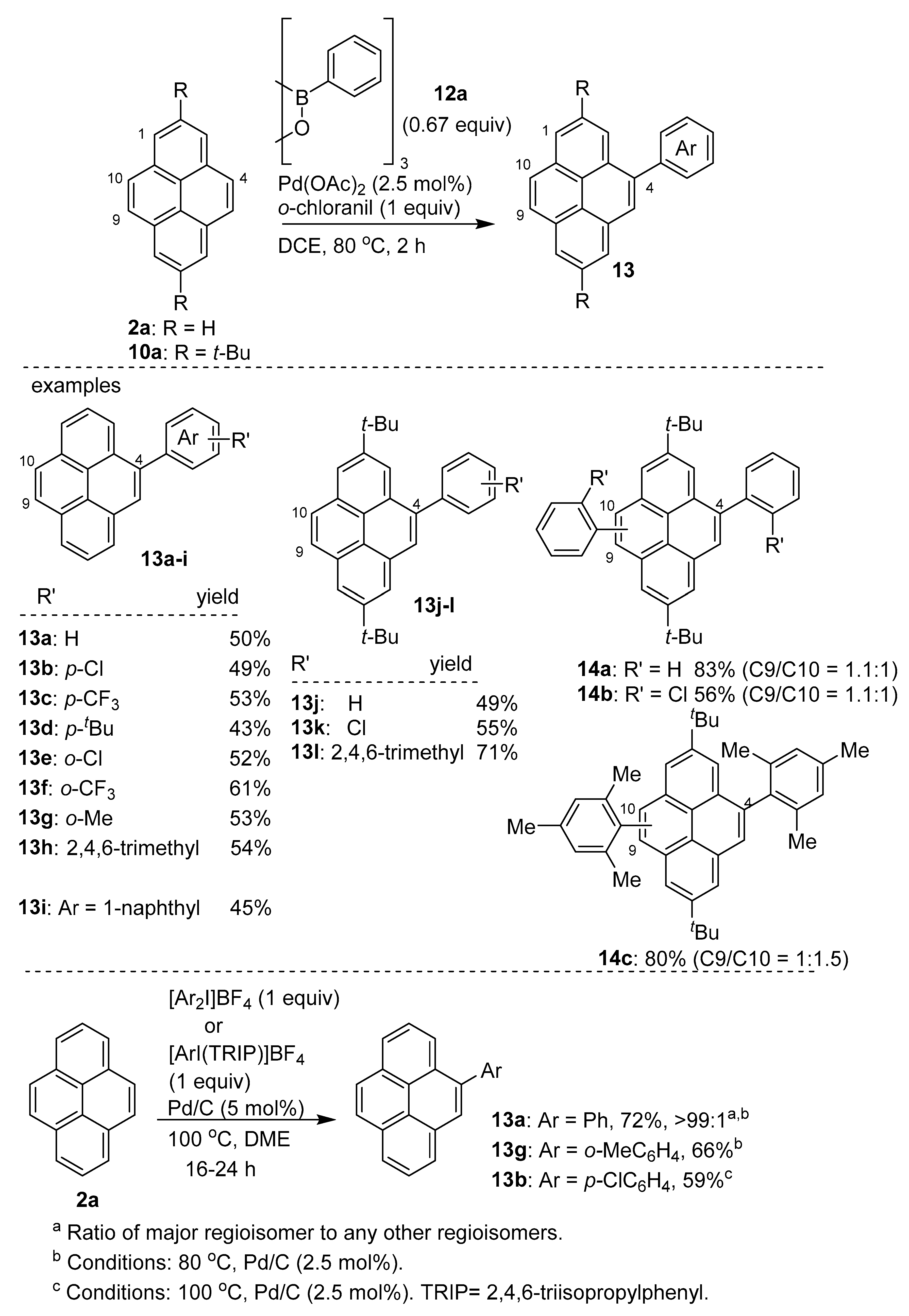
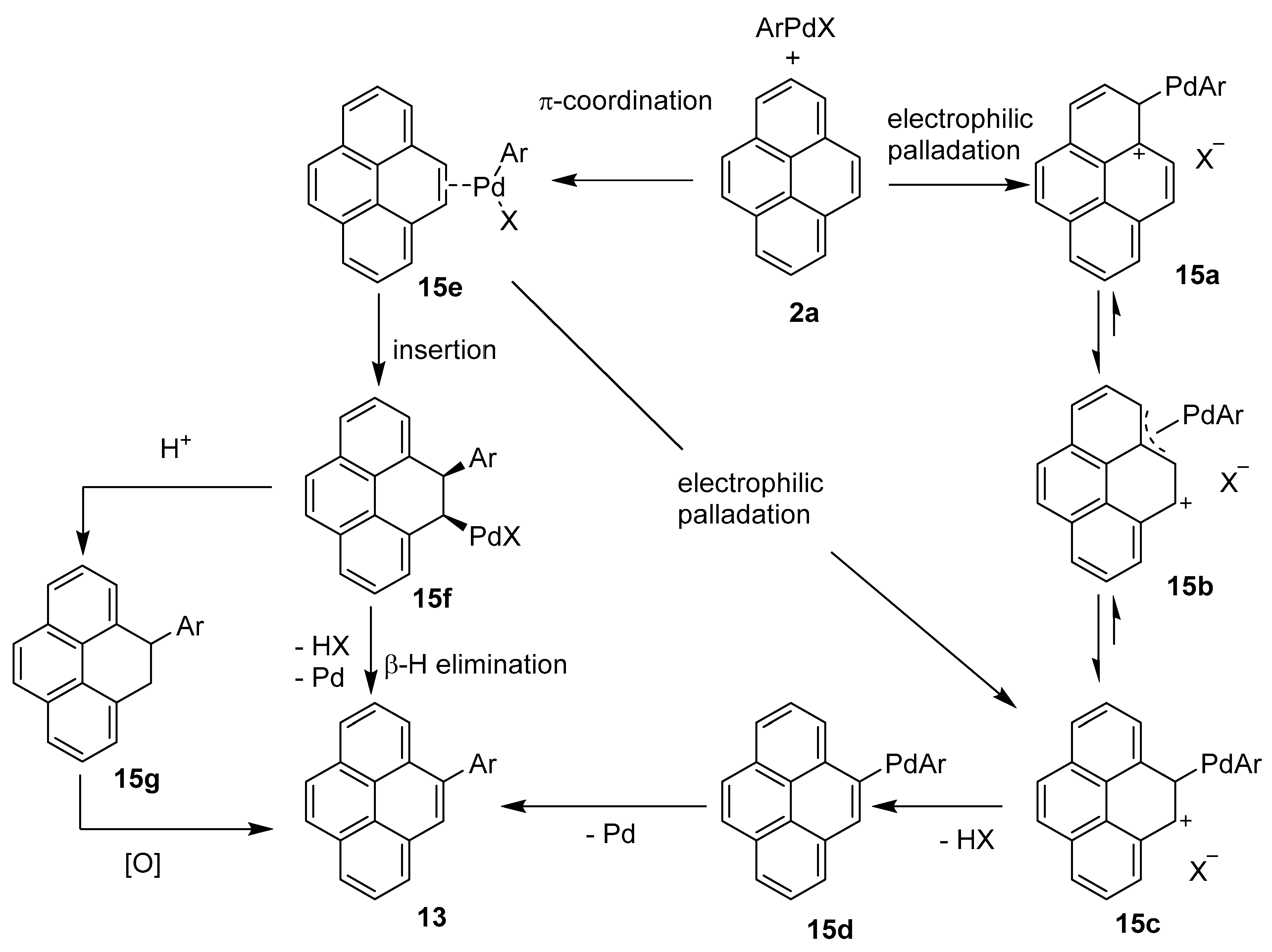
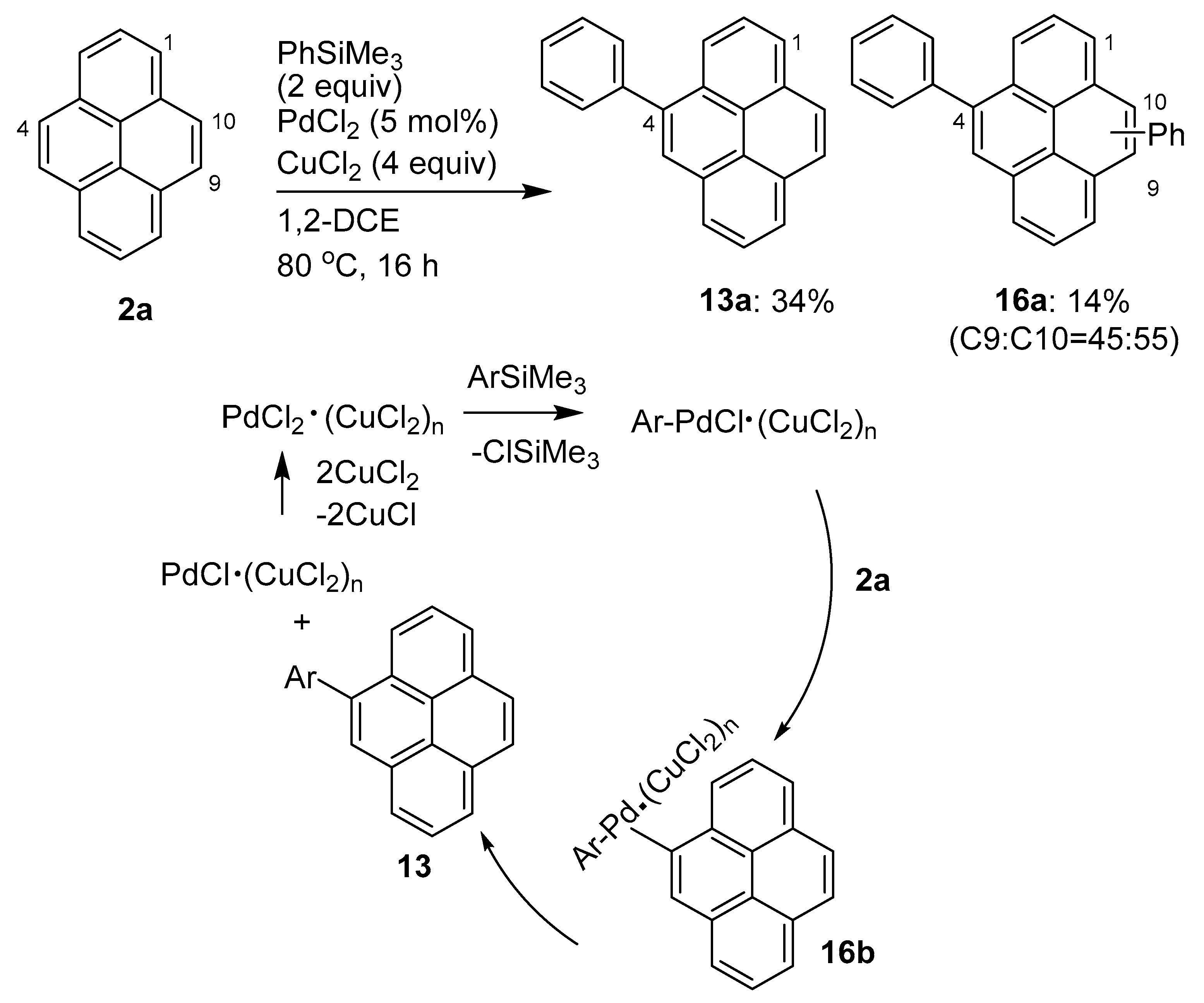
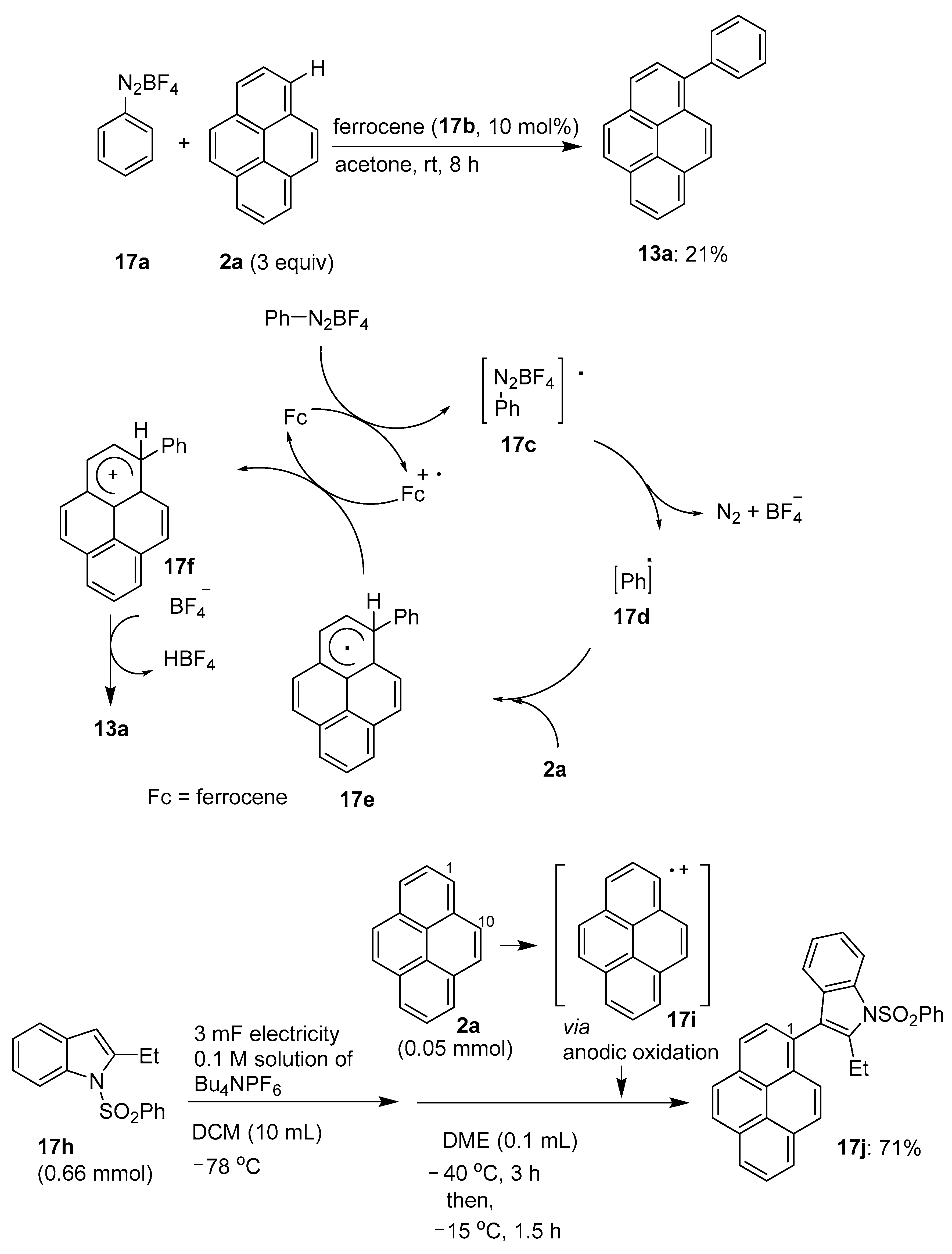
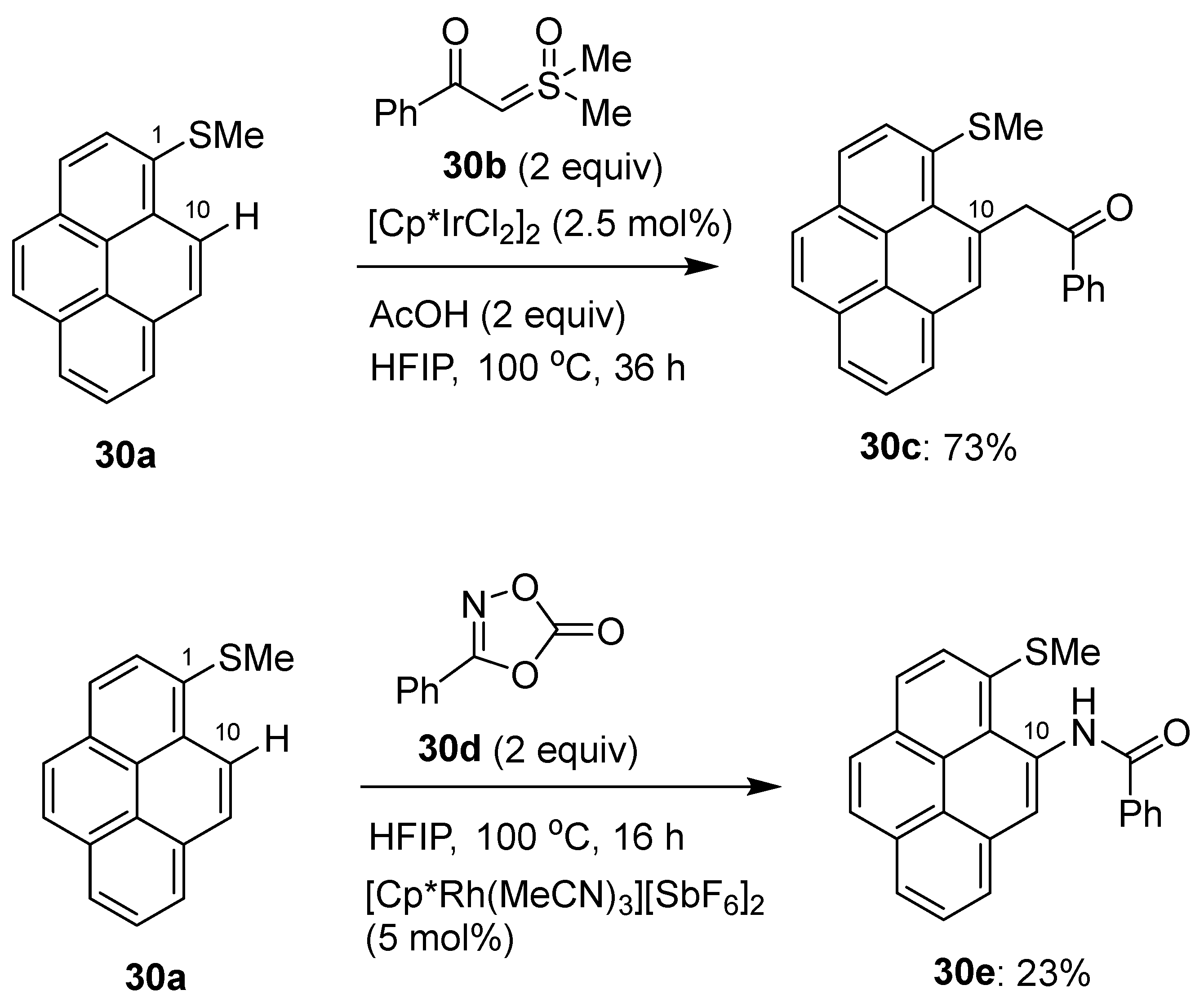
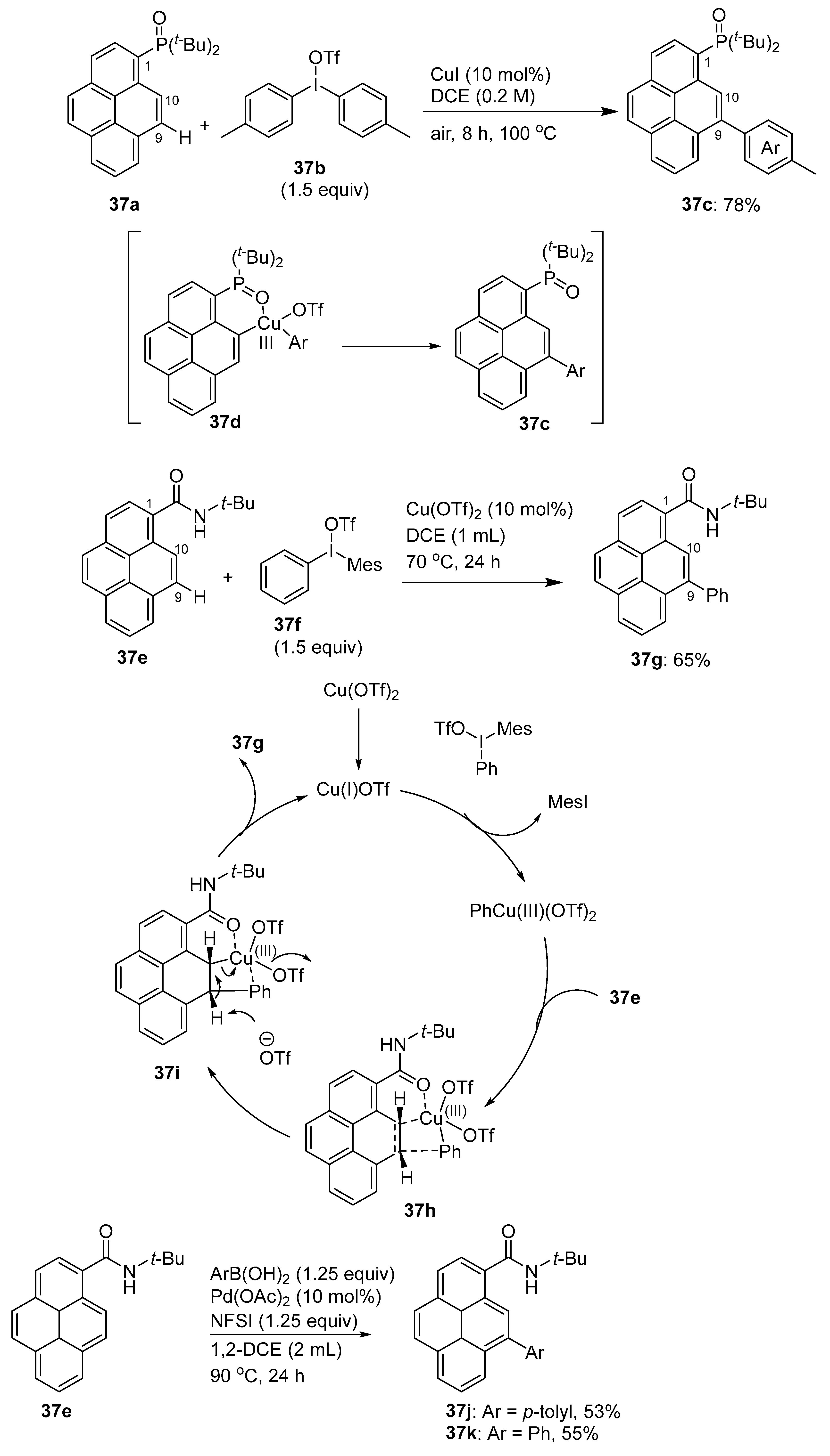
2.4. Direct Functionalization of the C9 and C10 Positions of Pyrenes
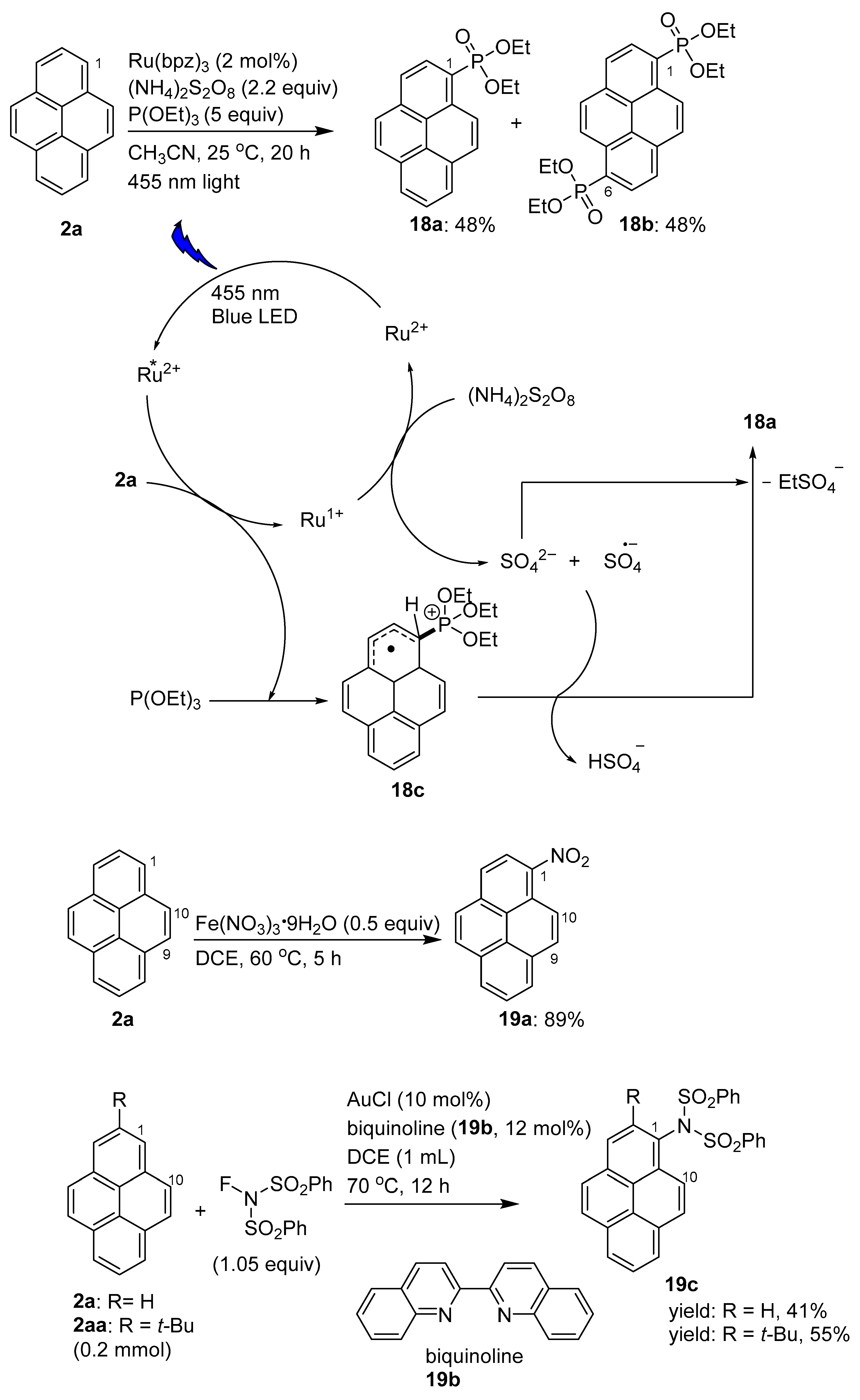
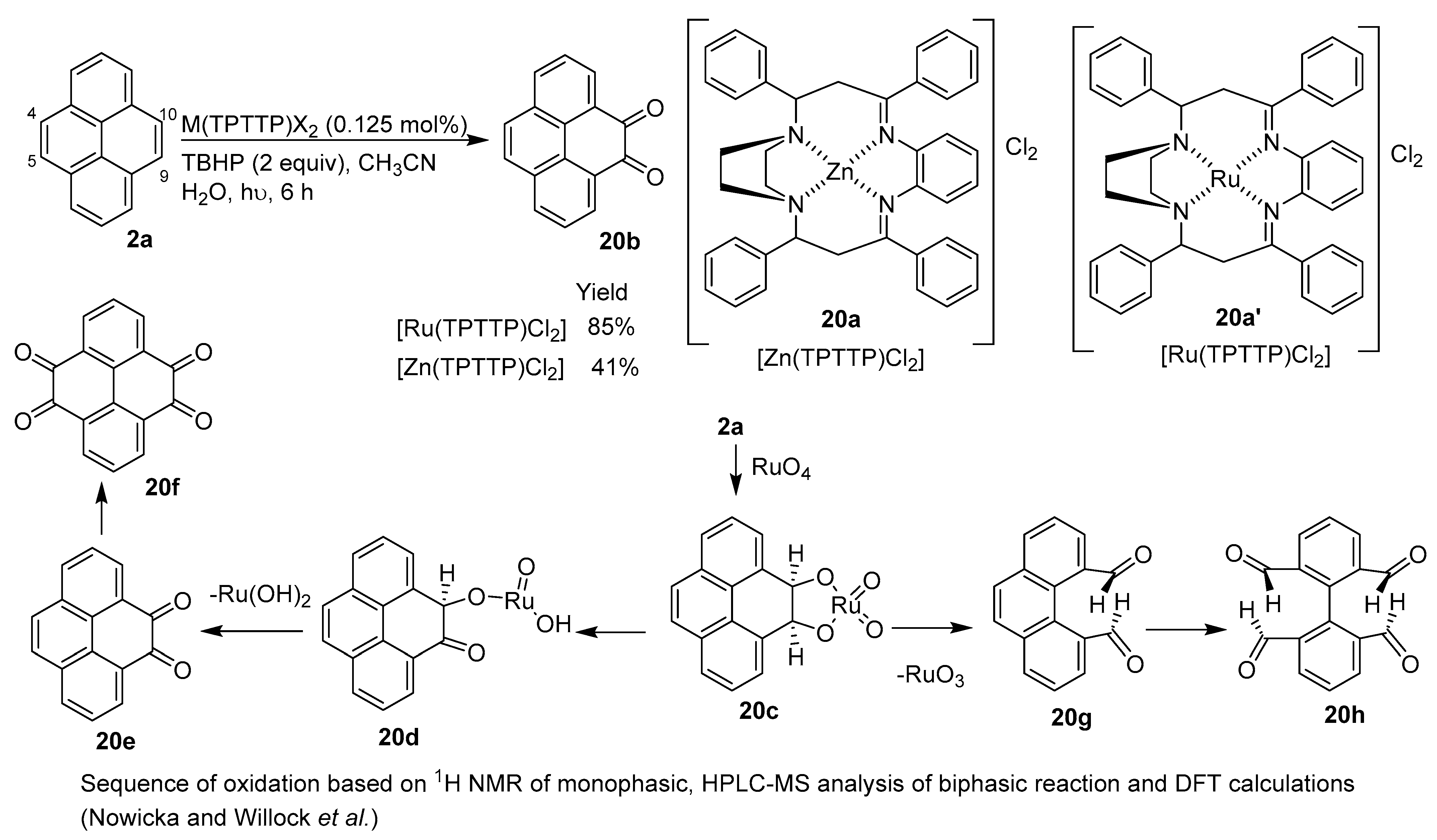
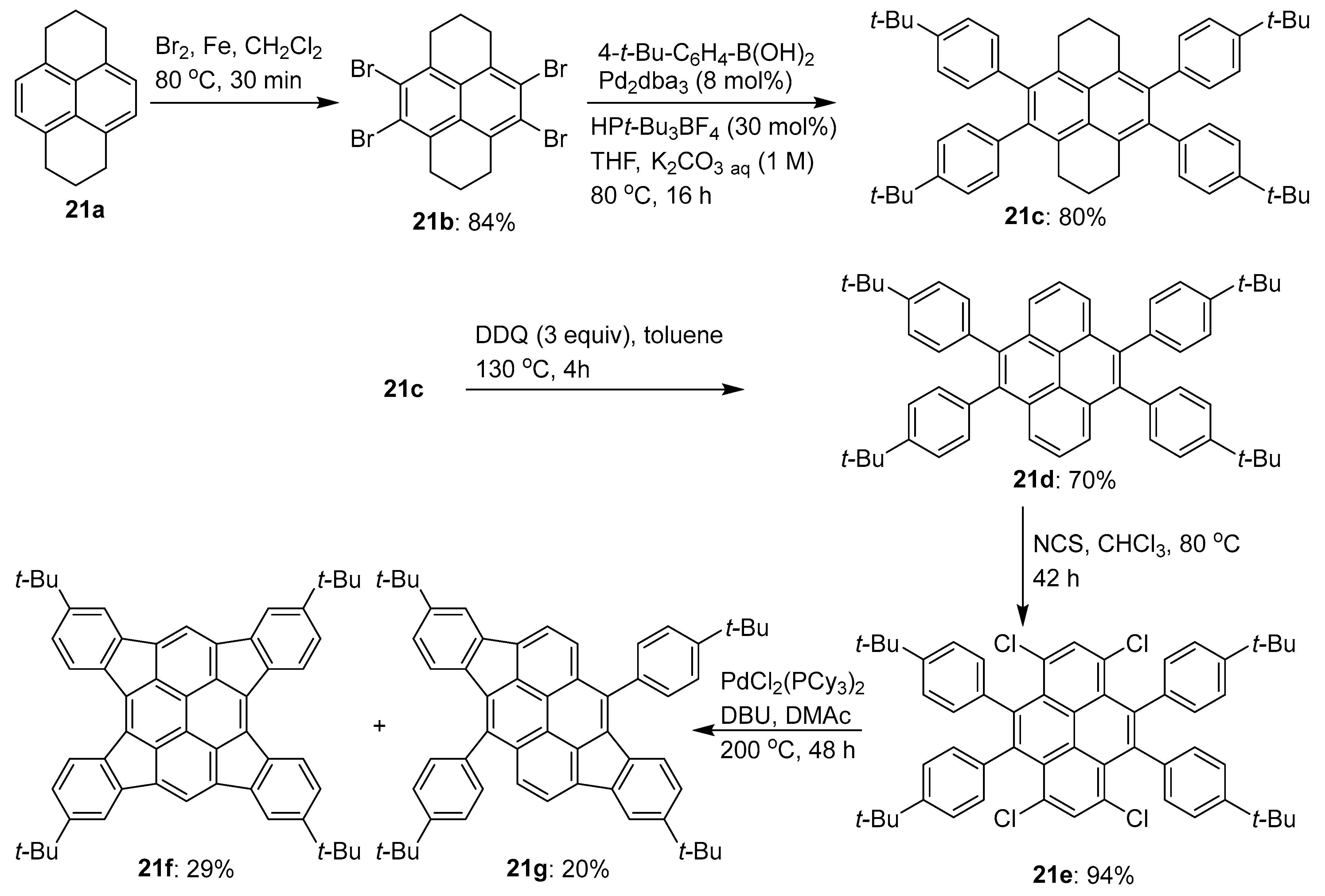
2.5. C–H Functionalization and Involving the Pyrene Backbone
3. Directing Group-Assisted C–H Activation and Functionalization of Pyrenes, Affording Functionalized Pyrenes
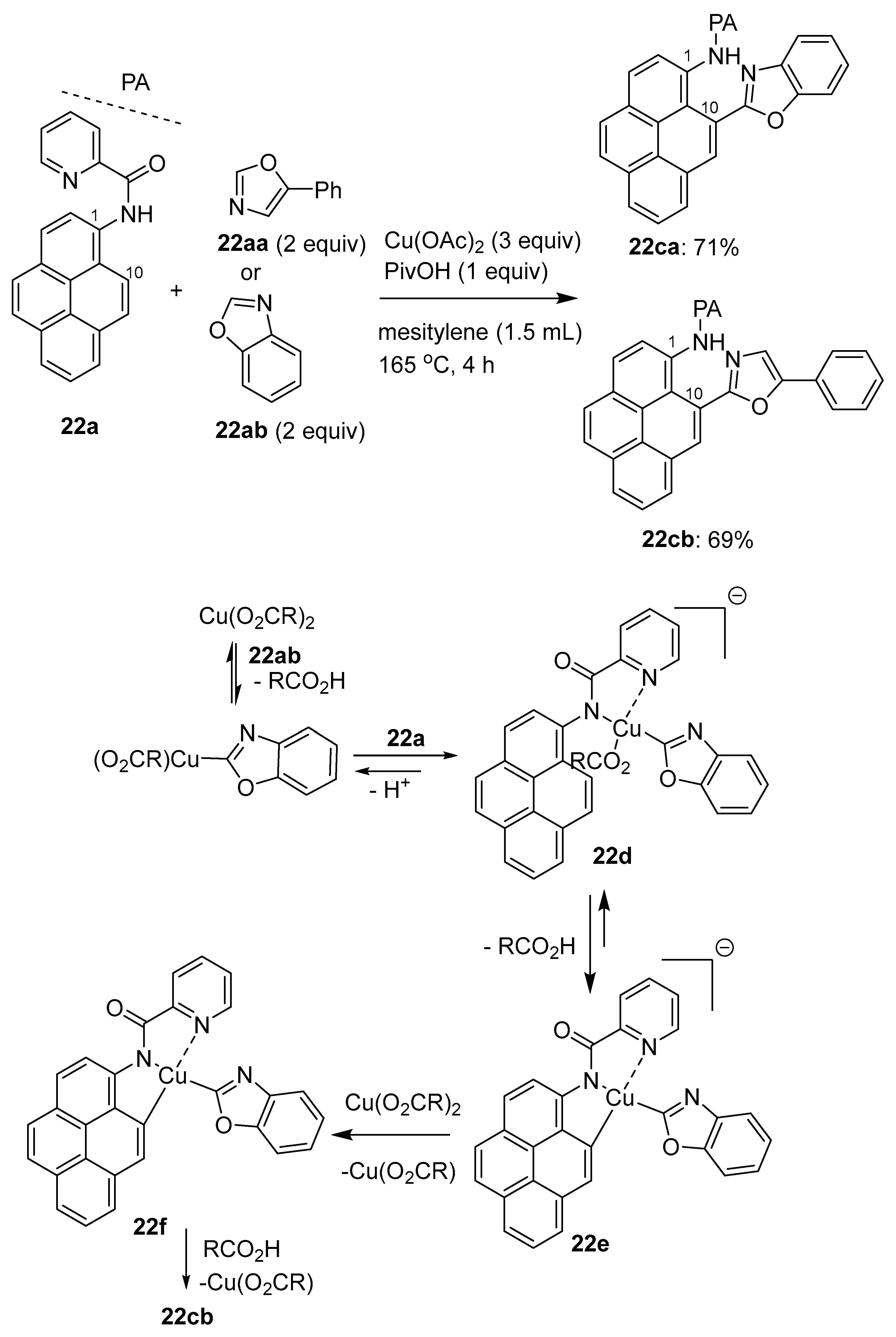
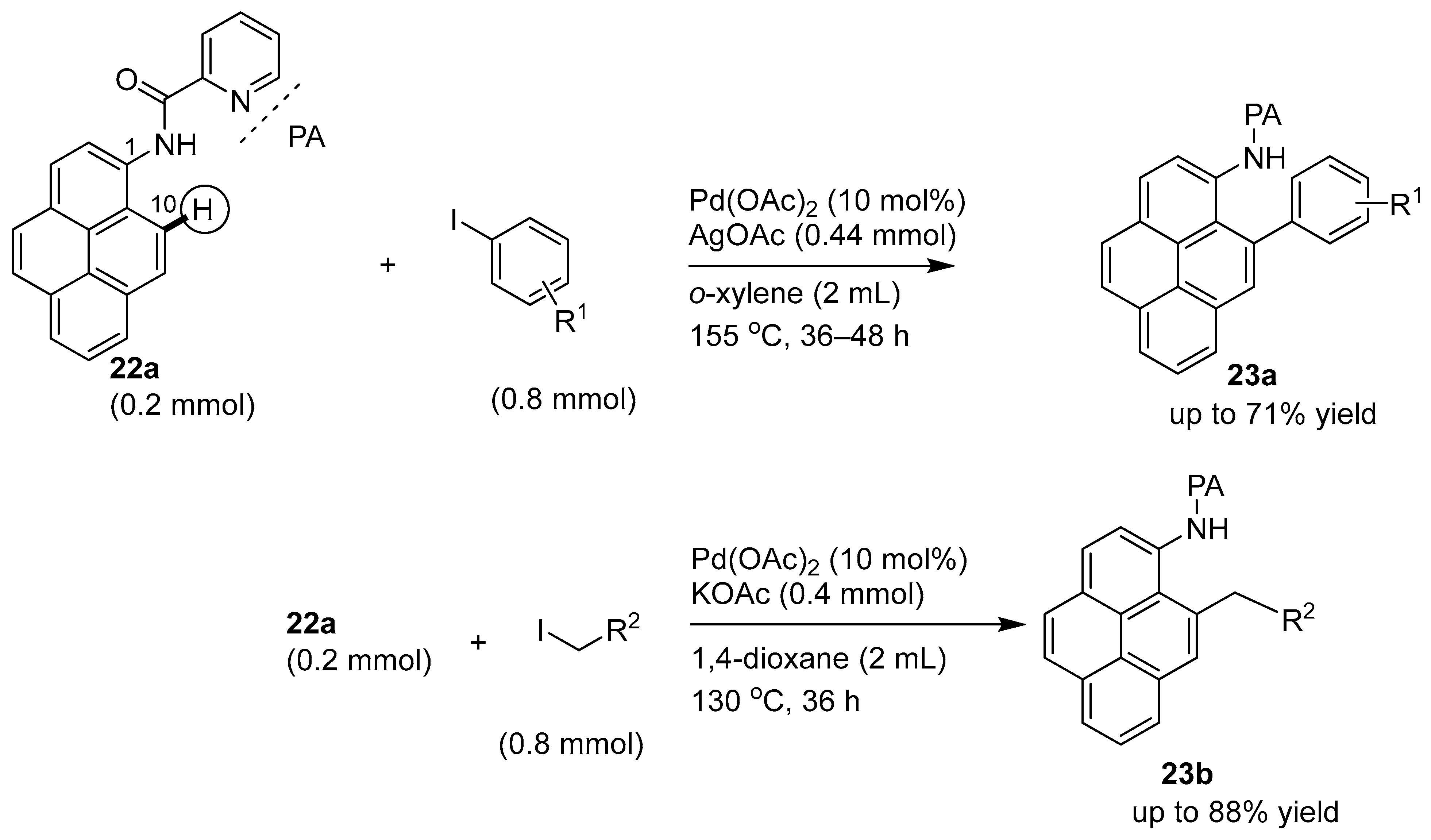
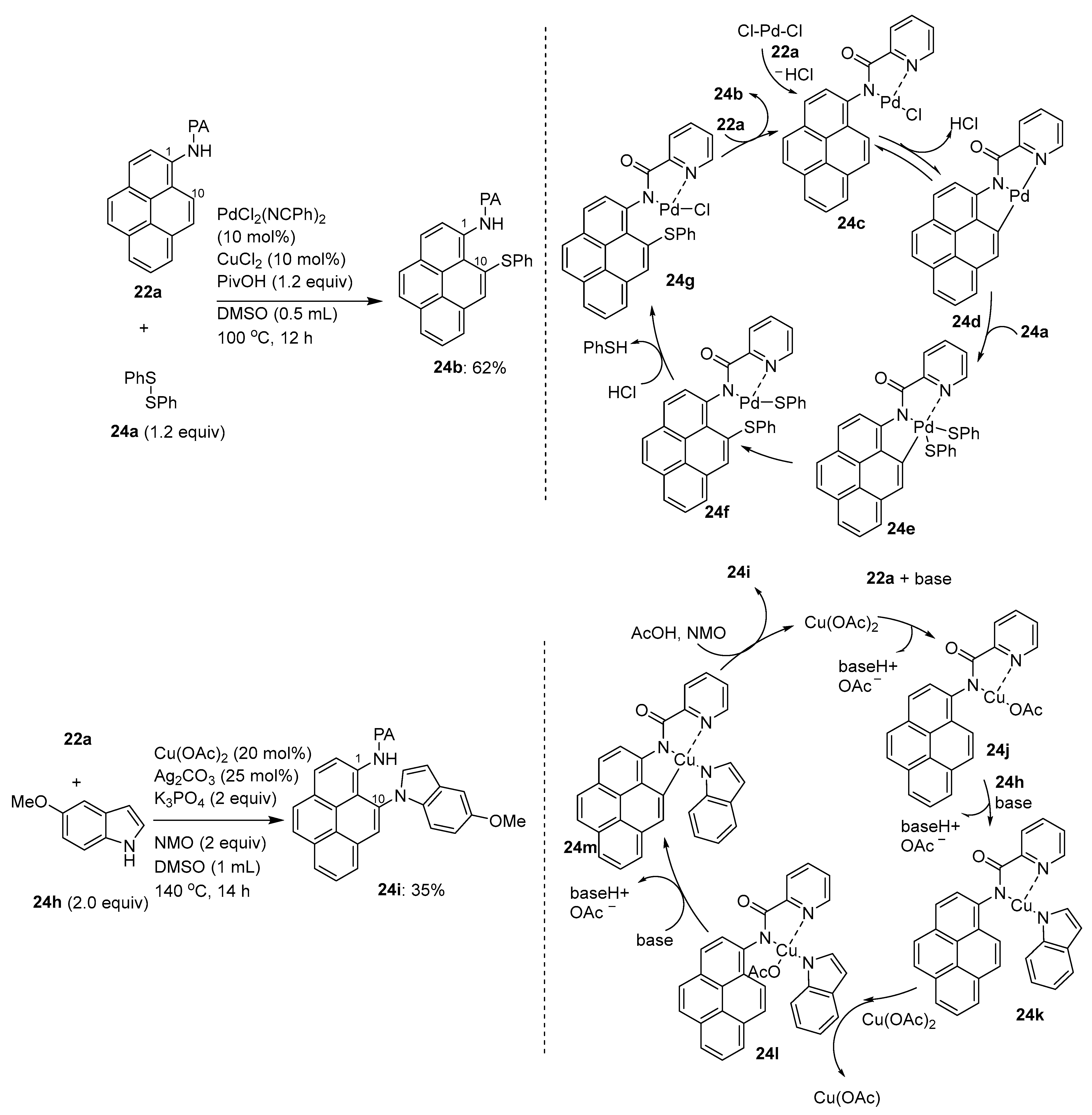
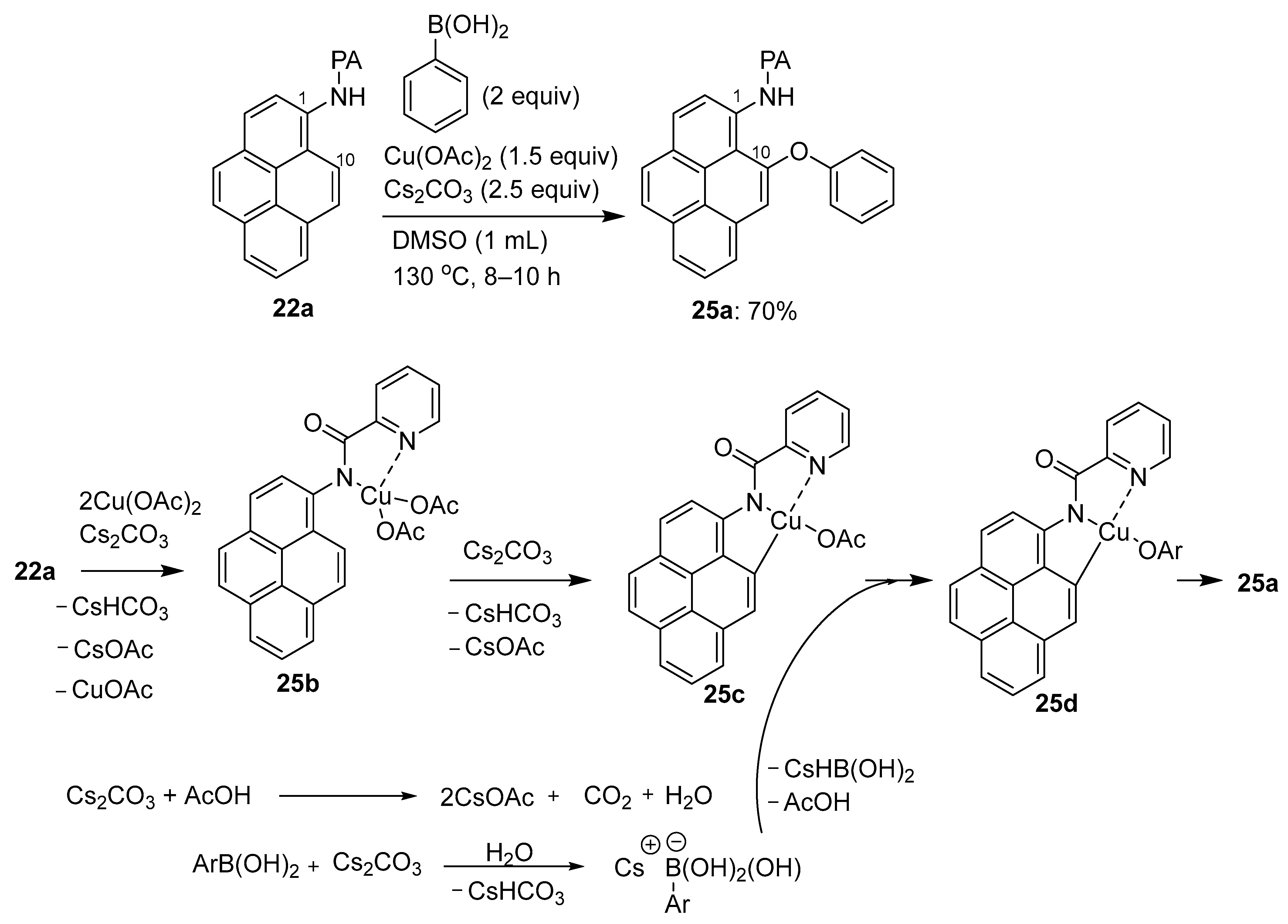
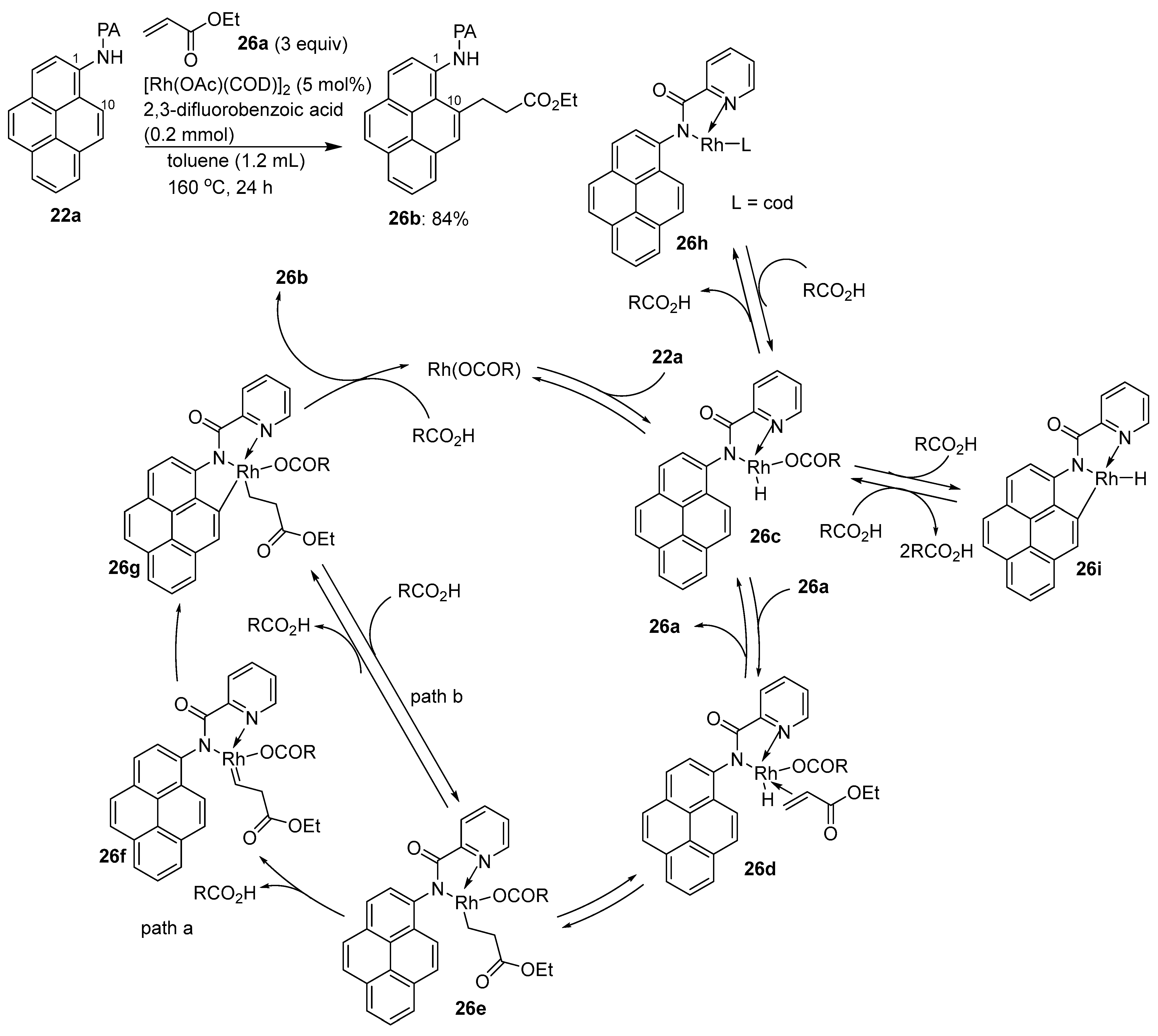
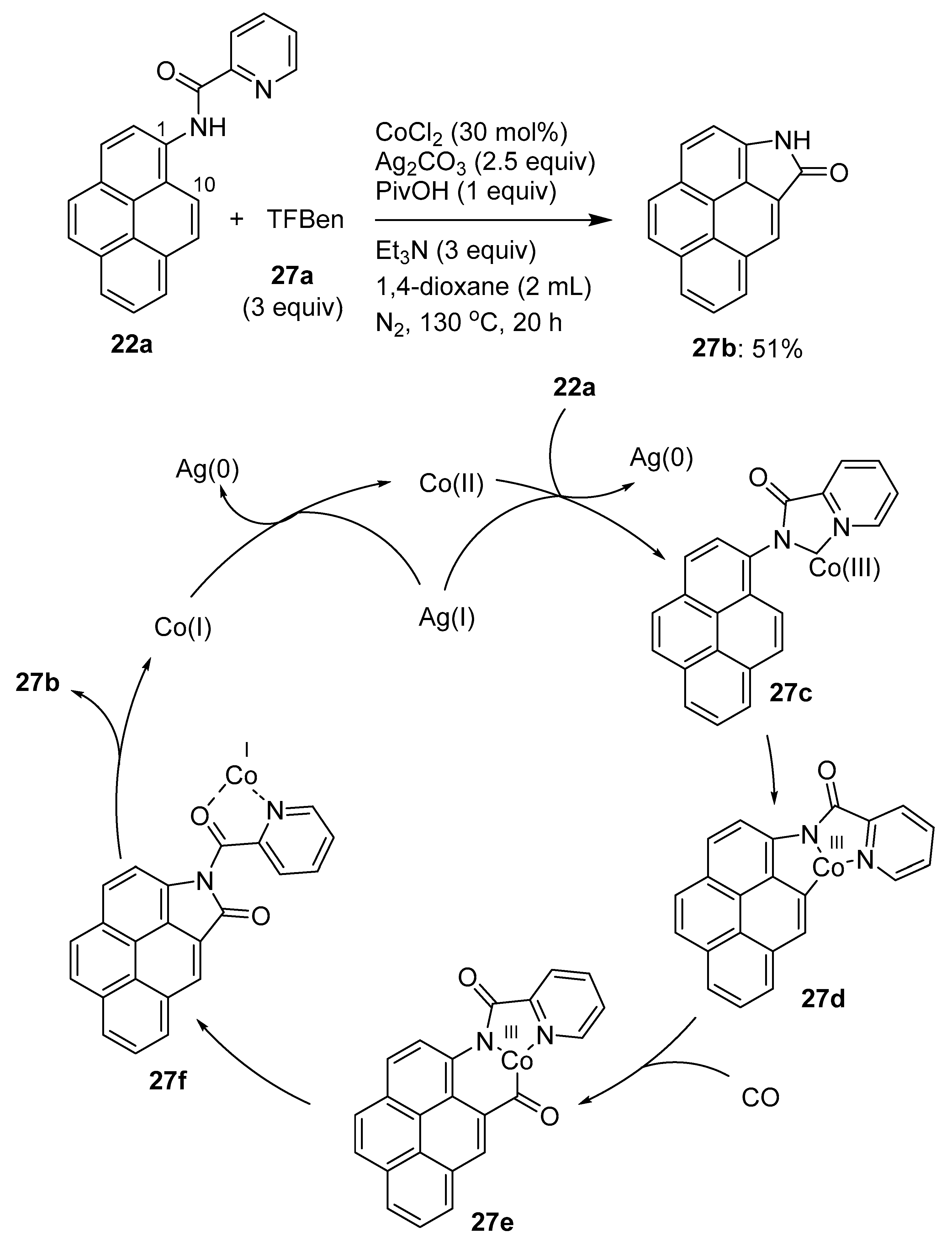
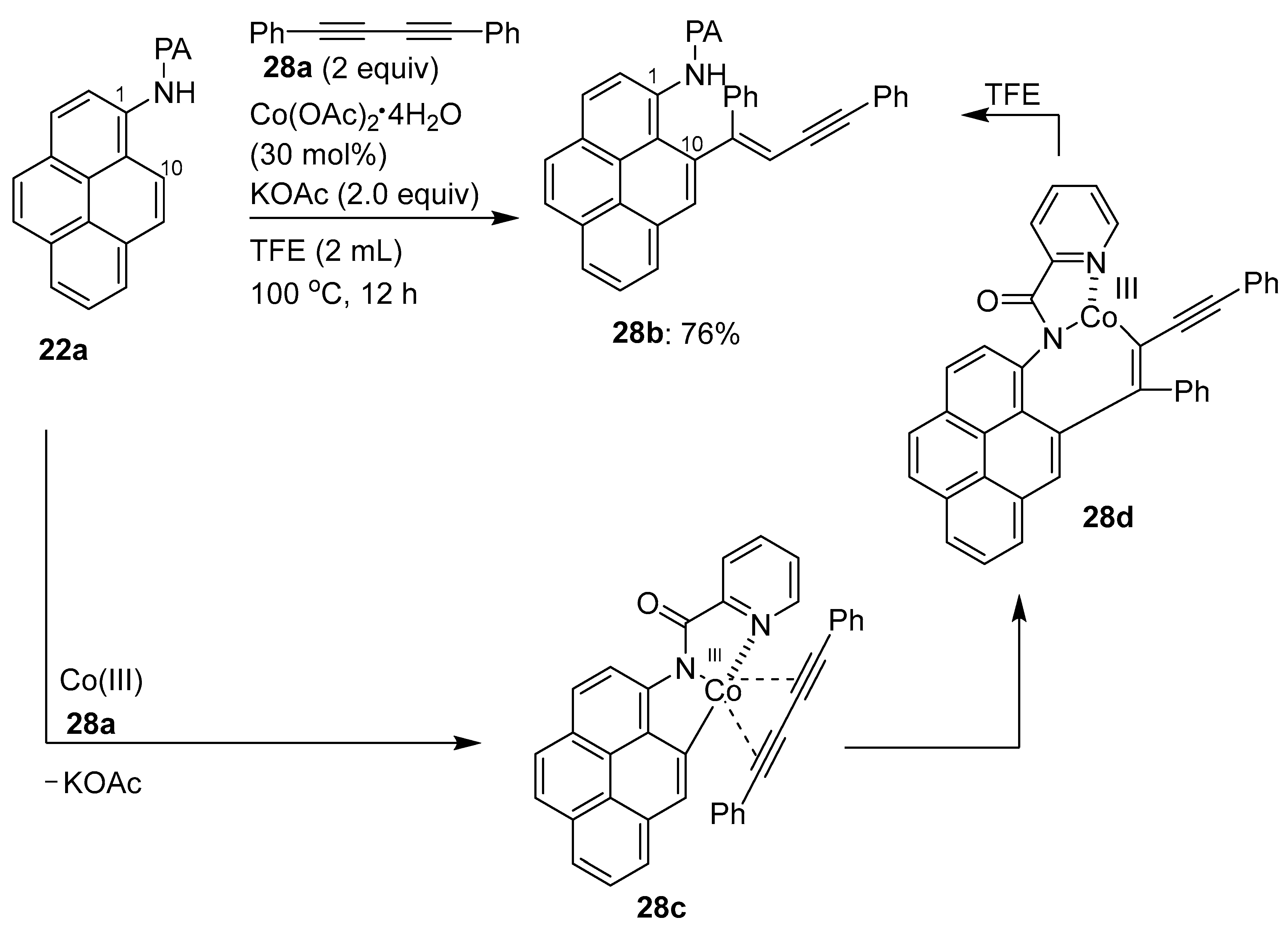
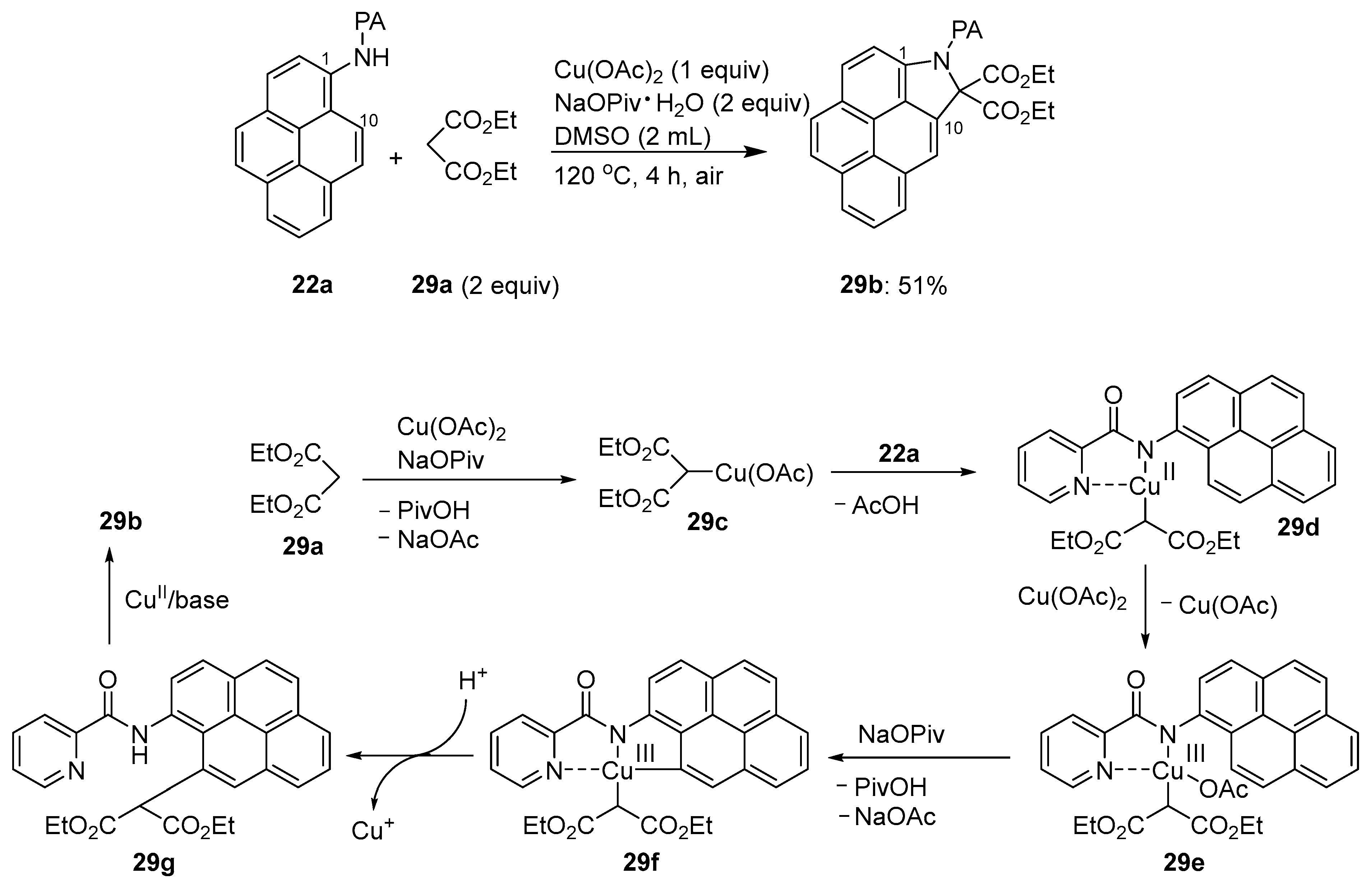
3.1. Directing Group-Assisted C–H Functionalization of the C10 Position of Pyrenes
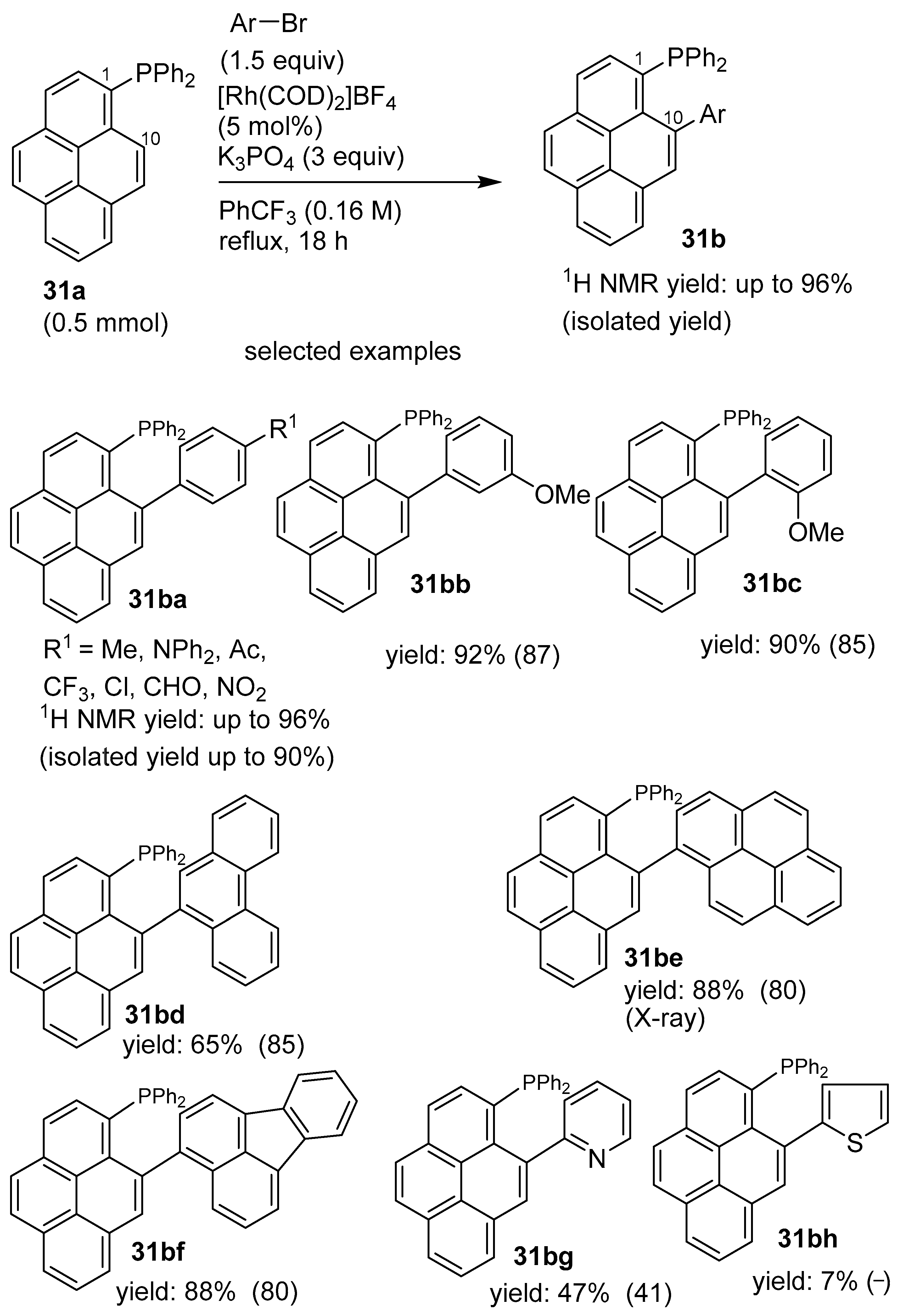
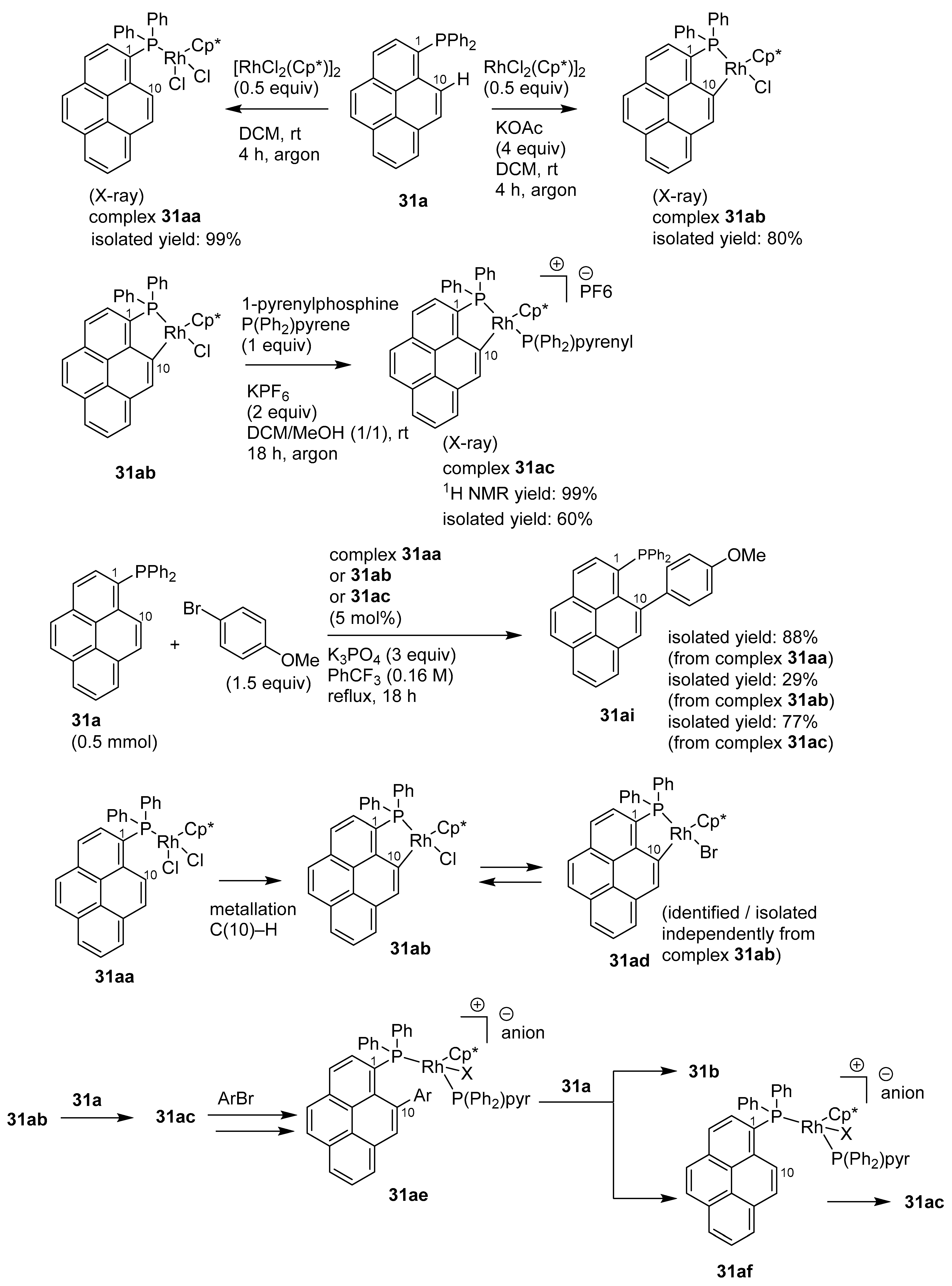
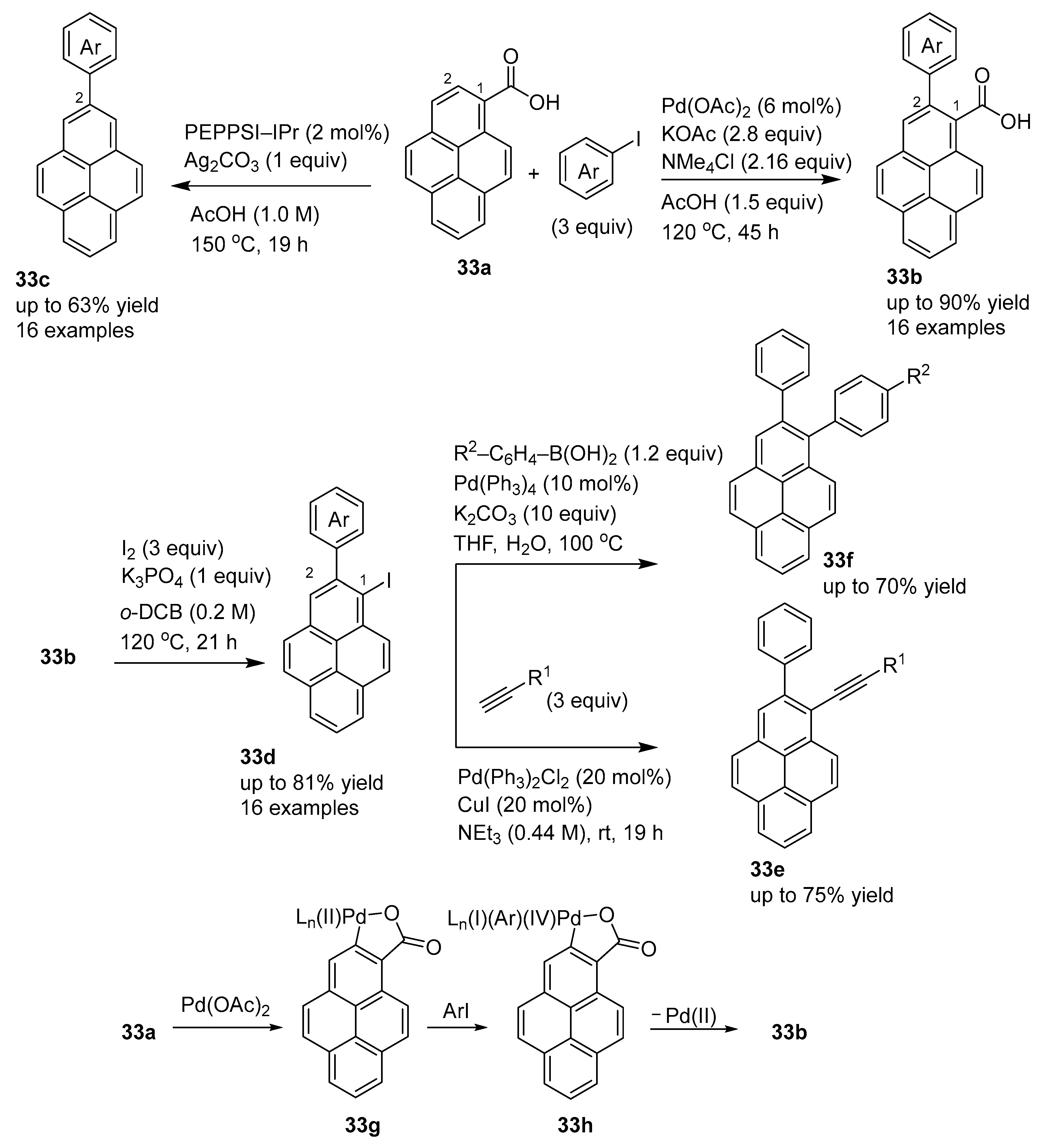
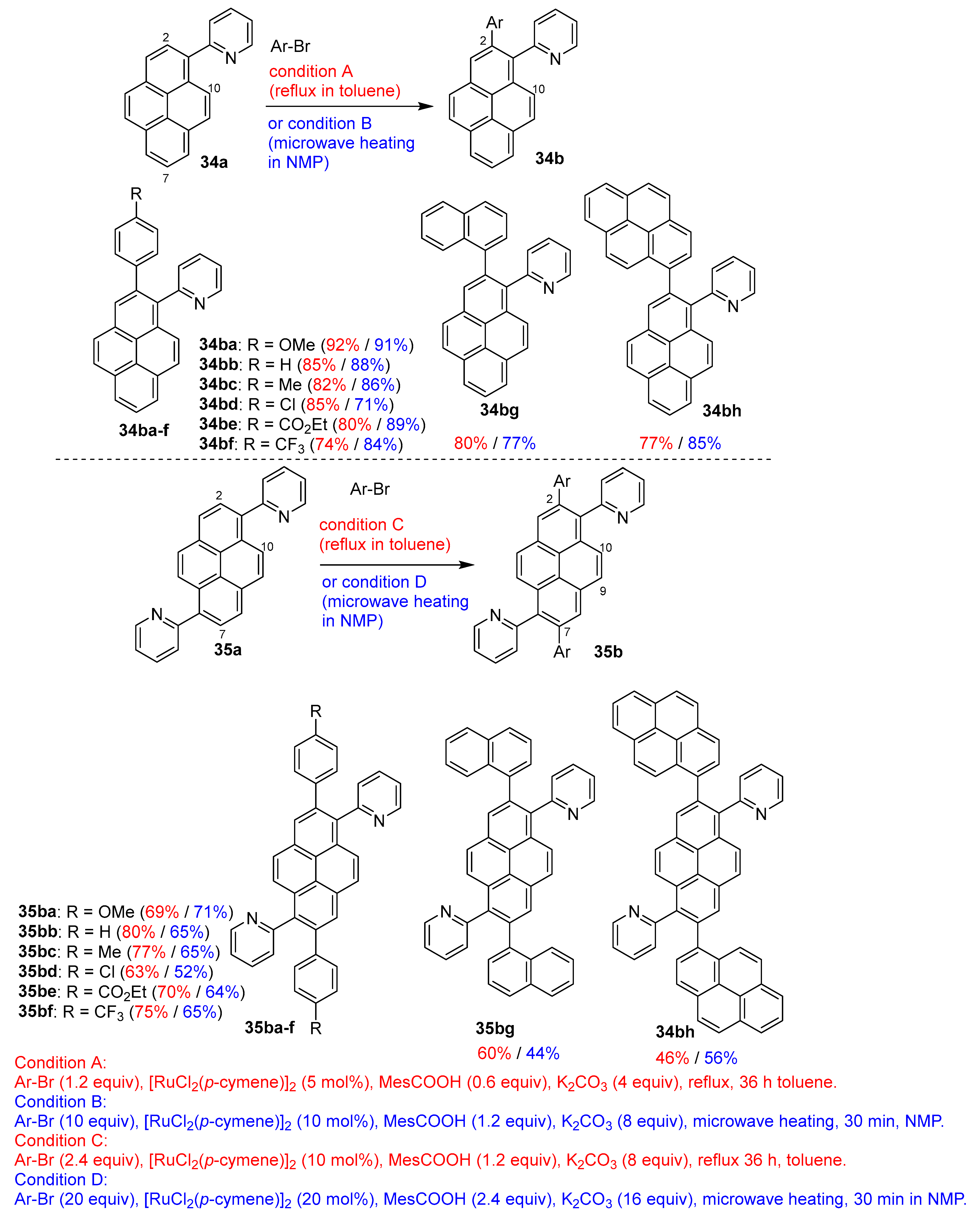
3.2. Directing Group-Assisted C–H Functionalization of the C2 and C7 Positions of Pyrenes
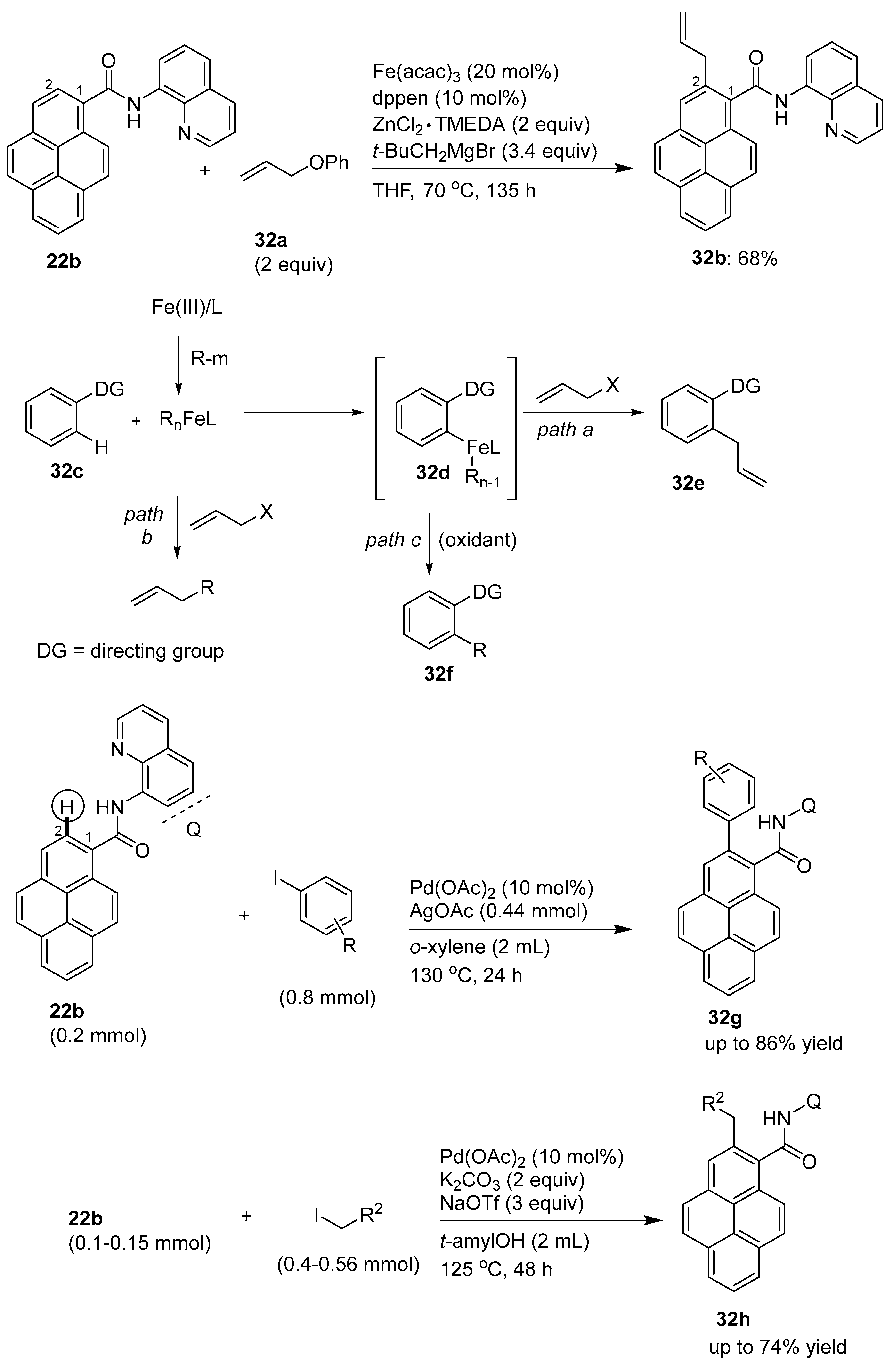
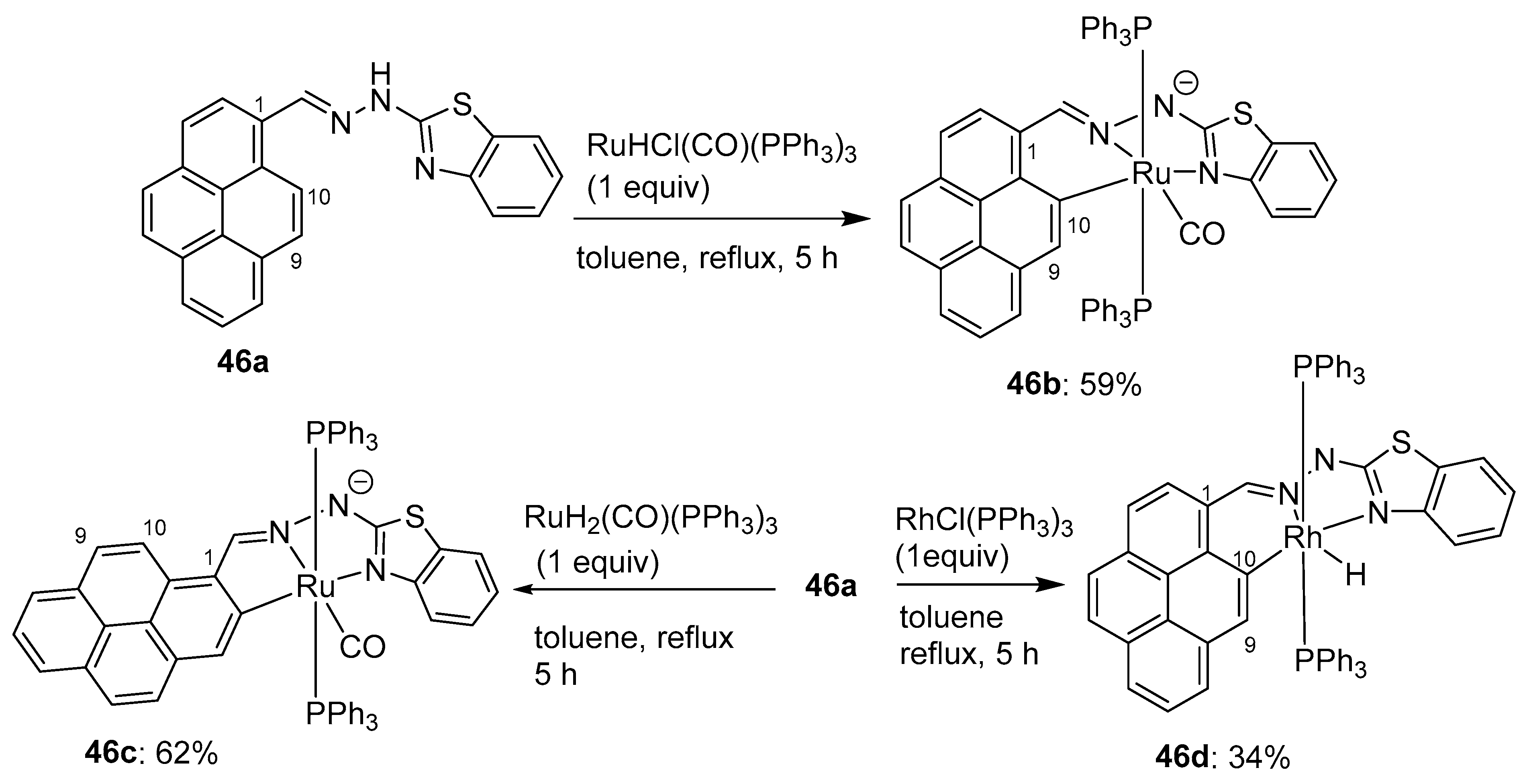
3.3. Directing Group-Assisted C–H Functionalization of the C9 Position of Pyrenes
4. Annulation of C–H Bonds of Pyrenes, Affording Pyrenes Appended with Additional Rings
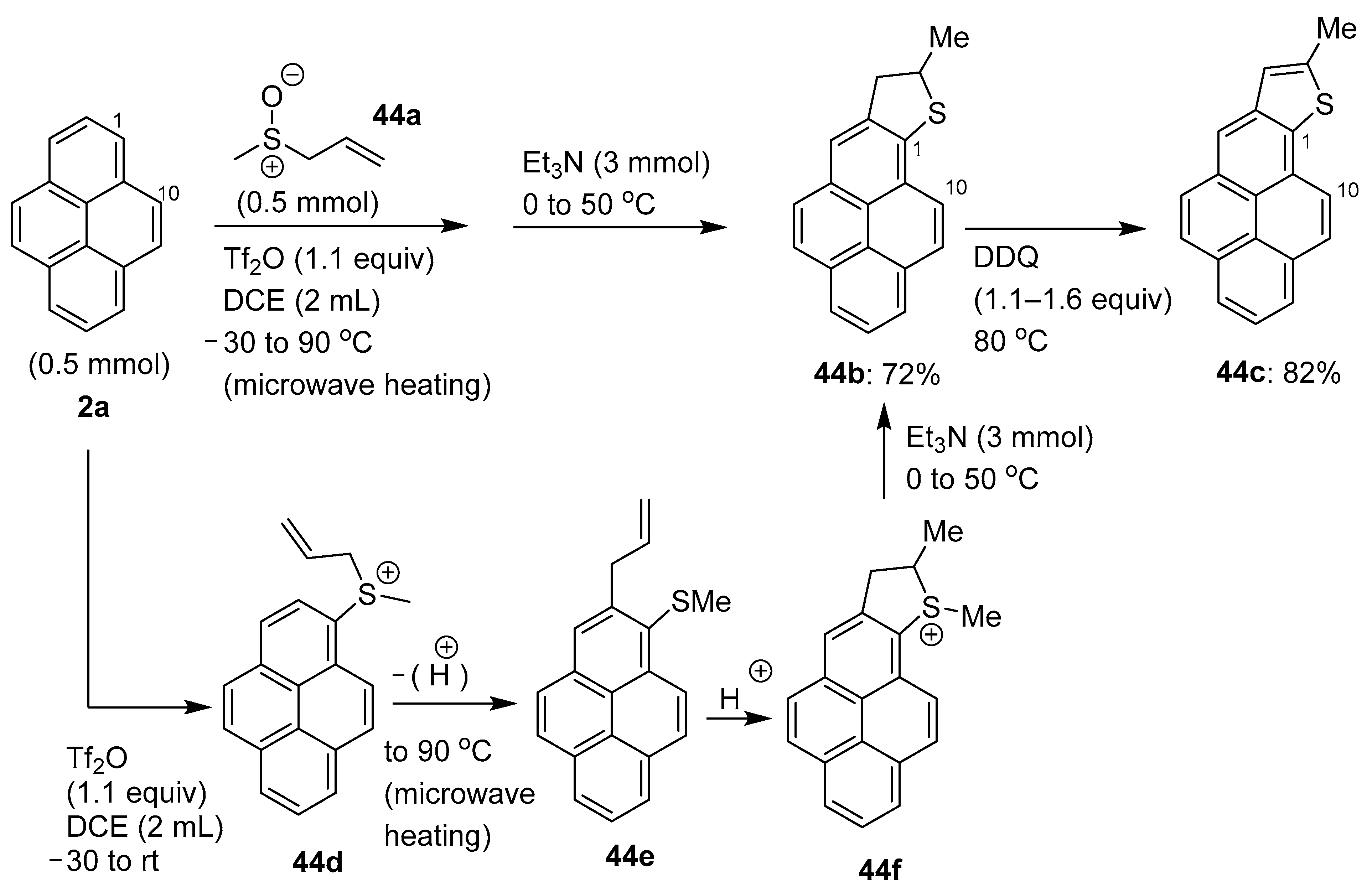
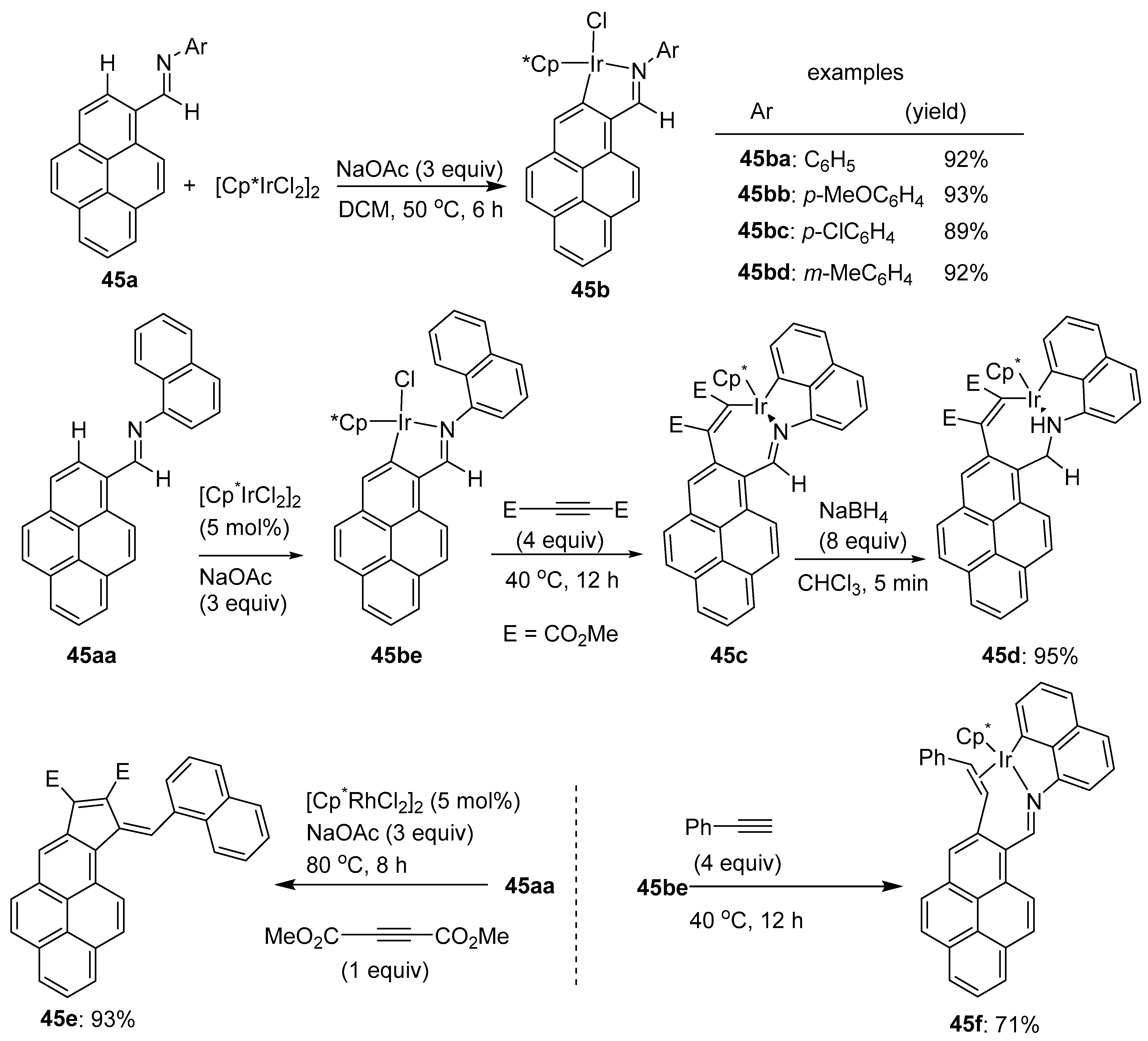
5. Miscellaneous C–H Functionalization Transformations Involving Pyrenes
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Figueira-Duarte, T.M.; Müllen, K. Pyrene-based materials for organic electronics. Chem. Rev. 2011, 111, 7260–7314. [Google Scholar] [CrossRef] [PubMed]
- Bains, G.; Patel, A.B.; Narayanaswami, V. Pyrene: A probe to study protein conformation and conformational changes. Molecules 2011, 16, 7909–7935. [Google Scholar] [CrossRef]
- Manandhar, E.; Wallace, K.J. Host−guest chemistry of pyrene-based molecular receptors. Inorg. Chim. Acta 2012, 381, 15–43. [Google Scholar] [CrossRef]
- Mateo-Alonso, A. Synthetic approaches to pyrene-fused twistacenes. Eur. J. Org. Chem. 2017, 2017, 7006–7011. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef]
- Koch, N. Supramolecular Materials for Opto-Electronics; The Royal Society of Chemistry: Cambridge, UK, 2015. [Google Scholar]
- Zhao, Z.; Lam, J.W.Y.; Tang, B.Z. Tetraphenylethane: A versatile AIE building block for construction of efficient luminescent materials for organic light-emitting diodes. J. Mater. Chem. 2012, 22, 23726–23740. [Google Scholar] [CrossRef]
- Casas-Solvas, J.M.; Howgego, J.D.; Davis, A.P. Synthesis of substituted pyrenes by indirect methods. Org. Biomol. Chem. 2014, 12, 212–232. [Google Scholar] [CrossRef]
- Zych, D. Non-K region disubstituted pyrenes (1,3-, 1,6- and 1,8-) by (hetero)aryl groups-review. Molecules 2019, 24, 2551. [Google Scholar] [CrossRef]
- Feng, X.; Hu, J.-Y.; Redshaw, C.; Yamato, T. Functionalization of pyrene to prepare luminescent materials−typical examples of synthetic methodology. Chem.-Eur. J. 2016, 22, 11898–11916. [Google Scholar] [CrossRef]
- Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chemistry of carbon nanotubes. Chem. Rev. 2006, 106, 1105–1136. [Google Scholar] [CrossRef]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current progress on the chemical modification of carbon nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef]
- Le Goff, A.; Gorgy, K.; Holzinger, M.; Haddad, R.; Zimmerman, M.; Cosnier, S.A. Supramolecular bridge for the biofunctionalization of carbon nanotubes via π-stacking and pyrene/β-cyclodextrin host−guest interactions. Chem.-Eur. J. 2011, 17, 10216–10221. [Google Scholar] [CrossRef]
- Chen, Y.L.; Zhu, B.; Han, Y.; Bo, Z.S. Self-assembly of cationic pyrene nanotubes. J. Mater. Chem. 2012, 22, 4927–4931. [Google Scholar] [CrossRef]
- Xu, Y.; Bai, H.; Lu, G.; Li, C.; Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 2008, 18, 5856–5867. [Google Scholar] [CrossRef]
- Kodali, V.K.; Scrimgeour, J.; Kim, S.; Hankinson, J.H.; Carroll, K.M.; de Heer, W.A.; Berger, C.; Curtis, J.E. Nonperturbative chemical modification of graphene for protein micropatterning. Langmuir 2011, 27, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.A.; Rodríguez-López, J.; Abruña, H.D.; Dichtel, W.R. Multivalent binding motifs for the noncovalent functionalization of graphene. J. Am. Chem. Soc. 2011, 133, 17614–17617. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, H.M.; Zhu, Z.; Williams, D.J.; Drew, M.G.B.; Cardin, C.J.; Gan, Y.; Crawford, A.G.; Marder, T.B. Induced-fit binding of π-electron-donor substrates to macrocyclic aromatic ether imide sulfones: A versatile approach to molecular assembly. Chem. -Eur. J. 2010, 16, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zou, Y.; Li, C.; Sicking, W.; Piantanida, I.; Yi, T.; Schmuck, C. A molecular peptide beacon for the ratiometric sensing of nucleic acids. J. Am. Chem. Soc. 2012, 134, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Takahashi, Y.; Tanabe, S.; Nishizawa, S. Conjugating pyrene onto PNA-based fluorescent probes for improved detection selectivity toward double-stranded siRNA. Org. Biomol. Chem. 2020, 18, 4009–4013. [Google Scholar] [CrossRef]
- Endo, M.; Sugiyama, H. Chemical approaches to DNA nanotechnology. ChemBioChem 2009, 10, 2420–2443. [Google Scholar] [CrossRef]
- Astakhova, I.V.; Korshun, V.A.; Wengel, J. Highly fluorescent conjugated pyrenes in nucleic acid probes: (phenylethynyl) pyrenecarbonyl-functionalised locked nucleic acids. Chem.-Eur. J. 2008, 14, 11010–11026. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M.; Fujimoto, K.; Furusyo, M.; Nakazumi, H. Molecular recognition abilities of a new class of water-soluble cyclophanes capable of encompassing a neutral cavity. J. Am. Chem. Soc. 1999, 121, 1452–1458. [Google Scholar] [CrossRef]
- Escobar, L.; Ballester, P. Molecular recognition in water using macrocyclic synthetic receptors. Chem. Rev. 2021, 121, 2445–2514. [Google Scholar] [CrossRef] [PubMed]
- Benito, J.M.; Meldal, M. Bicyclic organo-peptides as selective carbohydrate receptors: Design, solid-phase synthesis, and on-bead binding capability. QSAR Comb. Sci. 2004, 23, 117–129. [Google Scholar] [CrossRef]
- Vollmann, H.; Becker, H.; Corell, M.; Streeck, H. Beitrage zur kenntnis des pyrene und seiner derivate. Liebigs Ann. 1937, 531, 1–159. [Google Scholar] [CrossRef]
- Coventry, D.N.; Batsanov, A.S.; Goeta, A.E.; Howard, J.A.K.; Marder, T.B.; Perutz, R.N. Selective Ir-catalysed borylation of polycyclic aromatic hydrocarbons: Structures of naphthalene-2,6-bis(boronate), pyrene-2,7-bis(boronate) and perylene-2,5,8,11-tetra(boronate) esters. Chem. Commun 2005, 2172–2174. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.G.; Liu, Z.; Mkhalid, I.A.I.; Thibault, M.-H.; Schwarz, N.; Alcaraz, G.; Steffen, A.; Collings, J.C.; Batsanov, A.S.; Howard, J.A.K.; et al. Synthesis of 2- and 2,7-functionalized pyrene derivatives: An application of selective C−H borylation. Chem.-Eur. J. 2012, 18, 5022–5035. [Google Scholar] [CrossRef]
- Pataki, J.; Konieczny, M.; Harvey, R.G. Synthesis of tert-butylarenes from acetylarenes. J. Org. Chem. 1982, 47, 1133–1136. [Google Scholar] [CrossRef]
- Ji, L.; Lorbach, A.; Edkins, M.R.; Marder, T.B. Synthesis and photophysics of a 2,7-disubstituted donor−acceptor pyrene derivative: An example of the application of sequential Ir-catalyzed C−H borylation and substitution chemistry. J. Org. Chem. 2015, 80, 5658–5665. [Google Scholar] [CrossRef]
- Chen, H.; Hu, X.; Ng, S.J. Synthesis and characterization of soluble conjugated polymers having pyrene moiety in the main chain. Polym. Sci. Part A Polym. Chem. 2010, 48, 5562–5569. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, J.; Xu, W.; Zhu, D. Novel 2,7-substituted pyrene derivatives: Syntheses, solid-state structures, and properties. Tetrahedron 2011, 67, 3395–3405. [Google Scholar] [CrossRef]
- Lee, O.P.; Yiu, A.T.; Beaujuge, P.M.; Woo, C.H.; Holcombe, T.W.; Millstone, J.E.; Douglas, J.D.; Chen, M.S.; Fréchet, J.M.J. Efficient small molecule bulk heterojunction solar cells with high fill factors via pyrene-directed molecular self-assembly. Adv. Mater. 2011, 23, 5359–5363. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Anbarasan, P.; Neumann, H.; Beller, M. From noble metal to Nobel prize: Palladium catalyzed coupling reactions as key methods in organic synthesis. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef]
- Grimshaw, J.; Trocha-Grimshaw, J. Characterisation of 1,6- and 1,8-dibromopyrenes. J. Chem. Soc. Perkin 1 1972, 1622–1623. [Google Scholar] [CrossRef]
- Sharif, M.; Reimann, S.; Wittler, K.; Knöpke, L.R.; Surkus, A.-E.; Roth, C.; Villinger, A.; Ludwig, R.; Langer, P. 1-(Arylalkenyl)pyrenes—Synthetic, structural, photophysical, theoretical, and electrochemical investigations. Eur. J. Org. Chem. 2011, 2011, 5261–5271. [Google Scholar] [CrossRef]
- Ogino, K.; Iwashima, S.; Inokuchi, H.; Harada, Y. Photoelectric emission and electrical conductivity of the cesium complex with pyrene derivatives. Bull. Chem. Soc. Jpn. 1965, 38, 473–477. [Google Scholar] [CrossRef]
- Yamato, T.; Fujimoto, M.; Miyazawa, A.; Matsuo, K. Selective preparation of polycyclic aromatic hydrocarbons. Part 5.1 Bromination of 2,7-di-tert-butylpyrene and conversion into pyrenoquinones and their pyrenoquinhydrones. J. Chem. Soc. Perkin Trans. 1 1997, 1, 1201–1208. [Google Scholar] [CrossRef]
- Tashiro, M.; Yamato, T. Metacyclophanes and related compounds. 4. Halogenations of 8,16-dialkyl-anti-5,13-di-tert-butyl[2.2]metacyclophan-1-enes and 2,7-di-tert-butyl trans-10b, 10c-dialkyl-10b, 10c-dihydropyrenes. J. Am. Chem. Soc. 1982, 104, 3701–3707. [Google Scholar] [CrossRef]
- Yamato, T.; Miyazawa, A.; Tashiro, M. Medium-sized cyclophanes. Part 31. Synthesis and electrophilic substitution of 8-substituted [2]metacyclo[2](1,3)pyrenophanes. J. Chem. Soc. Perkin Trans. 1 1993, 3127–3137. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, D.; Harris, F.W. Ruthenium(III) chloride catalyzed oxidation of pyrene and 2,7-disubstitued pyrenes: An efficient, one-step synthesis of pyrene-4,5-diones and pyrene-4,5,9,10-tetraones. J. Org. Chem. 2005, 70, 707–708. [Google Scholar] [CrossRef]
- Zöphel, L.; Beckmann, D.; Enkelmann, V.; Chercka, D.; Rieger, R.; Müllen, K. Asymmetric pyrene derivatives for organic field-effect transistors. Chem. Commun. 2011, 47, 6960–6962. [Google Scholar] [CrossRef] [PubMed]
- Young, E.R.; Funk, R.L. A practical synthesis of pyrene-4, 5-dione. J. Org. Chem. 1998, 63, 9995–9996. [Google Scholar] [CrossRef]
- Crabtree, R.H. The organometallic chemistry of alkane. Chem. Rev. 1985, 85, 245–269. [Google Scholar] [CrossRef]
- Kakiuchi, F.; Murai, S. Catalytic C–H/olefin coupling. Acc. Chem. Res. 2002, 35, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Bergman, R.G. C–H Activation. Nature 2007, 446, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Engle, K.M.; Wang, D.H.; Yu, J.-Q. Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: Versatility and practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C–H Activation for the construction of C–B bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium-catalysed ligand directed C–H functionalization. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef]
- Sun, C.L.; Li, B.J.; Shi, Z.J. Direct C–H transformation via iron catalysis. Chem. Rev. 2011, 111, 1293–1314. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S. If C–H bonds could talk: Selective C–H bond oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374. [Google Scholar] [CrossRef]
- Neufeldt, S.R.; Sanford, M.S. Controlling site selectivity in palladium-catalysed C–H bond functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef]
- Kuhl, N.; Hopkinson, M.N.; Delord, J.W.; Glorius, F. Beyond directing groups: Transition metal catalysed C–H activation of simple arenes. Angew. Chem. Int. Ed. 2012, 51, 10236–10254. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic functionalization of C(sp2)–H and C(sp3)–H bonds by using bidentate directing groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef] [PubMed]
- Daugulis, O.; Roane, J.; Tran, L.D. Bidentate, monoanionic auxiliary-directed functionalization of carbon-hydrogen bonds. Acc. Chem. Res. 2015, 48, 1053–1064. [Google Scholar] [CrossRef]
- Rit, R.K.; Yadav, M.R.; Ghosh, K.; Sahoo, A.K. Reusable directing groups [8-aminoquinoline, picolinamide, sulfoximine] in C(sp3)–H bond activation: Present and future. Tetrahedron 2015, 71, 4450–4459. [Google Scholar] [CrossRef]
- Castro, L.C.M.; Chatani, N. Nickel catalysts/N,N′-bidentate directing groups: An excellent partnership in directing C–H activation reactions. Chem. Lett. 2015, 44, 410–421. [Google Scholar] [CrossRef]
- Yang, X.; Shan, G.; Wang, L.; Rao, Y. Recent advances in transition metal (Pd, Ni)-catalysed C(sp3)–H bond activation with bidentate directing groups. Tetrahedron Lett. 2016, 57, 819–836. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-catalysed transformation of alkyl C–H bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C–H activation: Toward sustainability and applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
- Saini, G.; Kapur, M. Palladium-catalysed functionalization of acidic and non-acidic C(sp3)–H bonds—Recent advances. Chem. Commun. 2021, 57, 1693–1714. [Google Scholar] [CrossRef]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate directing groups: An efficient tool in C–H bond functionalization chemistry for the expedient construction of C-C bonds. Chem. Rev. 2020, 120, 1788–1887. [Google Scholar] [CrossRef]
- Babu, S.A.; Aggarwal, Y.; Patel, P.; Tomar, R. Diastereoselective palladium-catalyzed functionalization of prochiral C(sp3)–H bonds of aliphatic and alicyclic compounds. Chem. Commun. 2022, 58, 2612–2633. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.K.; Guin, S.; Maiti, S.; Biswas, J.P.; Porey, S.; Maiti, D. Toolbox for distal C–H bond functionalizations in organic molecules. Chem. Rev. 2022, 122, 5682–5841. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.M.; Dong, L.; Duckett, S.B.; Partridge, M.G.; Jones, W.D.; Perutz, R.N. Control of η2–coordination vs C–H bond activation by rhodium. J. Am. Chem. Soc. 1993, 115, 7685–7695. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Chen, Y.; Liu, J.; Fang, Q.; Kleeberg, C.; Marder, T.B. Ir-catalysed direct borylation at the 4-position of pyrene. J. Org. Chem. 2012, 77, 7124–7128. [Google Scholar] [CrossRef]
- Ren, H.; Zhou, Y.P.; Bai, Y.; Cui, C.; Driess, M. Cobalt-catalyzed regioselective borylation of arenes: N-heterocyclic silylene as an electron donor in the metal-mediated activation of C−H bonds. Chem.-Eur. J. 2017, 23, 5663–5667. [Google Scholar] [CrossRef]
- Murai, M.; Takami, K.; Takai, K. Iridium-catalyzed intermolecular dehydrogenative silylation of polycyclic aromatic compounds without directing groups. Chem.-Eur. J. 2015, 21, 4566. [Google Scholar] [CrossRef]
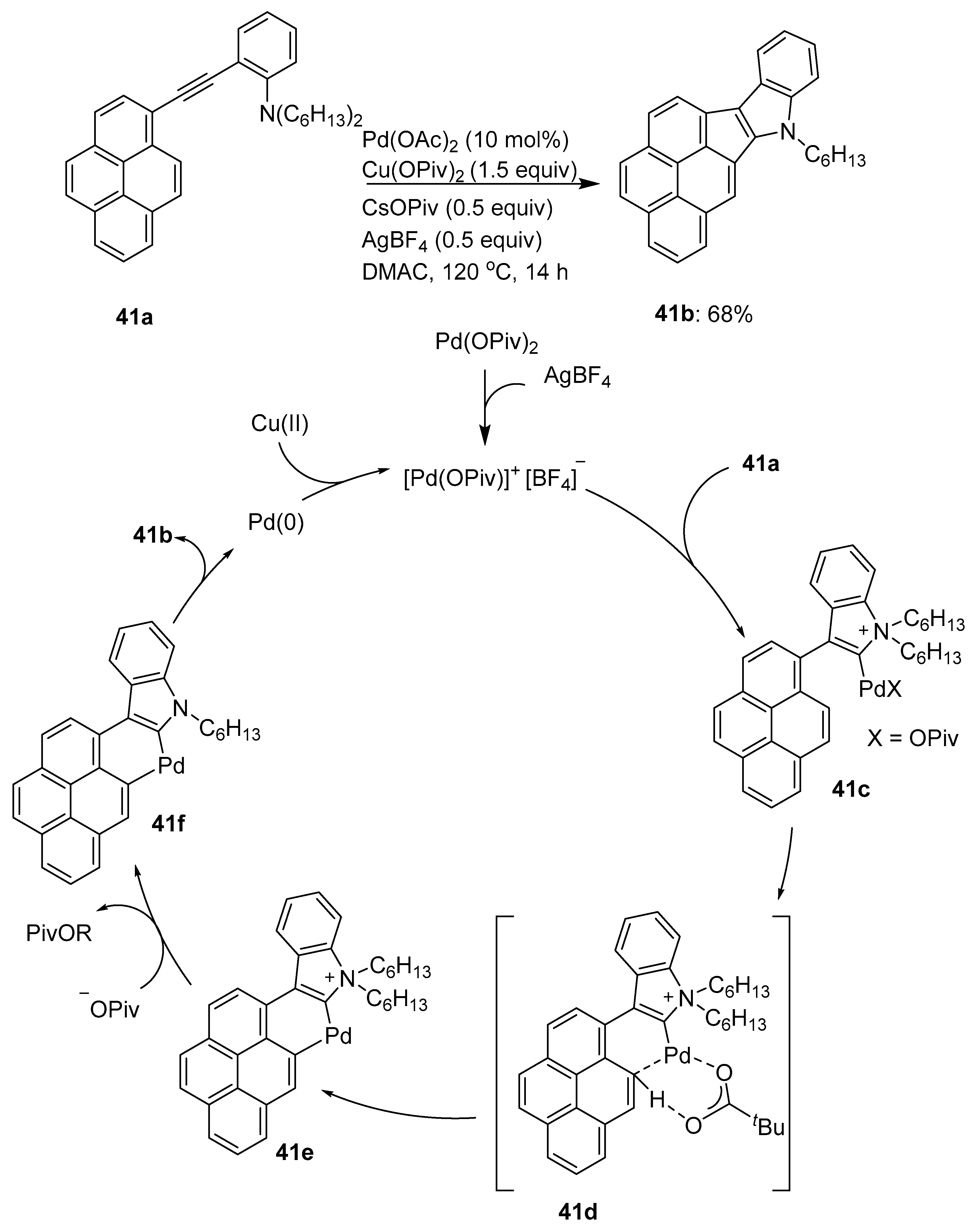
- Mochida, K.; Kawasumi, K.; Segawa, Y.; Itami, K. Direct arylation of polycyclic aromatic hydrocarbons through palladium catalysis. J. Am. Chem. Soc. 2011, 133, 10716–10719. [Google Scholar] [CrossRef]
- Kawasumi, K.; Mochida, K.; Kajino, T.; Segawa, Y.; Itami, K. Pd(OAc)2/o-chloranil/M(OTf)n: A catalyst for the direct C–H arylation of polycyclic aromatic hydrocarbons with boryl-, silyl-, and unfunctionalized arenes. Org. Lett. 2012, 14, 418–421. [Google Scholar] [CrossRef]
- Collins, K.D.; Honeker, R.; Vasquez-Cespedes, S.; Tang, D.-T.D.; Glorius, F. C–H Arylation of triphenylene, naphthalene and related arenes using Pd/C. Chem. Sci. 2015, 6, 1816–1824. [Google Scholar] [CrossRef]
- Funaki, K.; Kawai, H.; Sato, T.; Oi, S. Palladium-catalysed direct C–H bond arylation of simple arenes with aryltrimethylsilanes. Chem. Lett. 2011, 40, 1050–1052. [Google Scholar] [CrossRef]
- Dixit, S.; Siddiqui, Q.T.; Muneer, M.; Agarwal, N. Ferrocene catalysed C–H arylation of arenes and reaction mechanism study using cyclic voltammetry. Tetrahedron Lett. 2016, 57, 4228–4231. [Google Scholar] [CrossRef]
- Morofuji, T.; Shimizu, A.; Yoshida, J.-i. Metal- and chemical-oxidant-free C–H/C–H cross-coupling of aromatic compounds: The use of radical-cation pools. Angew. Chem. Int. Ed. 2012, 51, 7259–7262. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.S.; Ghosh, I.; König, B. Direct C–H phosphonylation of electron-rich arenes and heteroarenes by visible-light photoredox catalysis. Chem. Eur. J. 2017, 23, 12120–12124. [Google Scholar] [CrossRef]
- Murai, M.; Nishinaka, N.; Kimura, M.; Takai, K. Regioselective functionalization of 9,9-dimethyl-9-silafluorenes by borylation, bromination, and nitration. J. Org. Chem. 2019, 84, 5667–5676. [Google Scholar] [CrossRef]
- Yip, S.J.; Kawakami, T.; Murakami, K.; Itami, K. Gold-catalyzed C−H imidation of polycyclic aromatic hydrocarbons. Asian J. Org. Chem. 2018, 7, 1372–1375. [Google Scholar] [CrossRef]
- Nowicka, E.; Hickey, N.W.; Sankar, M.; Jenkis, R.L.; Knight, D.H.; Willock, D.J.; Hutchings, G.J.; Francisco, M.; Taylor, S.H. Mechanistic insights into selective oxidation of polyaromatic compounds using RICO chemistry. Chem.-Eur. J. 2018, 24, 12359–12369. [Google Scholar] [CrossRef]
- Gugulothu, V.; Ahemed, J.; Subburu, M.; Yadagiri, B.; Mittal, R.; Prabhakar, C.; Pola, S. Evolution of physical and photocatalytic properties of Zn(II) and Ru(II) complexes. Polyhedron 2019, 170, 412–423. [Google Scholar] [CrossRef]
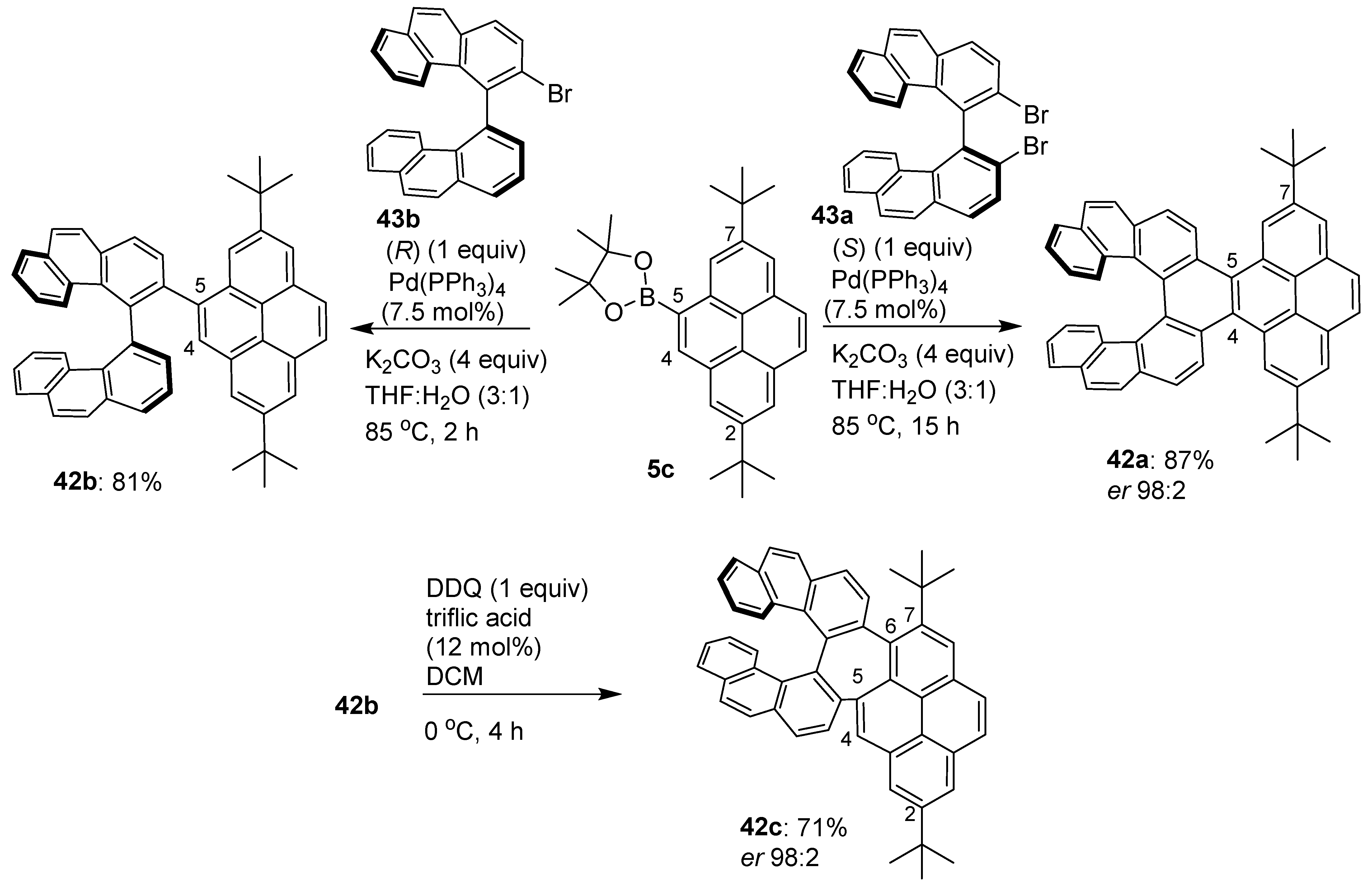
- Elbert, S.M.; Haidish, A.; Kirshbaum, T.; Rominger, F.; Zschieschang, U.; Kliauk, H.; Mastalerz, M. 2,7,11,16-Tetra-tert-butyl tetraindenopyrene revisited by “inverse” synthetic approach. Chem.-Eur. J. 2020, 26, 10585–10590. [Google Scholar] [CrossRef]
- Odani, R.; Hirano, K.; Satoh, T.; Miura, M. Copper-mediated dehydrogenative biaryl coupling of naphthylamines and 1,3-azoles. J. Org. Chem. 2013, 78, 11045–11052. [Google Scholar] [CrossRef]
- Shabashov, D.; Daugulis, O. Auxiliary-assisted palladium-catalyzed arylation and alkylation of sp2 and sp3 carbon−hydrogen bonds. J. Am. Chem. Soc. 2010, 132, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Han, B.; Chen, Z.; Zhang, X.; He, G.; Chen, G. Construction of natural-product-like cyclophane-braced peptide macrocycles via sp3 C–H arylation. J. Am. Chem. Soc. 2019, 141, 9401–9407. [Google Scholar] [CrossRef] [PubMed]
- Parella, R.; Babu, S.A. Pd(II)-Catalyzed, picolinamide-assisted, Z-selective γ-arylation of allylamines to construct Z-cinnamylamines. J. Org. Chem. 2017, 82, 6550–6567. [Google Scholar] [CrossRef] [PubMed]
- Nadres, E.T.; Santos, G.I.F.; Shabashov, D.; Daugulis, O. Scope and limitations of auxiliary-assisted, palladium-catalyzed arylation and alkylation of sp2 and sp3 C–H bonds. J. Org. Chem. 2013, 78, 9689–9714. [Google Scholar] [CrossRef] [PubMed]
- Bisht, N.; Singh, P.; Babu, S.A. Pd(II)-Catalyzed, picolinamide-aided γ-(sp2)-C–H functionalization of racemic and enantiopure α-methylbenzylamine and phenylglycinol scaffolds. Synthesis 2022, 54, 4059–4094. [Google Scholar] [CrossRef]
- Aggarwal, Y.; Padmavathi, R.; Singh, P.; Babu, S.A. Pd(II)-Catalyzed, γ-C(sp2)−H alkoxylation in α-methylbenzylamine, phenylglycinol, 3-amino-3-phenylpropanol toward enantiopure aryl alkyl ethers. Asian J. Org. Chem. 2022, 11, e202200327. [Google Scholar] [CrossRef]
- Narang, U.; Singh, P.; Babu, S.A. Construction of β-phenylalanine derivatives through Pd-catalyzed, C(sp2)−H (ortho) functionalization. Eur. J. Org. Chem. 2023, 26, e202300463. [Google Scholar] [CrossRef]
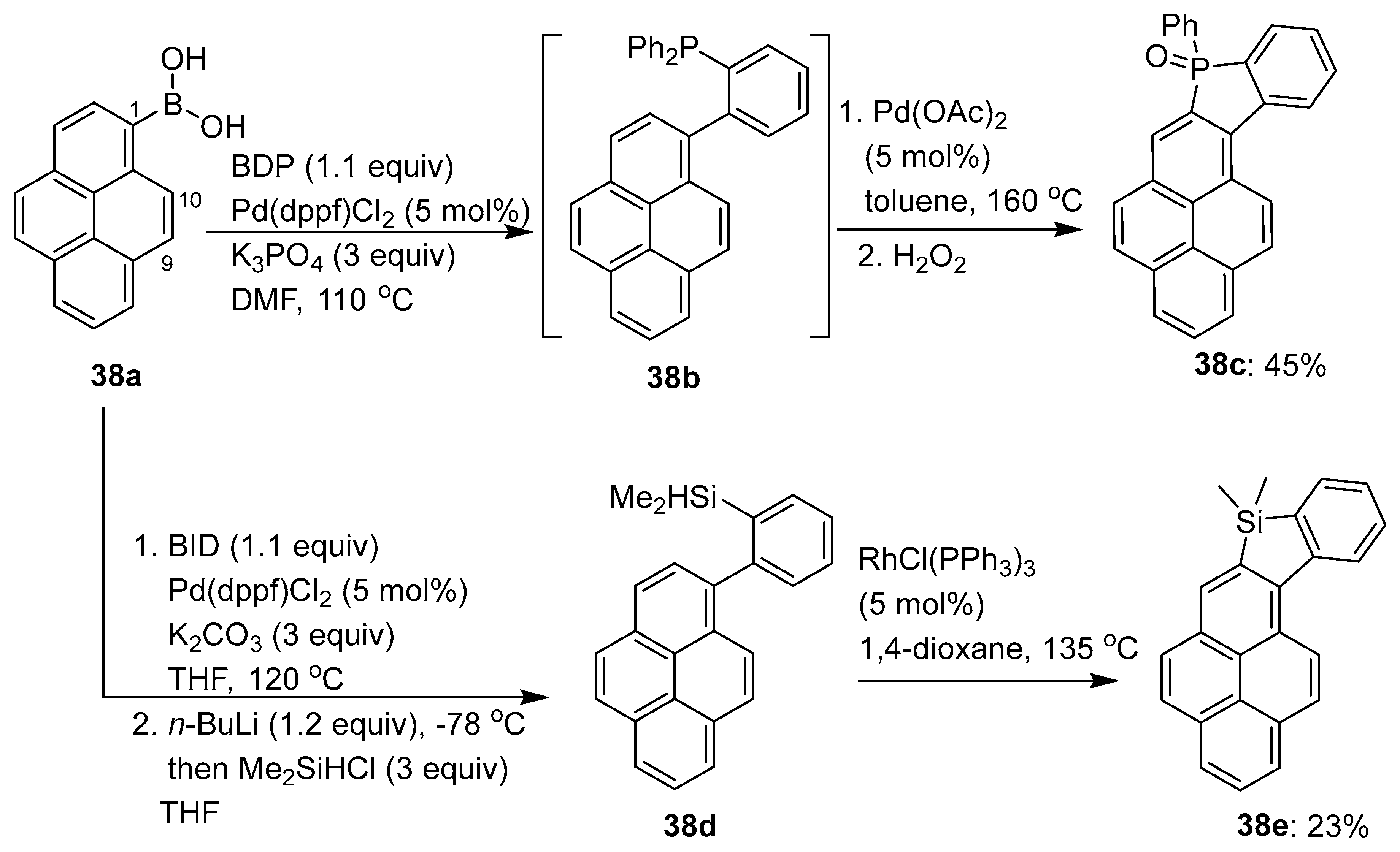
- Dalal, A.; Babu, S.A. Pd(II)-Catalyzed directing-group-aided C–H arylation and alkylation of pyrene core: Synthesis of C1,C2- and C1,C10-disubstituted pyrene motifs. Synthesis 2021, 53, 3307–3324. [Google Scholar]
- Iwasaki, M.; Kaneshika, W.; Tsuchiya, Y.; Nakajima, K.; Nishihara, Y. Palladium-catalysed peri-selective chalcogenation of naphthylamines with diaryl disulfides and diselenides via C–H bond cleavage. J. Org. Chem. 2014, 79, 11330–11338. [Google Scholar] [CrossRef]
- Pradhan, S.; De, P.B.; Punniyamurthy, T. Copper(II)-mediated chelation-assisted regioselective N-naphthylation of indoles, pyrazoles and pyrrole through dehydrogenative cross-coupling. J. Org. Chem. 2017, 82, 4883–4890. [Google Scholar] [CrossRef]
- Roy, S.; Pradhan, S.; Punniyamurthy, T. Copper-mediated regioselective C–H etherification of naphthylamides with arylboronic acids using water as an oxygen source. Chem. Commun. 2018, 54, 3899–3902. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Chatani, N. Rhodium(I)-catalyzed C8-alkylation of 1-naphthylamide derivatives with alkenes through a bidentate picolinamide chelation system. ACS Catal. 2018, 8, 6699–6706. [Google Scholar] [CrossRef]
- Ying, J.; Fu, L.Y.; Zhong, G.; Wu, X.F. Cobalt-catalyzed direct carbonylative synthesis of free (NH)-benzo[cd]indol-2(1H)-ones from naphthylamides. Org. Lett. 2019, 21, 5694–5698. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, M.; Wang, C.; Yang, Z.; Huang, X.; Feng, R.; Qi, C. Cobalt(II)-catalyzed hydroarylation of 1,3-diynes and internal alkynes with picolinamides promoted by alcohol. Chem. Commun. 2020, 56, 14231–14234. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Roy, S.; Banerjee, S.; De, P.B.; Punniyamuthy, T. Oxidative C−H/N−H annulation of aromatic amides with dialkyl malonates: Access to isoindolinones and dihydrobenzoindoles. J. Org. Chem. 2020, 85, 5741–5749. [Google Scholar] [CrossRef]
- Kona, C.N.; Oku, R.; Nishii, Y.; Miura, M. peri-Selective direct acylmethylation and amidation of naphthalene derivatives using iridium and rhodium catalysts. Synthesis 2021, 53, 3126–3136. [Google Scholar]
- Sire, C.; Cattey, H.; Tsivery, A.; Hierso, J.-C.; Roger, J. Phosphorus-directed rhodium-catalyzed C−H arylation of 1-pyrenylphosphines selective at the K-region. Adv. Synth. Catal. 2022, 364, 440–452. [Google Scholar] [CrossRef]
- Asako, S.; Ilies, L.; Nakamura, E. Iron-catalyzed ortho-allylation of aromatic carboxamides with allyl ethers. J. Am. Chem. Soc. 2013, 135, 17755–17757. [Google Scholar] [CrossRef]
- Just-Baringo, X.; Shin, Y.; Panigrahi, A.; Zarattini, M.; Nagyte, V.; Zhao, L.; Kostarelos, K.; Casiraghi, C.; Larrosa, I. Palladium catalysed C–H arylation of pyrenes: Access to a new class of exfoliating agents for water-based grapheme dispersions. Chem. Sci. 2020, 11, 2472–2478. [Google Scholar] [CrossRef]
- He, Y.Q.; Zhong, Y.W. The synthesis of 2- and 2,7-functionalized pyrene derivatives through Ru(II)-catalyzed C–H activation. Chem. Commun. 2015, 51, 3411–3414. [Google Scholar] [CrossRef]
- Niu, Y.; Qi, Z.; Lou, Q.; Bai, P.; Yang, S. Copper-catalyzed arylation of polycyclic aromatic hydrocarbons by the P=O group. Chem. Commun. 2020, 56, 14721–14724. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Zhang, M.; Fu, Z.; Lan, J.; Wu, D.; You, J. Copper-catalysed remote C–H arylation of polycyclic aromatic hydrocarbons (PAHs). Beilstein J. Org. Chem. 2020, 16, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, A.; Szűcs, R.; Caytan, E.; Roisnel, T.; Dorcet, V.; Bouit, A.P.; Nyulászi, L.; Hissler, M. Synthesis, optical, and redox properties of regioisomeric benzoheterocycles-fused pyrene. J. Org. Chem. 2019, 84, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Baba, K.; Tobisu, M.; Chatani, N. Palladium-catalyzed direct synthesis of phosphole derivatives from triarylphosphines through cleavage of carbon−hydrogen and carbon-phosphorus bonds. Angew. Chem. 2013, 125, 12108–12111. [Google Scholar] [CrossRef]
- Ureshino, T.; Yoshida, T.; Kuninobu, Y.; Takai, K. Rhodium-catalyzed synthesis of silafluorene derivatives via cleavage of silicon−hydrogen and carbon−hydrogen bonds. J. Am. Chem. Soc. 2010, 132, 14324–14326. [Google Scholar] [CrossRef]
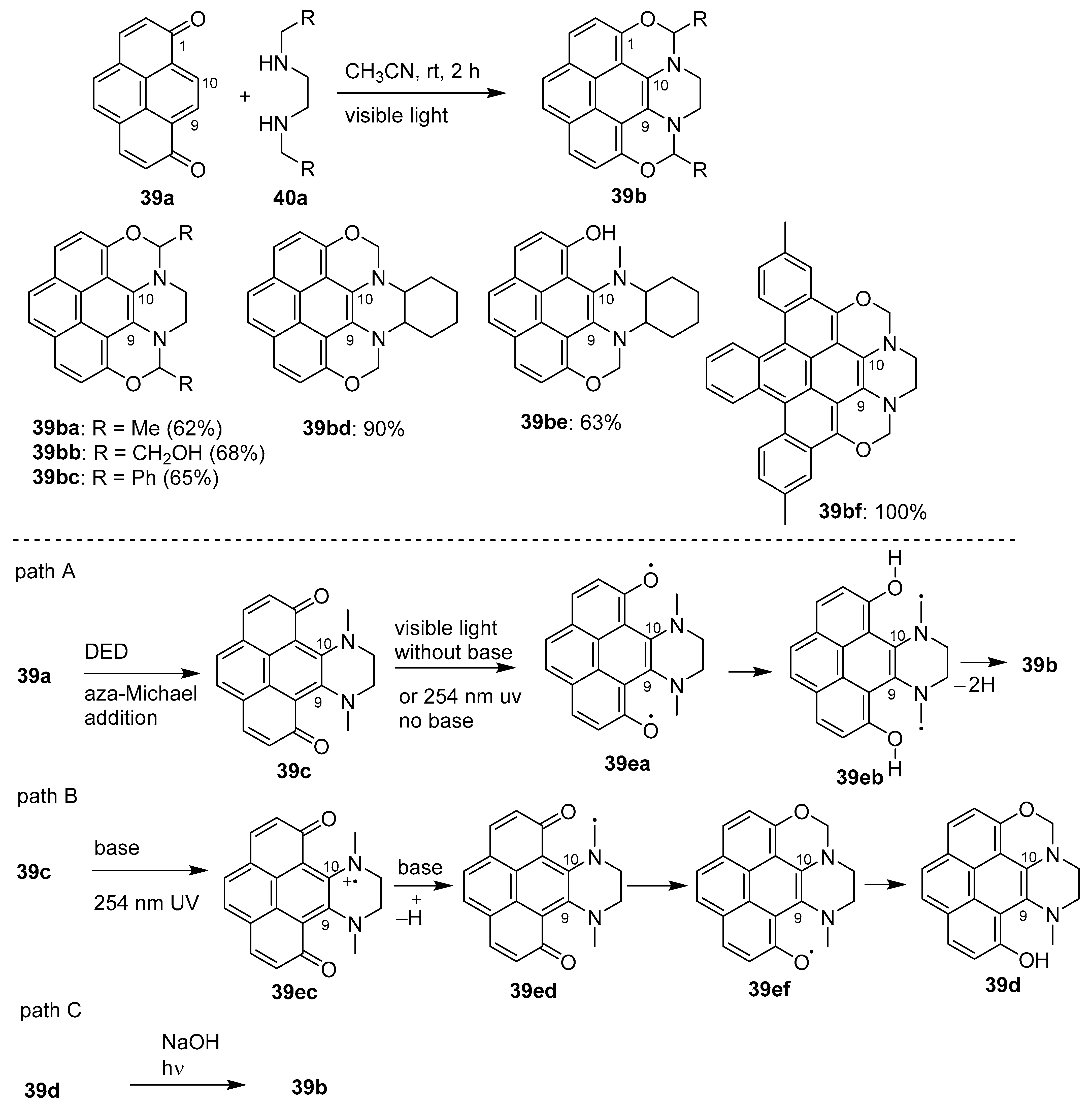
- Yao, W.; Sun, M.; Zhang, Y.; Yanh, H.; Liang, D.; Wu, R.; Wong, M.W.; Huang, D. Photo-induced C–H bond activation of N,N′-dialkylethylenediamine upon aza-Michael addition to1,8-pyrenedione: Facile synthesis of fluorescent pyrene derivatives. Org. Chem. Front. 2018, 5, 1679–1683. [Google Scholar] [CrossRef]
- Jin, T.; Suzuki, S.; Ho, H.E.; Mastsuyama, H.; Kawata, M.; Terada, M. Pd-catalyzed indolization/peri-C−H annulation/N-dealkylation cascade to cyclopenta-fused acenaphtho[1,2-b]indole scaffold. Org. Lett. 2021, 23, 9431–9435. [Google Scholar] [CrossRef]
- Swain, K.A.; Radacki, K.; Braunschweig, H.; Ravat, P. Pyrene-fused [7]helicenes connected via hexagonal and heptagonal rings: Stereospecific synthesis and chiroptical properties. J. Org. Chem. 2022, 87, 993–1000. [Google Scholar] [CrossRef]
- Yan, J.; Paulis, A.P.; Perry, G.J.P.; Procter, D.J. Metal-Free Synthesis of benzothiophenes by twofold C−H functionalization: Direct access to materials-oriented heteroaromatics. Angew. Chem. Int. Ed. 2019, 58, 15675–15679. [Google Scholar] [CrossRef]
- Han, Y.F.; Li, H.; Hu, P.; Jin, G.X. Alkyne insertion induced regiospecific C–H activation with [Cp*MCl2]2 (M = Ir, Rh). Organometallics 2011, 30, 905–911. [Google Scholar] [CrossRef]
- Dinda, S.; Patra, S.C.; Roy, S.; Halder, S.; Weyhermüller, T.; Pramanik, K.; Ganguly, S. Coligand driven diverse organometallation in benzothiazolyl-hydrazone derivatized pyrene: Ortho vs. peri C–H activation. New J. Chem. 2020, 44, 1407–1417. [Google Scholar] [CrossRef]
- Dinda, S.; Patra, S.C.; Samanta, T.; Basu, A.; Pramanik, K.; Ganguly, S. Rhodium assisted peri-C–H activation in benzothiazolyl-hydrazone derivatized pyrene. Polyhedron 2020, 179, 114352. [Google Scholar] [CrossRef]







































{kind=link}
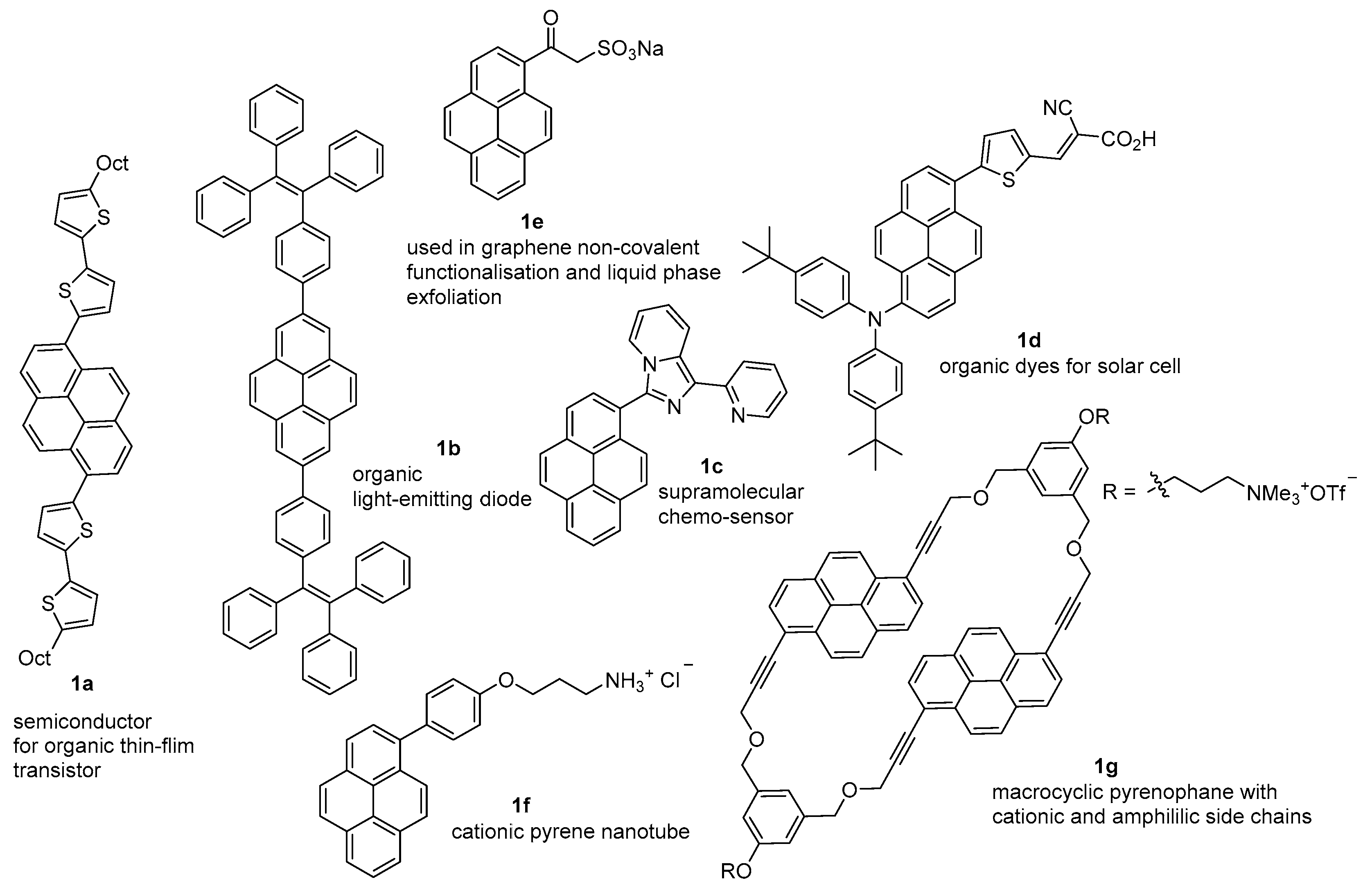{kind=link}
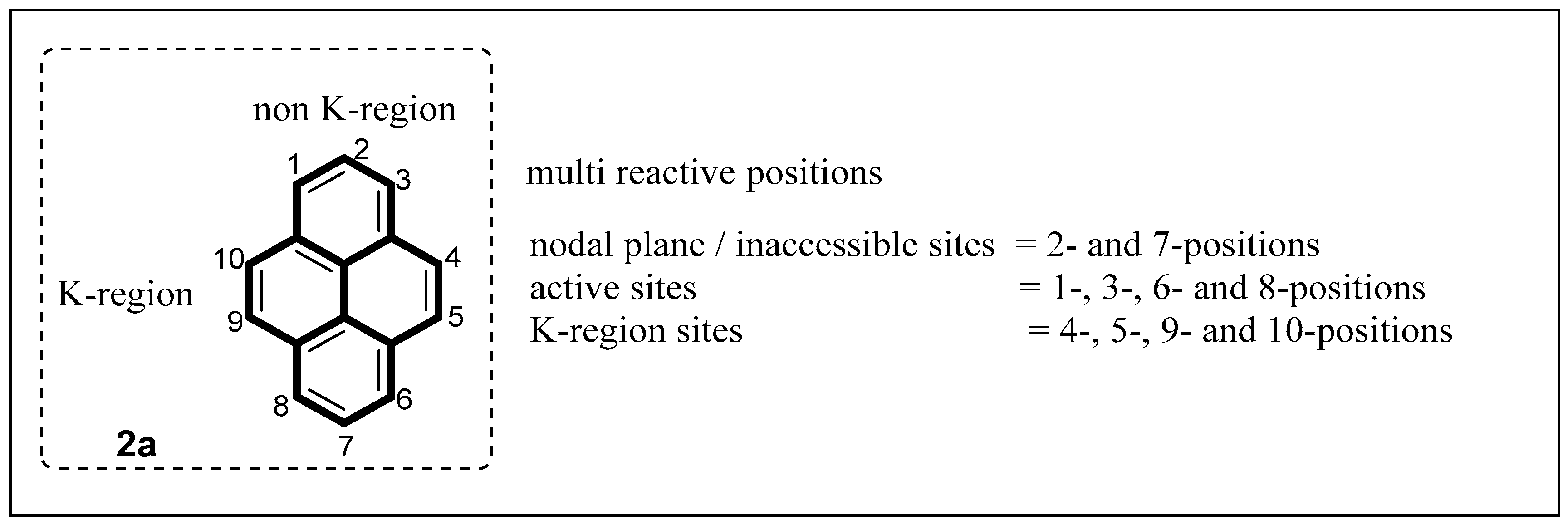{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arulananda Babu, S.; Dalal, A.; Bodak, S. Recent Advances in C–H Functionalization of Pyrenes. Chemistry 2023, 5, 2713-2755. https://doi.org/10.3390/chemistry5040175
Arulananda Babu S, Dalal A, Bodak S. Recent Advances in C–H Functionalization of Pyrenes. Chemistry. 2023; 5(4):2713-2755. https://doi.org/10.3390/chemistry5040175
Chicago/Turabian StyleArulananda Babu, Srinivasarao, Arup Dalal, and Subhankar Bodak. 2023. "Recent Advances in C–H Functionalization of Pyrenes" Chemistry 5, no. 4: 2713-2755. https://doi.org/10.3390/chemistry5040175