
Stereoselective Synthesis of a Novel Series of Dispiro-oxindolopyrrolizidines Embodying Thiazolo[3,2-a]benzimidazole Motif: A Molecular Electron Density Theory Study of the Mechanism of the [3 + 2] Cycloaddition Reaction

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
- Synthesis of 1H-benzo[d]imidazole-2-thiol 2
- Synthesis of compound (3) and acetyl derivative (4)
- 1-(3-Methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)ethan-1-one 4

- Synthesis of 3-hydroxy-3-(2-(3-methylbenzo[4,5]imidazo[2,1-b] thiazol-2-yl)-2-oxoethyl)indolin-2-one (6)
- 3-Hydroxy-3-(2-(3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)-2-oxoethyl)indolin-2-one 6

- Synthesis of 3-(2-(3-methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)-2-oxoethylidene)indolin-2-one (7)
- 3-(2-(3-Methylbenzo[4,5]imidazo[2,1-b]thiazol-2-yl)-2-oxoethylidene)indolin-2-one 7

- General procedure for synthesis of spiro compounds (9a–d)
- (3S,6′R,7′S)-7′-(3-Methylbenzo[4,5]imidazo[2,1-b]thiazole-2-carbonyl)-7’,7a′-dihydro-1′H,3′H-dispiro[indoline-3,5′-pyrrolo[1,2-c]thiazole-6′,3″-indoline]-2,2″-dione 9a

- (3S,6′R,7′S)-5-Chloro-7′-(3-methylbenzo[4,5]imidazo[2,1-b]thiazole-2-carbonyl)-7′,7a′-dihydro-1′H,3′H-dispiro[indoline-3,5′-pyrrolo[1,2-c]thiazole-6′,3″-indoline]-2,2″-dione 9b

- (3S,6′R,7′S)-7′-(3-Methylbenzo[4,5]imidazo[2,1-b]thiazole-2-carbonyl)-5-nitro-7′,7a′-dihydro-1′H,3′H-dispiro[indoline-3,5′-pyrrolo[1,2-c]thiazole-6′,3″-indoline]-2,2″-dione 9c

- (3S,6′R,7′S)-5-Bromo-7′-(3-methylbenzo[4,5]imidazo[2,1-b]thiazole-2-carbonyl)-7′,7a′-dihydro-1′H,3′H-dispiro[indoline-3,5′-pyrrolo[1,2-c]thiazole-6′,3″-indoline]-2,2″-dione 9d

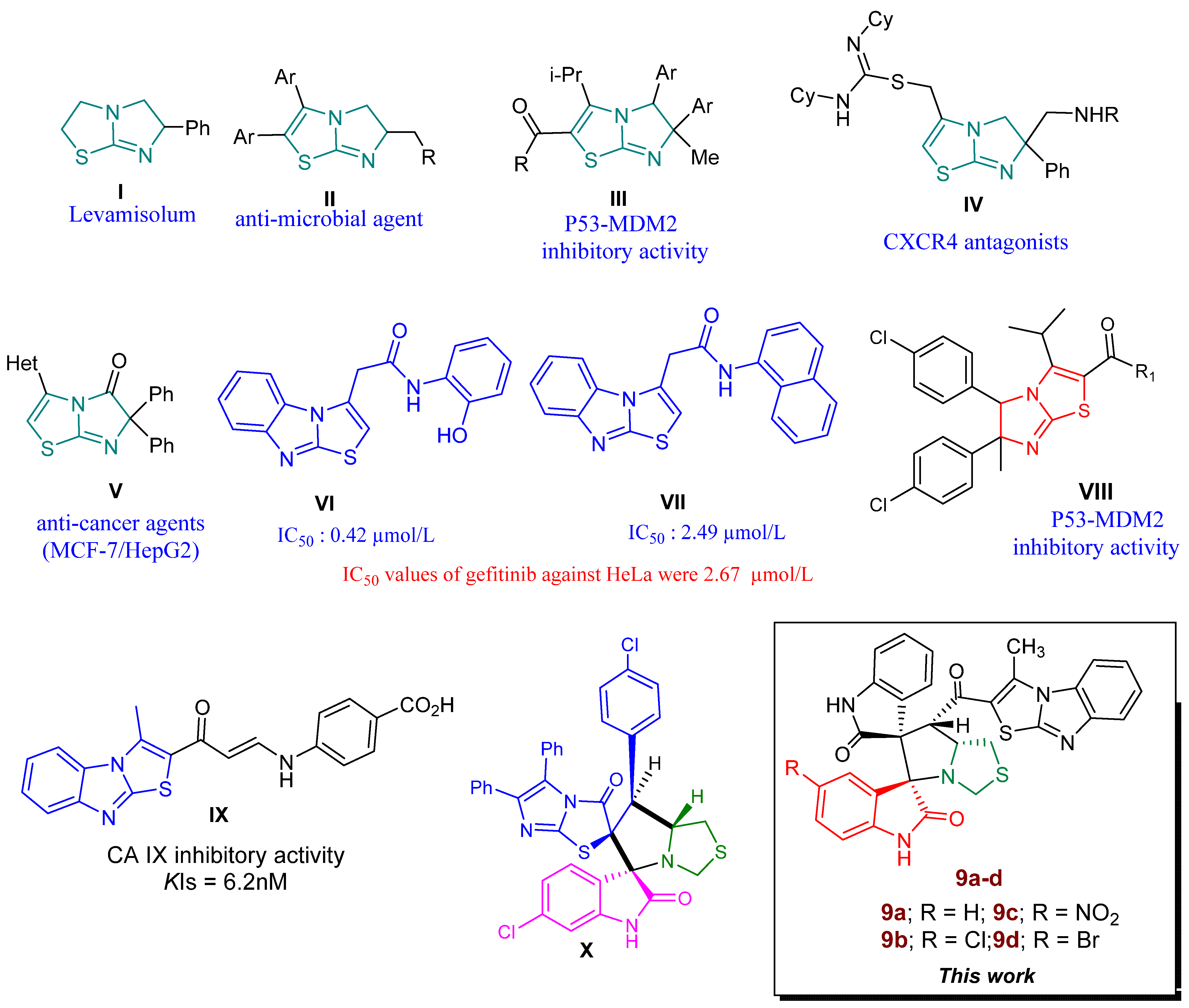
3. Results and Discussion
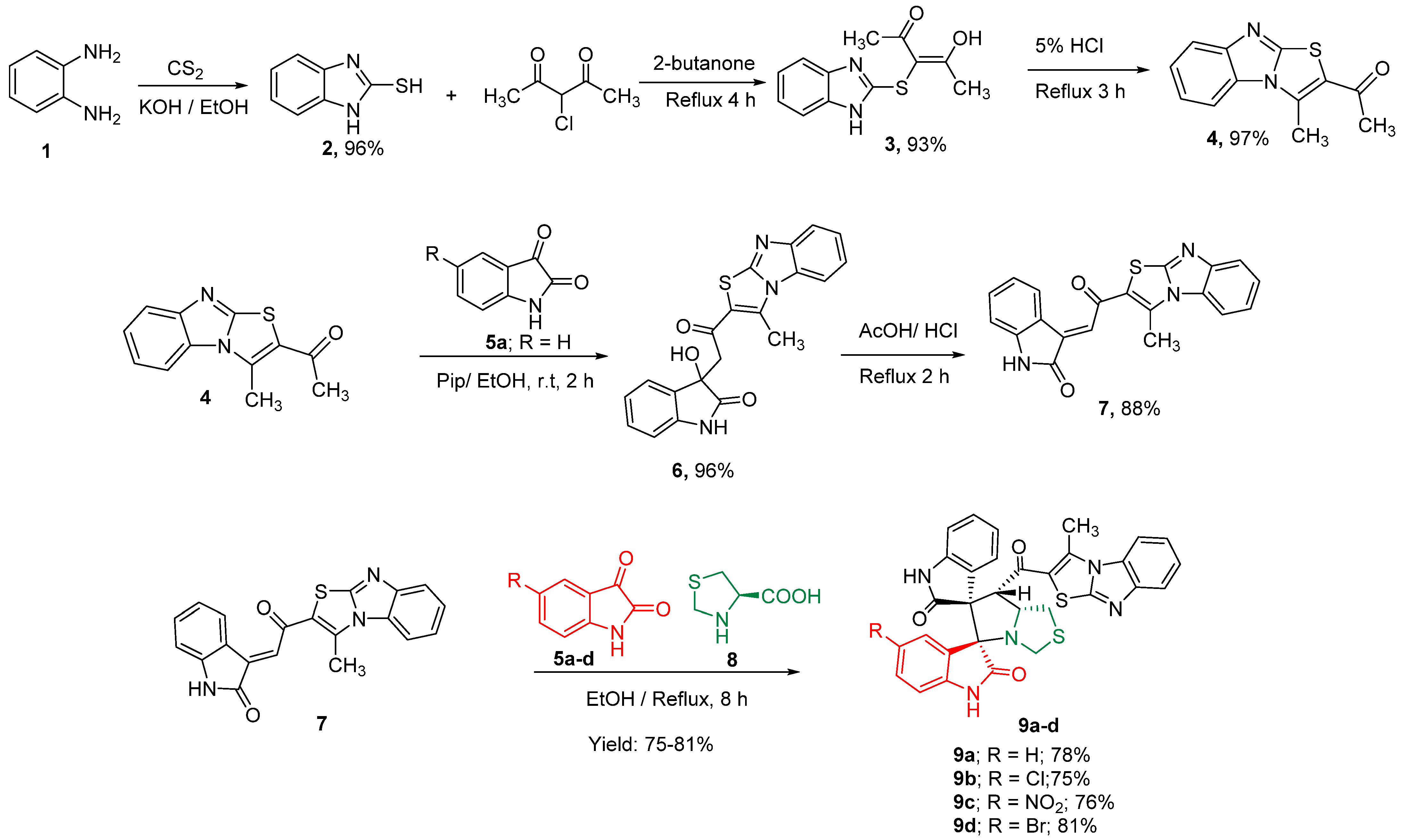
3.1. Synthesis of Spiro Compounds (9a–d)
3.1.1. Synthesis of Acetyl Derivative (4)
3.1.2. Synthesis of Spiro Compounds (9a–d)
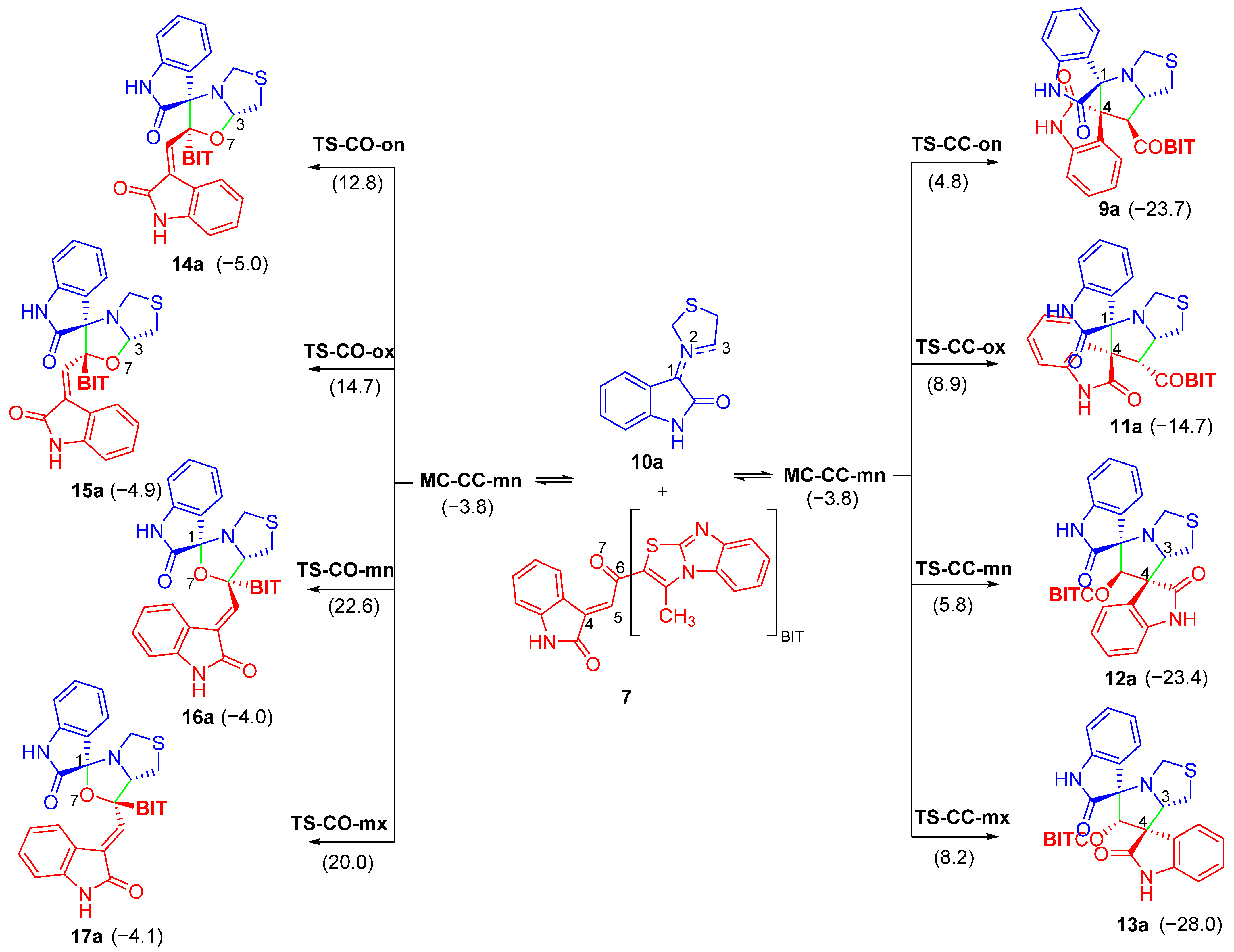
3.2. MEDT Study of the 32CA Reaction between AY 10a and Chalcone 7
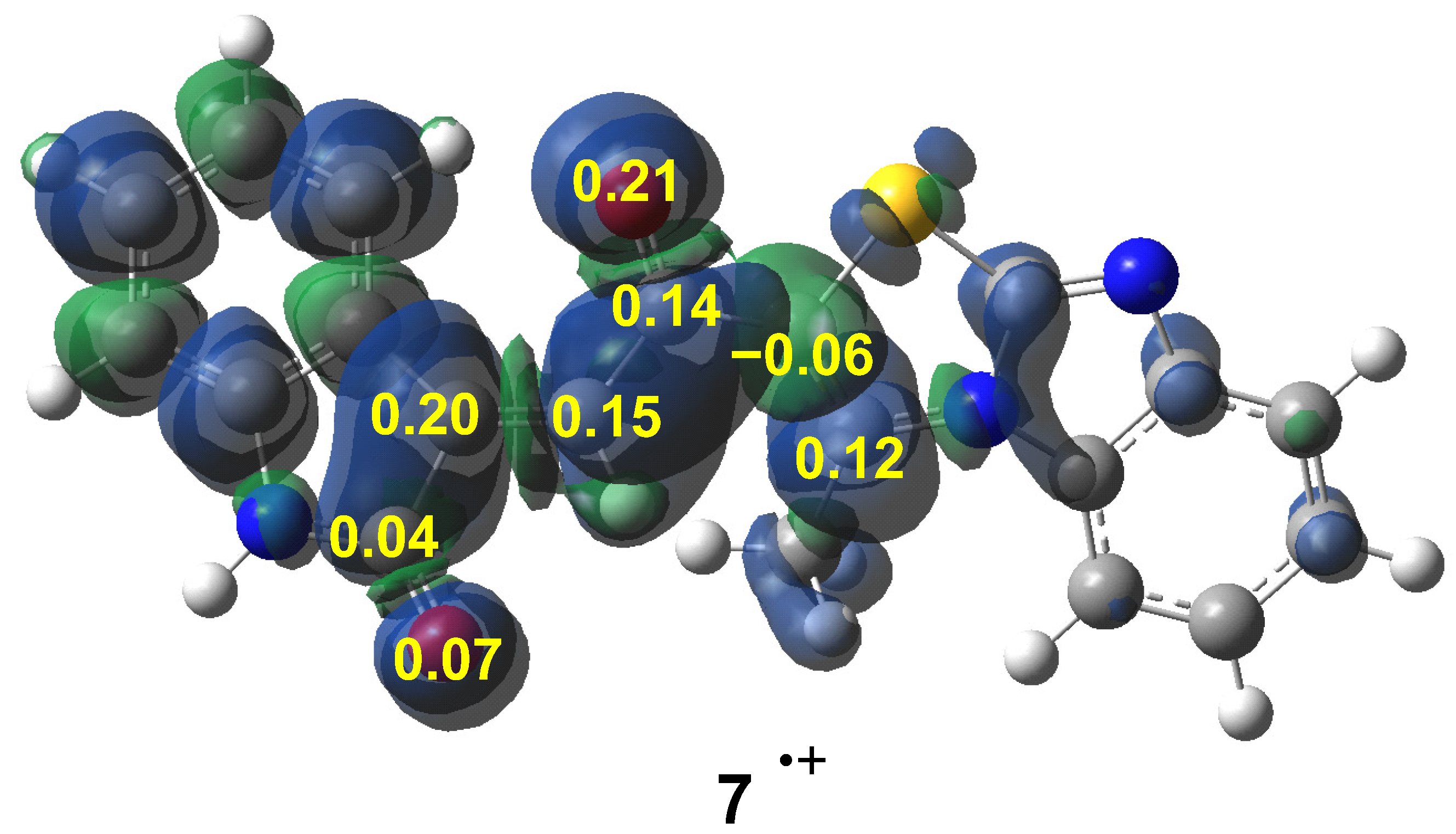
3.2.1. Analysis of Reactivity Indicators
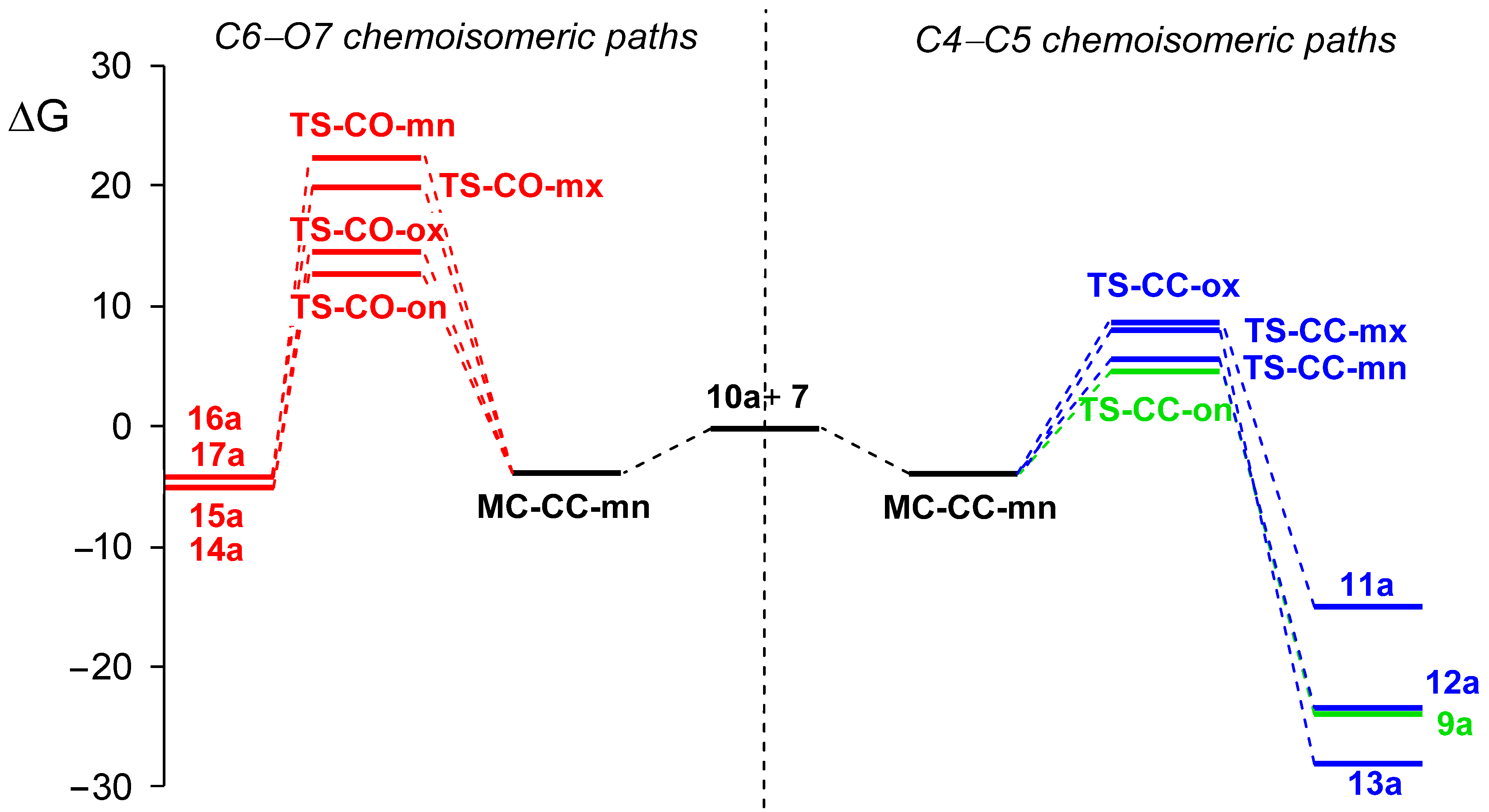
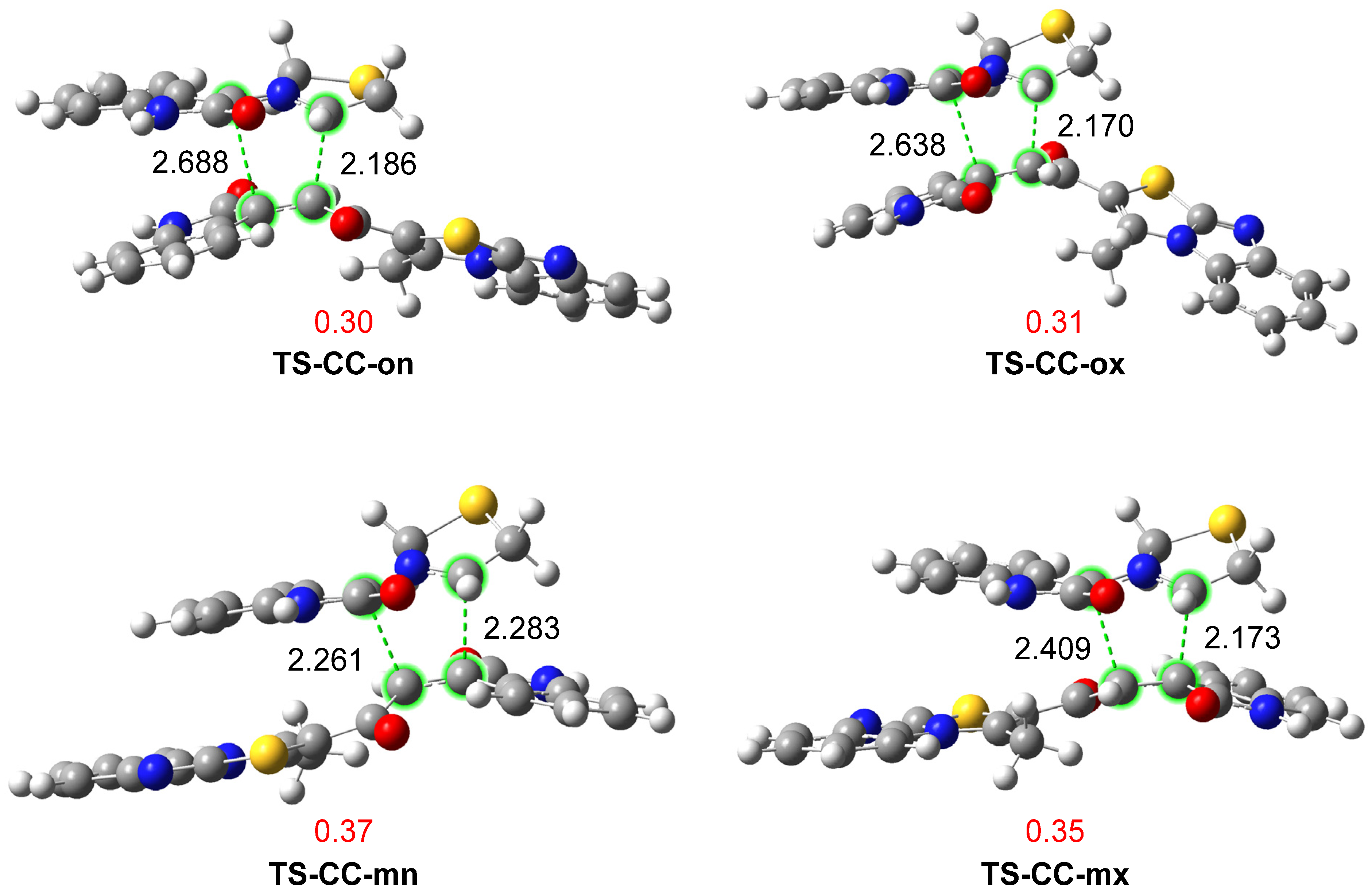
3.2.2. Study of the Competitive Reaction Paths
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.J. Heterocyclic Chemistry in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Shareef, M.A.; Khan, I.; Babu, B.N.; Kamal, A. A Comprehensive review on the therapeutic versatility of imidazo[2,1-b]thiazoles. Curr. Med. Chem. 2020, 27, 6864–6887. [Google Scholar] [CrossRef] [PubMed]
- Çapan, G.; Ulusoy, N.; Ergenç, N.; Kiraz, M. New 6-phenylimidazo[2,1-b]thiazole derivatives: Synthesis and antifungal activity. Monatsh. Chem. 1999, 130, 1399–1407. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Petri, G.L.; Cusimano, M.G.; Schillaci, D.; DI Sarno, V.; Musella, S.; Giovannetti, E.; Cirrincione, G.; Diana, P. 2,6-Disubstituted imidazo[2,1-b][1,3,4]thiadiazole derivatives as potent staphylococcal biofilm inhibitors. Eur. J. Med. Chem. 2019, 167, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bionda, N.; Fleeman, R.; Wang, H.; Ozawa, A.; Houghten, R.A.; Shaw, L. Identification of 5,6-dihydroimidazo[2,1-b]thiazoles as a new class of antimicrobial agents. Bioorg. Med. Chem. 2016, 24, 5633–5638. [Google Scholar] [CrossRef] [PubMed]
- Sbenati, R.M.; Semreen, M.H.; Semreen, A.M.; Shehata, M.K.; Alsaghir, F.M.; El-Gamal, M.I. Evaluation of imidazo[2,1–b]thiazole-based anticancer agents in one decade (2011–2020): Current status and future prospects. Bioorg. Med. Chem. 2021, 29, 115897. [Google Scholar] [CrossRef] [PubMed]
- Bașoğ lu, F.; Ulusoy-Güzeldemirci, N.; Akalın-Çiftçi, G.; Çetinkaya, S.; Ece, A. Novel imidazo[2,1-b] thiazole-based anticancer agents as potential focal adhesion kinase inhibitors: Synthesis, in silico, and in vitro evaluation. Chem. Biol. Drug. Des. 2021, 98, 270–282. [Google Scholar] [CrossRef]
- Potikha, L.M.; Brovarets, V.S. Synthesis of imidazo[2,1-b][1,3] thiazoles–potential anticancer agents derived from γ-bromodipnones. Chem. Heterocycl. Compd. 2020, 56, 1073–1077. [Google Scholar] [CrossRef]
- Miyazaki, M.; Naito, H.; Sugimoto, Y.; Kawato, H.; Okayama, T.; Shimizu, H.; Miyazaki, M.; Kitagawa, M.; Seki, T.; Fukutake, S.; et al. Lead optimization of novel p53-MDM2 interaction inhibitors possessing dihydroimidazothiazole scaffold. Bioorg. Med. Chem. Lett. 2013, 23, 728–732. [Google Scholar] [CrossRef]
- Mona, C.E.; Besserer-Offroy, É.; Cabana, J.; Leduc, R.; Lavigne, P.; Heveker, N.; Marsault, É.; Escher, E. Design, synthesis, and biological evaluation of CXCR4 ligands. Org. Biomol. Chem. 2016, 14, 10298–10311. [Google Scholar] [CrossRef]
- Alkhaldi, A.A.M.; Al-Sanea, M.M.; Nocentini, A.; Eldehna, W.M.; Elsayed, Z.M.; Bonardi, A.; Abo-Ashour, M.F.; El-Damasy, A.K.; Abdel-Maksoud, M.S.; Al-Warhi, T.; et al. 3-Methylthiazolo[3,2-a] benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: Design, synthesis, biological and molecular modeling studies. Eur. J. Med. Chem. 2020, 207, 112745. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Hassab, M.A.; Abo-Ashour, M.F.; Al-Warhi, T.; Elaasser, M.M.; Safwat, N.A.; Suliman, H.; Ahmed, M.F.; Al-Rashood, S.T.; Abdel-Aziz, H.A.; et al. Development of isatin-thiazolo[3,2-a] benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations. Bioorg. Chem. 2021, 110, 104748. [Google Scholar] [CrossRef] [PubMed]
- Kamat, V.; Yallur, B.C.; Poojary, B.; Patil, V.B.; Nayak, S.P.; Krishna, P.M.; Joshi, S.D. Synthesis, molecular docking, antibacterial, and anti-inflammatory activities of benzimidazole-containing tricyclic systems. J. Chin. Chem. Soc. 2021, 68, 1055–1066. [Google Scholar] [CrossRef]
- Ali, A.M.; Tawfik, S.S.; Mostafa, A.S.; Massoud, M.A.M. Benzimidazole-based protein kinase inhibitors: Current perspectives in targeted cancer therapy. Chem. Biol. Drug Des. 2022, 100, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Altowyan, M.S.; Soliman, S.M.; Haukka, M.; Al-Shaalan, N.H.; Alkharboush, A.A.; Barakat, A. [3+2] Cycloaddition Reaction for the Stereoselective Synthesis of a New Spirooxindole Compound Grafted Imidazo[2,1-b]thiazole Scaffold: Crystal Structure and Computational Study. Crystals 2022, 12, 5. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Ghawas, H.M.; Al-Majid, A.M.; Elsenduny, F.; Badria, F.A.; Elshaier, Y.; Ghabbour, H.A. Design and synthesis of new substituted spirooxindoles as potential inhibitors of the MDM2–p53 interaction. Bioorg. Chem. 2019, 86, 598–608. [Google Scholar] [CrossRef]
- Altowyan, M.S.; Barakat, A.; Al-Majid, A.M.; Al-Ghulikah, H. Spiroindolone analogues bearing benzofuran moiety as a selective cyclooxygenase COX-1 with TNF-α and IL-6 inhibitors. Saudi J. Biol. Sci. 2020, 27, 1208–1216. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; Azam, M.; Verma, V.P.; Barakat, A.; Haukka, M.; Elgazar, A.A.; Mira, A.; Badria, F.A. Construction of spirooxindole analogues engrafted with indole and pyrazole scaffolds as acetylcholinesterase inhibitors. ACS Omega 2021, 6, 31539–31556. [Google Scholar] [CrossRef]
- Zhou, L.-M.; Qu, R.-Y.; Yang, G.-F. An overview of spirooxindole as a promising scaffold for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 603–625. [Google Scholar] [CrossRef]
- Alshahrani, S.; Al-Majid, A.M.; Ali, M.; Alamary, A.S.; Abu-Serie, M.M.; Dömling, A.; Shafiq, M.; Ul-Haq, Z.; Barakat, A. Rational Design, Synthesis, Separation, and Characterization of New Spiroxindoles Combined with Benzimidazole Scaffold as an MDM2 Inhibitor. Separations 2023, 10, 225. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2019, 2019, 267–282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M. Application of Reactivity Indices in the Study of Polar Diels–Alder Reactions. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; Liu, S., Ed.; WILEY-VCH GmbH: Weinheim, Germany, 2022; Volume 2, pp. 481–502. [Google Scholar]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Based on the Flux of the Electron Density. Sci. Rad. 2023, 2, 1. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Saz Sousa, A.; Domingo, L.R. Electrophilicity and nucleophilicity scales at different DFF computational levels. J. Phys. Org. Chem. 2023, 36, e4503. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; Haukka, M.; Parveen, Z.; Ravaiz, N.; Wadood, A.; Rehman, A.U.; Ríos-Gutiérrez, M.; Domingo, L.R.; Barakat, A. A novel alpha-amylase inhibitor-based spirooxindole-pyrrolidine-clubbed thiochromene-pyrzaole pharmacophores: Unveiling the [3+2] cycloaddition reaction by molecular electron density theory. Chem. Biol. Drug. Des. 2023, 102, 972–995. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Trans. Faraday Soc. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar Cycloadditions toward Electrophilic Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| Chalcone 7 | −4.61 | 6.45 | 1.65 | 3.56 |
| AY 10a | −3.29 | 6.83 | 0.79 | 4.70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barakat, A.; Alshahrani, S.; Al-Majid, A.M.; Alamary, A.S.; Ali, M.; Ríos-Gutiérrez, M. Stereoselective Synthesis of a Novel Series of Dispiro-oxindolopyrrolizidines Embodying Thiazolo[3,2-a]benzimidazole Motif: A Molecular Electron Density Theory Study of the Mechanism of the [3 + 2] Cycloaddition Reaction. Chemistry 2023, 5, 2392-2405. https://doi.org/10.3390/chemistry5040158
Barakat A, Alshahrani S, Al-Majid AM, Alamary AS, Ali M, Ríos-Gutiérrez M. Stereoselective Synthesis of a Novel Series of Dispiro-oxindolopyrrolizidines Embodying Thiazolo[3,2-a]benzimidazole Motif: A Molecular Electron Density Theory Study of the Mechanism of the [3 + 2] Cycloaddition Reaction. Chemistry. 2023; 5(4):2392-2405. https://doi.org/10.3390/chemistry5040158
Chicago/Turabian StyleBarakat, Assem, Saeed Alshahrani, Abdullah Mohammed Al-Majid, Abdullah Saleh Alamary, M. Ali, and Mar Ríos-Gutiérrez. 2023. "Stereoselective Synthesis of a Novel Series of Dispiro-oxindolopyrrolizidines Embodying Thiazolo[3,2-a]benzimidazole Motif: A Molecular Electron Density Theory Study of the Mechanism of the [3 + 2] Cycloaddition Reaction" Chemistry 5, no. 4: 2392-2405. https://doi.org/10.3390/chemistry5040158