


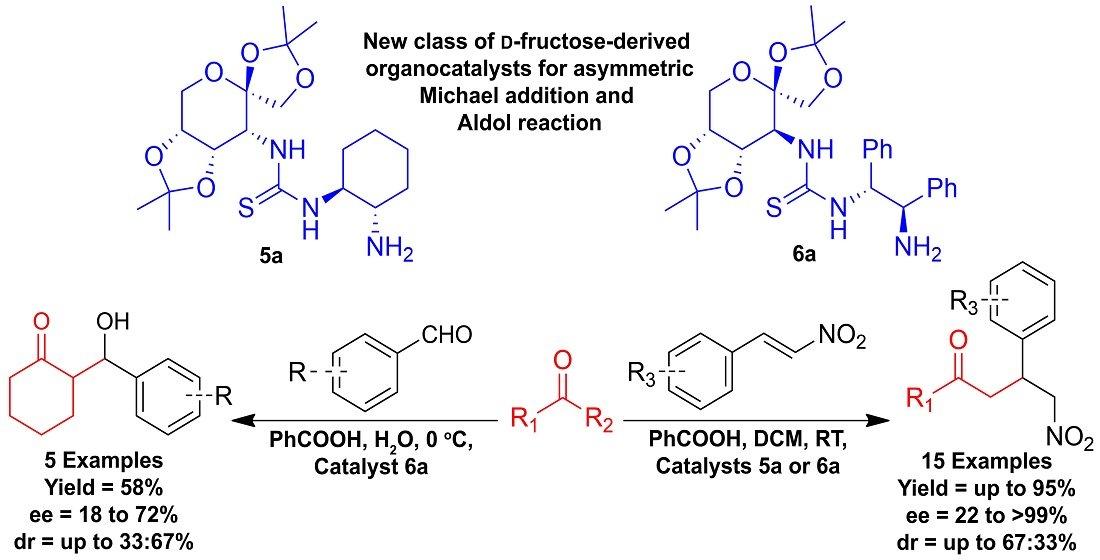
Synthesis of D-Fructose-Based Bifunctional Primary Amine-Thiourea Organocatalysts and Their Applications in Asymmetric Reactions
 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General
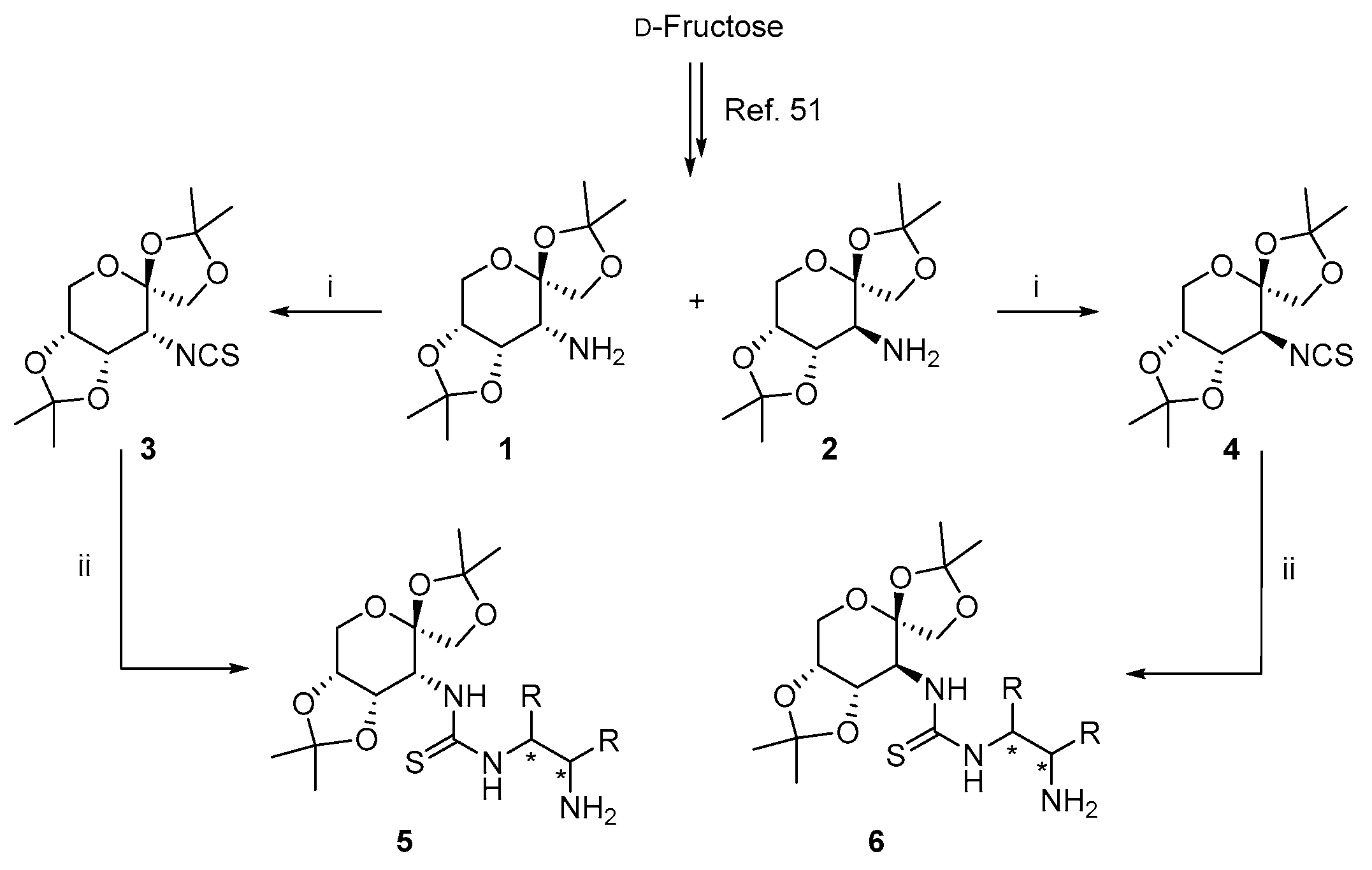
2.2. Synthesis of Saccharide-Based Isothiocyanates 3 and 4
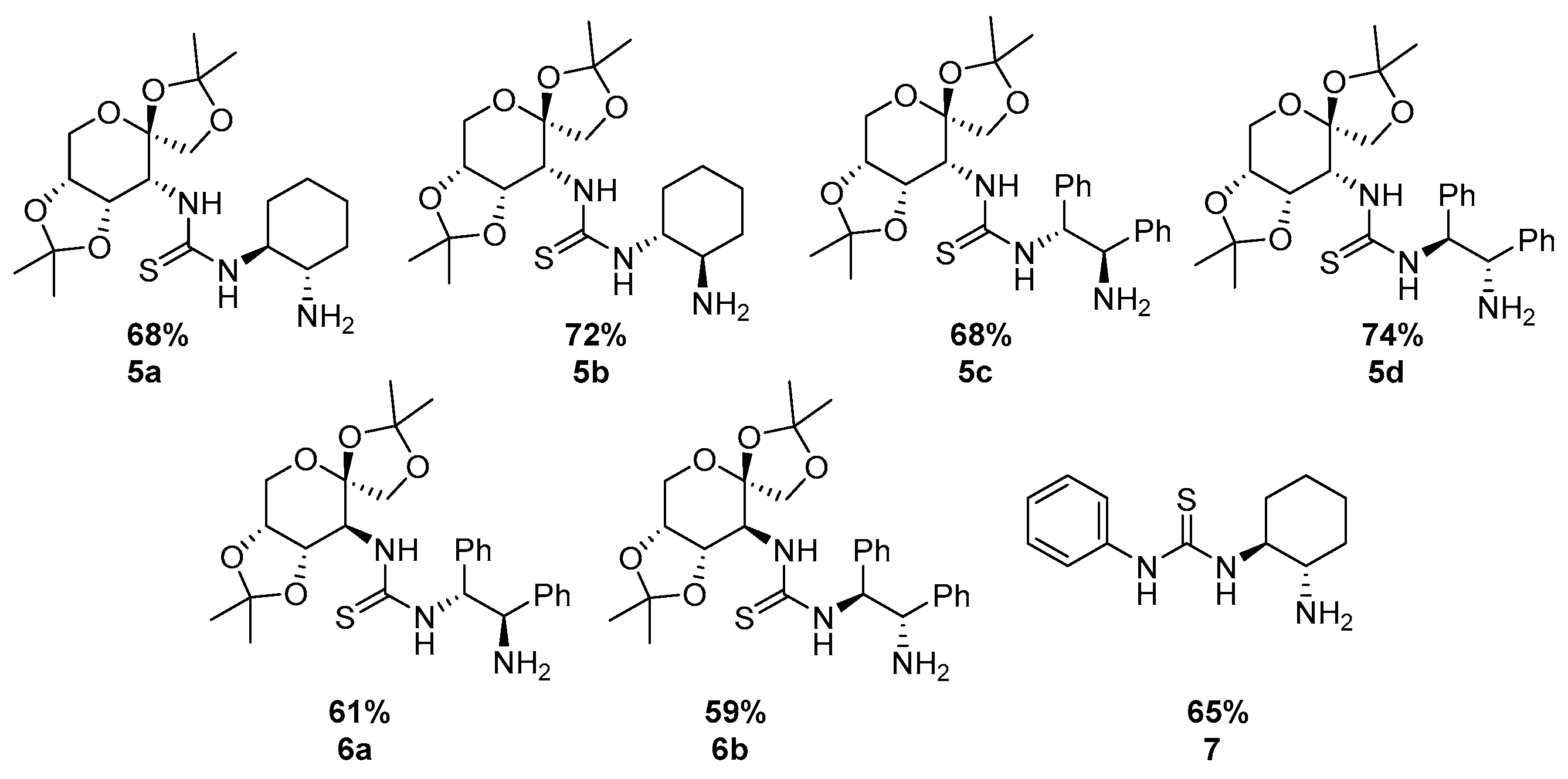
2.3. Synthesis of Saccharide-Derived Amine Thiourea 5a–d and 6a–b
2.4. Typical Procedure for Asymmetric Michael Addition Reaction
2.5. Typical Procedure for the Asymmetric Aldol Reaction
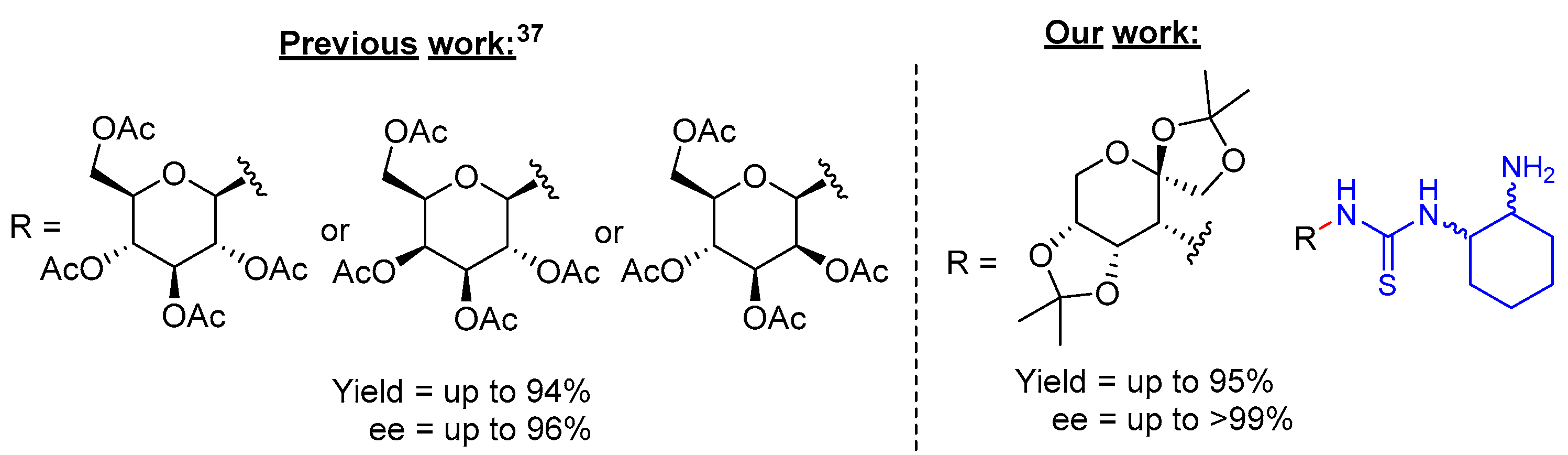
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheffler, U.; Mahrwald, R. Recent advances in organocatalytic methods for asymmetric C-C bond formation. Chem.-Eur. J. 2013, 19, 14346–14396. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.G.; Eger, E.; Kroutil, W. Building Bridges: Biocatalytic C-C-Bond Formation toward Multifunctional Products. ACS Catal. 2016, 6, 4286–4311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, D.M. Recent advances in organocatalytic asymmetric oxa-Michael addition triggered cascade reactions. Org. Chem. Front. 2020, 7, 3266–3283. [Google Scholar] [CrossRef]
- Gupta, V.; Singh, R.P. Enantioselective vinylogous Michael addition of β,γ-unsaturated butenolide to 2-iminochromenes. New J. Chem. 2019, 43, 9771–9775. [Google Scholar] [CrossRef]
- Ara, G.; Miran, M.S.; Islam, M.M.; Mollah, M.Y.A.; Rahman, M.M.; Susan, M.A.B.H. 1,8-Diazabicyclo[5.4.0]-undec-7-ene based protic ionic liquids and their binary systems with molecular solvents catalyzed Michael addition reaction. New J. Chem. 2020, 44, 13701–13706. [Google Scholar] [CrossRef]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef]
- Xiang, S.H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef] [PubMed]
- Aukland, M.H.; List, B. Organocatalysis emerging as a technology. Pure Appl. Chem. 2021, 93, 1371–1381. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Yalalov, D.A.; Hateley, M.J.; Weckbecker, C.; Huthmacher, K. Asymmetric organocatalysis with novel chiral thiourea derivatives: Bifunctional catalysts for the Strecker and nitro-Michael reactions. Eur. J. Org. Chem. 2005, 19, 4995–5000. [Google Scholar] [CrossRef]
- Parvin, T.; Yadav, R.; Choudhury, L.H. Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs). Org. Biomol. Chem. 2020, 18, 5513–5532. [Google Scholar] [CrossRef] [PubMed]
- Rufino, V.C.; Pliego, J.R. Bifunctional Primary Amino-thiourea Asymmetric Catalysis: The Imine-Iminium Ion Mechanism in the Michael Addition of Nitromethane to Enone. Asian J. Org. Chem. 2021, 10, 1472–1485. [Google Scholar] [CrossRef]
- Joshi, H.; Singh, V.K. Cinchona Derivatives as Bifunctional H-bonding Organocatalysts in Asymmetric Vinylogous Conjugate Addition Reactions. Asian J. Org. Chem. 2022, 11, e202100053. [Google Scholar] [CrossRef]
- Zheng, Z.; Lin, J.; Sun, Y.; Zhang, S. Threonine-derived thioureas as bifunctional organocatalysts for enantioselective Michael addition. Tetrahedron Lett. 2020, 61, 151382. [Google Scholar] [CrossRef]
- Held, F.E.; Tsogoeva, S.B. Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: Recent advances. Catal. Sci. Technol. 2016, 6, 645–667. [Google Scholar] [CrossRef]
- Fang, X.; Wang, C.J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine-thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff base catalysts for the asymmetric strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Sigman, M.S.; Vachal, P.; Jacobsen, E.N. A general catalyst for the asymmetric strecker reaction. Angew. Chem. Int. Ed. 2000, 39, 1279–1281. [Google Scholar] [CrossRef]
- Vachal, P.; Jacobsen, E.N. Structure-based analysis and optimization of a highly enantioselective catalyst for the strecker reaction. J. Am. Chem. Soc. 2002, 124, 10012–10014. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Highly enantioselective catalytic acyl-Pictet-Spengler reactions. J. Am. Chem. Soc. 2004, 126, 10558–10559. [Google Scholar] [CrossRef]
- Yoon, T.P.; Jacobsen, E.N. Highly Enantioselective Thiourea-Catalyzed Nitro-Mannich Reactions. Angew. Chem. 2005, 117, 470–472. [Google Scholar] [CrossRef]
- Huang, H.; Jacobsen, E.N. Highly enantioselective direct conjugate addition of ketones to nitroalkenes promoted by a chiral primary amine-thiourea catalyst. J. Am. Chem. Soc. 2006, 128, 7170–7171. [Google Scholar] [CrossRef]
- Lalonde, M.P.; Chen, Y.; Jacobsen, E.N. A Chiral Primary Amine Thiourea Catalyst for the Highly Enantioselective Direct Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes. Angew. Chem. Int. Ed. 2006, 45, 6366–6370. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Thiourea-catalyzed nucleophilic addition of TMSCN and ketene silyl acetals to nitrones and aldehydes. Tetrahedron Lett. 2003, 44, 2817–2821. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio- and diastereoselective michael reaction of 1,3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, Y.; Okino, T.; Takemoto, Y. Enantioselective Michael Addition to α,β-Unsaturated Imides Catalyzed by a Bifunctional Organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 4032–4035. [Google Scholar] [CrossRef]
- Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Enantioselective Aza-Henry Reaction Catalyzed by a Bifunctional Organocatalyst. Org. Lett. 2004, 6, 625–627. [Google Scholar] [CrossRef]
- Hoashi, Y.; Yabuta, T.; Yuan, P.; Miyabe, H.; Takemoto, Y. Enantioselective tandem Michael reaction to nitroalkene catalyzed by bifunctional thiourea: Total synthesis of (−)-epibatidine. Tetrahedron 2006, 62, 365–374. [Google Scholar] [CrossRef]
- Inokuma, T.; Hoashi, Y.; Takemoto, Y. Thiourea-catalyzed asymmetric michael addition of activated méthylene compounds to α,β-unsaturated imides: Dual activation of imide by intra- and intermolecular hydrogen bonding. J. Am. Chem. Soc. 2006, 128, 9413–9419. [Google Scholar] [CrossRef] [PubMed]
- Faísca Phillips, A.M. Applications of Carbohydrate-Based Organocatalysts in Enantioselective Synthesis. Eur. J. Org. Chem. 2014, 2014, 7291–7303. [Google Scholar] [CrossRef]
- Henderson, A.S.; Bower, J.F.; Galan, M.C. Carbohydrates as enantioinduction components in stereoselective catalysis. Org. Biomol. Chem. 2016, 14, 4008–4017. [Google Scholar] [CrossRef]
- Baslé, O.; Raimondi, W.; Duque, M.D.M.S.; Bonne, D.; Constantieux, T.; Rodriguez, J. Highly diastereo- and enantioselective organocatalytic michael addition of α-ketoamides to nitroalkenes. Org. Lett. 2010, 12, 5246–5249. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Vera, S.; Mielgo, A.; Gómez-Bengoa, E. Highly efficient asymmetric Michael addition of aldehydes to nitroalkenes catalyzed by a simple trans-4-hydroxyprolylamide. Angew. Chem. Int. Ed. 2006, 45, 5984–5987. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Kutzscher, C.; Drache, F.; Senkovska, I.; Kaskel, S. Chiral Functionalization of a Zirconium Metal-Organic Framework (DUT-67) as a Heterogeneous Catalyst in Asymmetric Michael Addition Reaction. Inorg. Chem. 2018, 57, 1483–1489. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael additions to nitroalkenes. Eur. J. Org. Chem. 2002, 2002, 1877–1894. [Google Scholar] [CrossRef]
- Das, T.; Mohapatra, S.; Mishra, N.P.; Nayak, S.; Raiguru, B.P. Recent Advances in Organocatalytic Asymmetric Michael Addition Reactions to α, β-Unsaturated Nitroolefins. ChemistrySelect 2021, 6, 3745–3781. [Google Scholar] [CrossRef]
- Liu, K.; Cui, H.F.; Nie, J.; Dong, K.Y.; Li, X.J.; Ma, J.A. Highly enantioselective Michael addition of aromatic ketones to nitroolefins promoted by chiral bifunctional primary amine-thiourea catalysts based on saccharides. Org. Lett. 2007, 9, 923–925. [Google Scholar] [CrossRef]
- Gu, Q.; Guo, X.T.; Wu, X.Y. Highly enantioselective Michael addition of acetone to nitroolefins catalyzed by chiral bifunctional primary amine-thiourea catalysts with acetic acid. Tetrahedron 2009, 65, 5265–5270. [Google Scholar] [CrossRef]
- Lu, A.; Gao, P.; Wu, Y.; Wang, Y.; Zhou, Z.; Tang, C. Highly enantio- and diastereoselective Michael addition of cyclohexanone to nitroolefins catalyzed by a chiral glucose-based bifunctional secondary amine-thiourea catalyst. Org. Biomol. Chem. 2009, 7, 3141–3147. [Google Scholar] [CrossRef]
- Hai, M.; Liu, K.; Zhang, F.G.; Zhu, C.L.; Nie, J.; Jun-An, M. Chiral bifunctional thiourea-catalyzed enantioselective michael addition of ketones to nitrodienes. J. Org. Chem. 2010, 75, 1402–1409. [Google Scholar] [CrossRef]
- Li, X.J.; Liu, K.; Ma, H.; Nie, J.; Ma, J.A. Highly enantioselective Michael addition of malonates to nitroolefins catalyzed by chiral bifunctional tertiary amine-thioureas based on saccharides. Synlett 2008, 20, 3242–3246. [Google Scholar] [CrossRef]
- Gao, P.; Wang, C.; Wu, Y.; Zhou, Z.; Tang, C. Sugar-derived bifunctional thiourea organocatalyzed asymmetric Michael addition of acetylacetone to nitroolefins. Eur. J. Org. Chem. 2008, 2008, 4563–4566. [Google Scholar] [CrossRef]
- Pu, X.W.; Peng, F.Z.; Zhang, H.B.; Shao, Z.H. Doubly stereocontrolled asymmetric conjugate addition of acetylacetone to nitroolefins catalyzed by bifunctional tertiary amine-thiourea catalysts derived from both acyclic α-amino acids and carbohydrates. Tetrahedron 2010, 66, 3655–3661. [Google Scholar] [CrossRef]
- Puglisi, A.; Benaglia, M.; Raimondi, L.; Lay, L.; Poletti, L. Novel carbohydrate-based bifunctional organocatalysts for nucleophilic addition to nitroolefins and imines. Org. Biomol. Chem. 2011, 9, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Rončák, R.; Tvrdoňová, M.; Gonda, J.; Elečko, J. Novel carbohydrate-based thioureas as organocatalysts for asymmetric michael addition of 1,3-dicarbonyl compounds to nitroolefins. Tetrahedron 2020, 76, 131339. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Reddy, S.M.; Swain, M. Sugar thiourea catalyzed highly enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinone to β-nitroalkenes. RSC Adv. 2013, 3, 930–936. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, J.; Liu, S.; Yu, C.; Miao, Z. Asymmetric synthesis of spiro[chroman-3,3′-pyrazol] scaffolds with an all-carbon quaternary stereocenter via a oxa-Michael-Michael cascade strategy with bifunctional amine-thiourea organocatalysts. RSC Adv. 2015, 5, 91108–91113. [Google Scholar] [CrossRef]
- Yuan, H.-N.; Wang, S.; Nie, J.; Meng, W.; Yao, Q.; Ma, J.-A. Hydrogen-Bond-Directed Enantioselective Decarboxylative Mannich Reaction of β-Ketoacids with Ketimines: Application to the Synthesis of Anti-HIV Drug DPC 083. Angew. Chem. 2013, 125, 3961–3965. [Google Scholar] [CrossRef]
- Yuan, H.N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.A. Highly enantioselective decarboxylative Mannich reaction of malonic acid half oxyesters with cyclic trifluoromethyl ketimines: Synthesis of β-amino esters and anti-HIV drug DPC 083. Chem.-Eur. J. 2013, 19, 15856–15860. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Huang, Y.J.; Nie, J.; Ma, J.A. Highly Regio-, Diastereo-, and Enantioselective Mannich Reaction of Allylic Ketones and Cyclic Ketimines: Access to Chiral Benzosultam. Org. Lett. 2015, 17, 4608–4611. [Google Scholar] [CrossRef]
- Vanlaldinpuia, K.; Bora, P.; Basumatary, G.; Mohanta, R.; Bez, G. Enantioselective aminocatalysis: Michael addition of unactivated ketones to nitroolefins catalyzed by d-fructose derived monofunctional primary amine. J. Chem. Sci. 2017, 129, 1603–1610. [Google Scholar] [CrossRef]
- Vanlaldinpuia, K.; Bora, P.; Bez, G. Monofunctional primary amine: A new class of organocatalyst for asymmetric Aldol reaction. J. Chem. Sci. 2017, 129, 301–312. [Google Scholar] [CrossRef]
- Nie, J.; Li, X.J.; Zheng, D.H.; Zhang, F.G.; Cui, S.; Ma, J.A. Chiral bifunctional thiourea-catalyzed enantioselective aldol reaction of trifluoroacetaldehyde hemiacetal with aromatic ketones. J. Fluor. Chem. 2011, 132, 468–473. [Google Scholar] [CrossRef]
- Munch, H.; Hansen, J.S.; Pittelkow, M.; Christensen, J.B.; Boas, U. A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 2008, 49, 3117–3119. [Google Scholar] [CrossRef]
- Vural, U.; Durmaz, M.; Sirit, A. A novel calix[4]arene-based bifunctional squaramide organocatalyst for enantioselective Michael addition of acetylacetone to nitroolefins. Org. Chem. Front. 2016, 3, 730–736. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Shi, X.; Ji, N.; He, W.; Zhang, S.; Zhang, B. Modular bifunctional chiral thioureas as versatile organocatalysts for highly enantioselective aza-Henry reaction and michael addition. Adv. Synth. Catal. 2012, 354, 2264–2274. [Google Scholar] [CrossRef]
- Ban, S.; Du, D.M.; Liu, H.; Yang, W. Synthesis of Binaphthyl Sulfonimides and Their Application in the Enantioselective Michael Addition of Ketones to Nitroalkenes. Eur. J. Org. Chem. 2010, 2010, 5160–5164. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Liu, X.; Wang, Z.; Feng, X.; Su, Z.; Hua, C. Highly efficient amine organocatalysts based on bispidine for the asymmetric Michael addition of ketones to nitroolefins. Adv. Synth. Catal. 2008, 350, 2001–2006. [Google Scholar] [CrossRef]
- Shim, J.H.; Ahn, B.K.; Lee, J.Y.; Kim, H.S.; Ha, D.C. Organocatalysis for the asymmetric michael addition of cycloketones and α, β-unsaturated nitroalkenes. Catalysts 2021, 11, 1004. [Google Scholar] [CrossRef]
- Jiang, Z.; Yang, H.; Han, X.; Luo, J.; Wong, M.W.; Lu, Y. Direct asymmetric aldol reactions between aldehydes and ketones catalyzed by l-tryptophan in the presence of water. Org. Biomol. Chem. 2010, 8, 1368–1377. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, X.; Armstrong, D.W. Highly Efficient Asymmetric Direct Stoichiometric Aldol Reactions on/in Water. Angew. Chem. 2007, 119, 9231–9235. [Google Scholar] [CrossRef]
- Gao, J.; Bai, S.; Gao, Q.; Liu, Y.; Yang, Q. Acid controlled diastereoselectivity in asymmetric aldol reaction of cycloketones with aldehydes using enamine-based organocatalysts. Chem. Commun. 2011, 47, 6716–6718. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Additives (mol%) | Time (h) | Yield (%) [b] | ee (%) [c] |
| 1 | CH2Cl2 | 5a (10) | - | 96 | 48 | 57 |
| 2 | CH2Cl2 | 5a (10) | AcOH (10) | 56 | 76 | 92 |
| 3 | CH2Cl2 | 5a (10) | PhCO2H (10) | 54 | 89 | 96 |
| 4 | CH2Cl2 | 5a (10) | CF3CO2H (10) | 77 | 54 | 76 |
| 5 | CH2Cl2 | 5a (10) | 4-NO2C6H4CO2H (10) | 48 | 87 | 32 |
| 6 | CH2Cl2 | 5a (10) | 4-BrC6H4CO2H (10) | 52 | 83 | 43 |
| 7 | CH2Cl2 | 5b (10) | PhCO2H (10) | 52 | 82 | 90 |
| 8 | CH2Cl2 | 5c (10) | PhCO2H (10) | 66 | 77 | 93 |
| 9 | CH2Cl2 | 5d (10) | PhCO2H (10) | 96 | 81 | 86 |
| 10 | CH2Cl2 | 6a (10) | PhCO2H (10) | 49 | 88 | 89 |
| 11 | CH2Cl2 | 6b (10) | PhCO2H (10) | 72 | 81 | 92 |
| 12 | CH2Cl2 | 7 (10) | PhCO2H (10) | 24 | 78 | 88 |
| 13 | CH2Cl2 | 5a (5) | PhCO2H (10) | 72 | 73 | 95 |
| 14 | CH2Cl2 | 5a (15) | PhCO2H (10) | 52 | 92 | 97 |
| 15 | CH2Cl2 | 5a (20) | PhCO2H (10) | 44 | 93 | 96 |
| 16 | CH2Cl2 | 5a (15) | PhCO2H (5) | 58 | 92 | 93 |
| 17 | CH2Cl2 | 5a (15) | PhCO2H (15) | 48 | 95 | >99 |
| 18 | CH2Cl2 | 5a (15) | PhCO2H (20) | 44 | 96 | 98 |
| 19 | Toluene | 5a (15) | PhCO2H (15) | 72 | 60 | 88 |
| 20 | THF | 5a (15) | PhCO2H (15) | 96 | 43 | 93 |
| 21 | CH3CN | 5a (15) | PhCO2H (15) | 120 | 51 | 66 |
| 22 | Neat | 5a (15) | PhCO2H (15) | 48 | 78 | 82 |
| 23 [d] | CH2Cl2 | 5a (15) | PhCO2H (15) | 120 | 66 | 98 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Time (h) | Yield (%) [b] | ee (%) [c,d] |
| 1 | 4-Bromo | Me | H | 48 | 95 (8a) | >99 (S) |
| 2 | 4-Chloro | Me | H | 48 | 90 (8b) | 91 (S) |
| 3 | 4-Methyl | Me | H | 68 | 81 (8c) | 85 (S) |
| 4 | 2-Chloro | Me | H | 48 | 95 (8d) | 83 (S) |
| 5 | 2-Methoxy | Me | H | 94 | 92 (8e) | 81 (S) |
| 6 | H | Me | H | 36 | 93 (8f) | 95 (S) |
| 7 | 4-Chloro | Me | COMe | 100 | 88 (8g) | 32 (S) |
| 8 | 4-Methyl | Me | COMe | 120 | 91 (8h) | 22 (S) |
| 9 | H | R2 = R3 = (CH2)4 | - | 168 | Nr [e] | Nd [f] |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | R1 | Ketone | Time (h) | Yield (%) [b] | dr (%) [c] syn/anti | ee (%) [d,e] |
| 1 | 5b (15) | 4-Cl |  | 92 | 82 (8g) | - | 12 (R) |
| 2 | 5c (15) | 4-Cl | | 96 | 81 (8g) | - | 79 (S) |
| 3 | 5d (15) | 4-Cl | | 76 | 66 (8g) | - | 43 (S) |
| 4 | 6a (15) | 4-Cl | | 72 | 80 (8g) | - | 84 (S) |
| 5 | 6b (15) | 4-Cl | | 88 | 84 (8g) | - | 72 (R) |
| 6 | 6a (15) | 4-Me | | 72 | 73 (8h) | - | 81 (S) |
| 7 | 6a (15) | 4-Br | | 81 | 78 (8i) | - | 81 (S) |
| 8 | 6a (15) | 2-Cl | | 96 | 85 (8j) | - | 76 (S) |
| 9 | 6a (15) | 2-MeO | | 120 | 77 (8k) | - | 71 (S) |
| 10 | 6a (15) | H | | 96 | 81 (8l) | - | 74 (S) |
| 11 | 6a (15) | 4-Me |  | 168 | 46 (8m) | 68/32 | 78 [f] (R,S) |
| 12 | 6a (15) | 4-Br | | 168 | 55 (8n) | 68/32 | 20 [f] (S,R) |
| 13 | 6a (15) | H | | 180 | 42 (8o) | 67/33 | 79 [f] (S,R) |
| Entry | Catalyst (mol%) | Temp | Additives (mol%) | Solvent | Time (h) | Yield (%) | dr (%) Syn:anti | er (%) Syn:anti |
|---|---|---|---|---|---|---|---|---|
| 1 | 5a (20) | RT | - | Neat | 12 h | 85 | 50:50 | Racemic |
| 2 | 5a (20) | RT | PhCO2H (20) | H2O | 24 h | 63 | 48:52 | 15:21 |
| 3 | 5b (20) | RT | PhCO2H (20) | H2O | 36 h | 65 | 56:44 | 8:16 |
| 4 | 5c (20) | RT | PhCO2H (20) | H2O | 38 h | 55 | 51:49 | 14:24 |
| 5 | 5d (20) | RT | PhCO2H (20) | H2O | 38 h | 34 | 58:42 | 6:25 |
| 6 | 6a (20) | RT | PhCO2H (20) | H2O | 58 h | 54 | 43:57 | 13:39 |
| 7 | 6b (20) | RT | PhCO2H (20) | H2O | 48 h | 44 | 48:52 | 12:35 |
| 8 | 6a (20) | RT | CH3CO2H (20) | H2O | 72 h | 65 | 52:48 | 9:28 |
| 9 | 6a (20) | RT | DNP (20) | H2O | 48 h | 72 | 47:53 | 11:3 |
| 10 | 6a (20) | RT | TFA (20) | H2O | 56 h | 66 | 43:57 | 19:12 |
| 11 | 6a (20) | RT | PhCO2H (20) | DMSO | 96 h | 48 | 44:56 | Racemic |
| 12 | 6a (20) | RT | PhCO2H (20) | DCM | 126 h | trace | - | - |
| 13 | 6a (20) | 0 °C | PhCO2H (20) | H2O | 144 h | 62 | 47:53 | 30:47 |
| 14 | 6a (20) | 0 °C | PhCO2H (20) | Neat | 110 h | 72 | 45:55 | 9:38 |
 | |||||
|---|---|---|---|---|---|
| Entry | Aldehydes | Reaction Time (h) | Yield (%) [b] | dr (%) [c] Syn:anti | ee (%) [d,e] |
| 1 | 4-Nitro | 144 | 58 (9a) | 47:53 | 30 (R,R) |
| 2 | 3-Nitro | 120 | 52 (9b) | 46:54 | 15 (R,R) |
| 3 | 2-Nitro | 168 | 46 (9c) | 55:45 | 40 (R,R) |
| 4 | 3-Chloro | 128 | 54 (9d) | 54:46 | 23 (R,R) |
| 5 | 4-Bromo | 120 | 50 (9e) | 33:67 | 69 (R,R) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalhmangaihzuala, S.; Khiangte, V.; Laldinpuii, Z.; Nunnemi, L.; Malsawmsanga, J.; Lallawmzuali, G.; Liana, T.; Lalhriatpuia, C.; Pachuau, Z.; Vanlaldinpuia, K. Synthesis of D-Fructose-Based Bifunctional Primary Amine-Thiourea Organocatalysts and Their Applications in Asymmetric Reactions. Chemistry 2023, 5, 2362-2375. https://doi.org/10.3390/chemistry5040156
Lalhmangaihzuala S, Khiangte V, Laldinpuii Z, Nunnemi L, Malsawmsanga J, Lallawmzuali G, Liana T, Lalhriatpuia C, Pachuau Z, Vanlaldinpuia K. Synthesis of D-Fructose-Based Bifunctional Primary Amine-Thiourea Organocatalysts and Their Applications in Asymmetric Reactions. Chemistry. 2023; 5(4):2362-2375. https://doi.org/10.3390/chemistry5040156
Chicago/Turabian StyleLalhmangaihzuala, Samson, Vanlalngaihawma Khiangte, Zathang Laldinpuii, Lal Nunnemi, Joute Malsawmsanga, Gospel Lallawmzuali, Thanhming Liana, Chhakchhuak Lalhriatpuia, Zodinpuia Pachuau, and Khiangte Vanlaldinpuia. 2023. "Synthesis of D-Fructose-Based Bifunctional Primary Amine-Thiourea Organocatalysts and Their Applications in Asymmetric Reactions" Chemistry 5, no. 4: 2362-2375. https://doi.org/10.3390/chemistry5040156