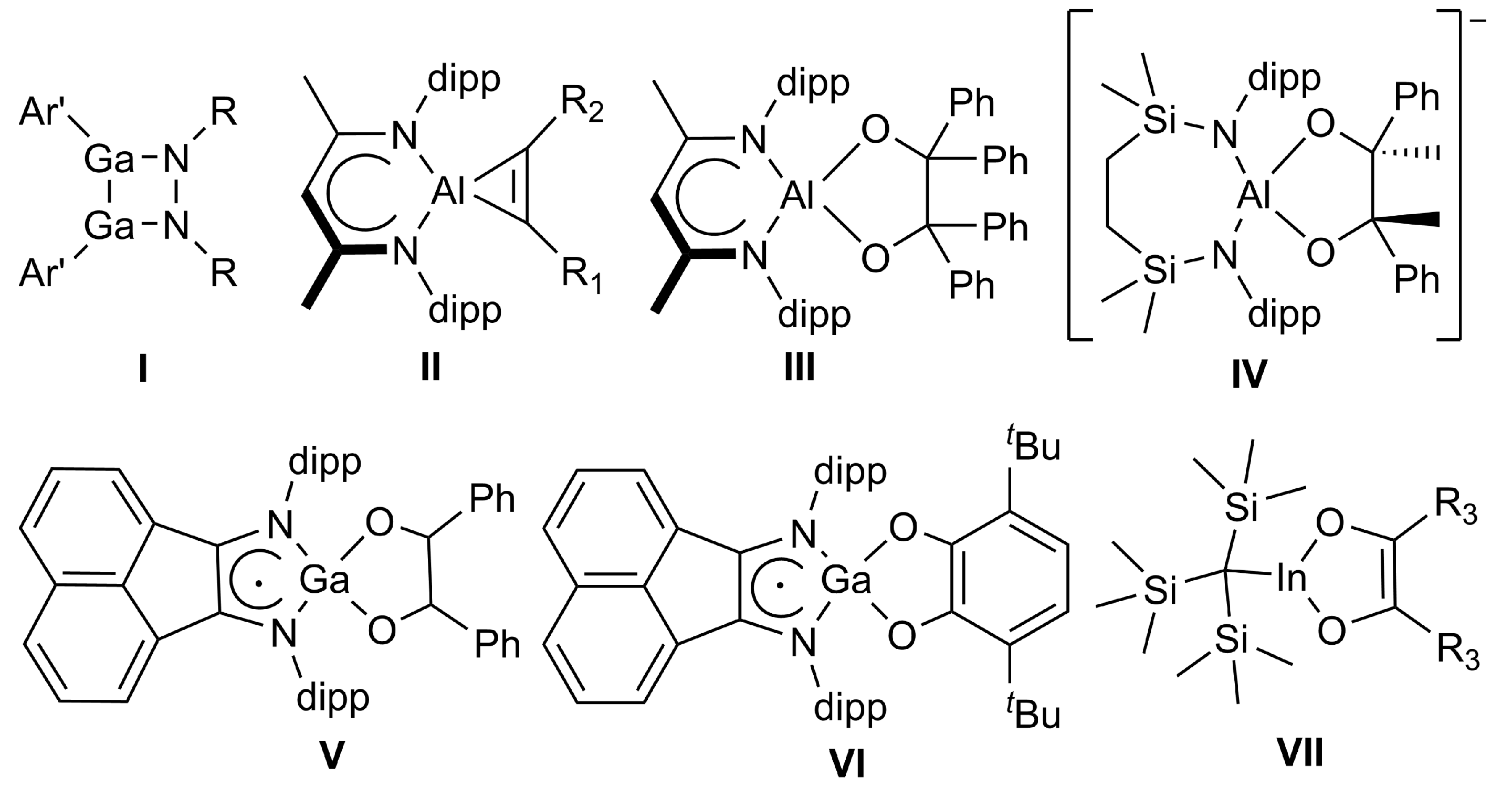
4. Materials and Methods
All manipulations were performed using standard Schlenk and glovebox techniques under an argon atmosphere, dried by passage through preheated Cu
2O pellets and molecular sieve columns. Toluene,
n-pentane, and
n-hexane were dried using an MBraun solvent purification system (SPS). Benzene, deuterated benzene, thf, 2-methyltetrahydrofuran and deuterated thf were distilled from Na/K alloy and acetonitrile from CaH
2. Solvents were degassed and stored over activated molecular sieves. Starting reagents L’BCl
2 [
34,
36], LAl [
1,
37], LGa [
2,
38,
39], LIn [
3], and LTl [
4] were prepared according to the (slightly modified) literature methods (LK was isolated and not prepared in situ).
1H (300 MHz, 400 MHz, 600 MHz),
11B{
1H} (128.5 MHz, 192.5 MHz),
13C{
1H} (75.5 MHz, 100.7 MHz, 150.9 MHZ), and
19F{
1H} (376.5 MHz) spectra were recorded with a Bruker Avance DPX-300, a Bruker Avance Neo 400 MHz or a Bruker Avance III HD 600 NMR spectrometer and are referenced to internal C
6D
6 (
1H: δ = 7.16,
13C: δ = 128.06), and thf-d
8 (
1H: δ = 3.58;
13C: δ = 67.21). Heteronuclear NMR measurements were performed protium decoupled unless otherwise noted. IR spectra were recorded in a glovebox using a BRUKER ALPHAT FT-IR spectrometer equipped with a single reflection ATR sampling module to ensure oxygen- and water-free conditions. Microanalysis was performed at the Elemental Analysis Laboratory of the University of Duisburg-Essen.
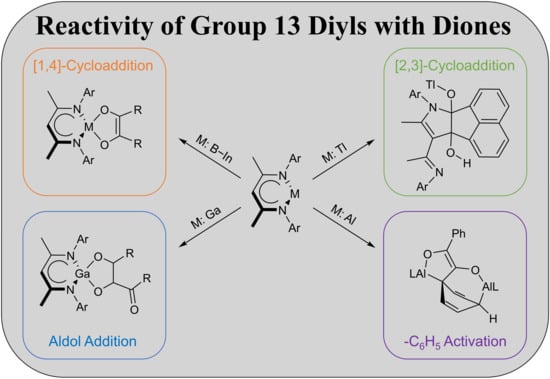
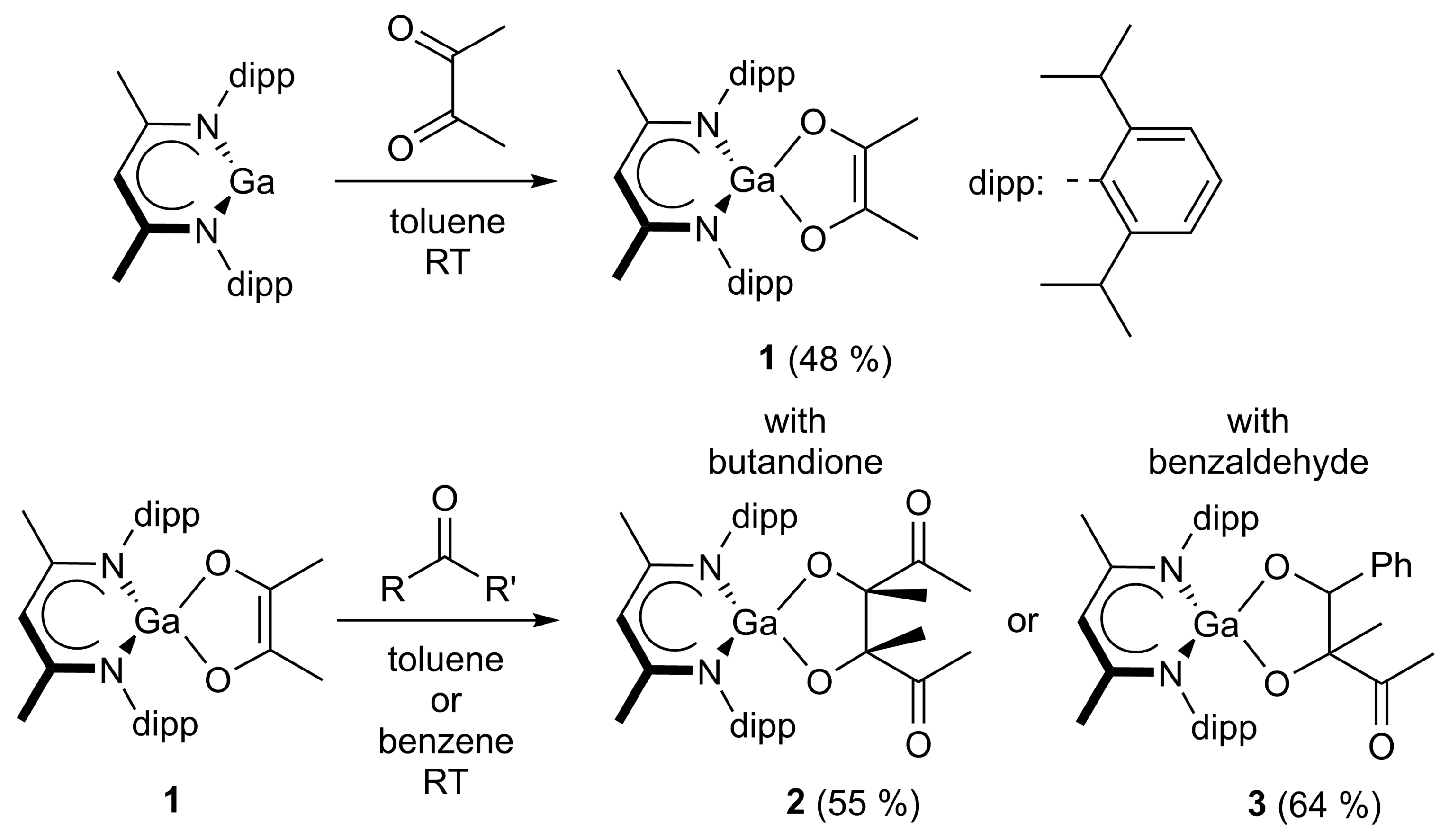
4.1. Synthesis of LGa(C4H6O2) (1)
A total of 35.3 mg of butanedione (410 μmol) was added to 200 mg of LGa (410 μmol) dissolved in 5 mL of toluene. The clear yellow solution turned orange and was stirred for 15 min. The solution was layered with 15 mL of n-hexane and stored overnight at room temperature. Small amounts of solids were separated by filtration and the filtrate was concentrated to about 0.5 mL. The product was obtained as an orange crystalline solid when stored at −30 °C. Yield: 112 mg (197 μmol, 48%).
Anal. Calcd. for C33H47GaN2O2: C, 69.12, H, 8.26; N, 4.88; Found: C, 69.3, H, 8.29; N, 4.86%. ATR-IR: υ 2960, 2866, 1660, 1587, 1555, 1529, 1462, 1440, 1382, 1316, 1256, 1214, 1176, 1125, 1101, 1022, 984, 932, 872, 803, 772, 761, 747, 668, 533, 533 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.04 (s, 6 H, C6H3-2,6iPr2), 4.86 (s, 1 H, γ-CH), 3.32 (sept, 3JHH = 6.8 Hz, 4 H, CH(CH3)2), 1.74 (s, 6 H, Ga(OCCH3)2), 1.54 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.54 (s, 6 H, ArNCCH3), 1.12 (d, 3JHH = 6.9 Hz, 12 H CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 171.4 (ArNCCH3), 144.2, 138.8, 128.1, 124.4 (ArC), 131.6 (Ga(OCCH3)2), 96.0 (γ-CH), 28.7 (CH(CH3)2), 24.8, 24.6 (CH(CH3)2), 23.3 (ArNCCH3), 16.1 (Ga(OCCH3)2).
4.2. Synthesis of LGa(C8H12O4) (2)
8.8 mg of butanedione (103 μmol) was added to 20 mg of LGa (41 μmol) dissolved in 0.5 mL of C6D6. A colorless precipitate formed shortly after, which was redissolved by heating the suspension. Storage at room temperature gave the product a colorless crystalline solid. Yield: 15 mg (23 μmol, 55%).
Anal. Calcd. for C37H53Ga1N2O4: C, 67.38, H, 8.10; N, 4.25; Found: C, 67.0, H, 8.88 N, 4.07%. ATR-IR: υ 2958, 2928, 2866, 1706, 1535, 1464, 1438, 1384, 1347, 1318, 1262, 1217, 1182, 1109, 1082, 1059, 1024, 995, 925, 875, 798, 761, 695, 668, 605, 542, 529 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.15-6.98 (m, 6 H, C6H3-2,6iPr2), 4.86 (s, 1 H, γ-CH), 3.32 (sept, 3JHH = 6.8 Hz, 1 H, CH(CH3)2), 3.18 (m, 3 H, CH(CH3)2), 1.96 (s, 4.5 H, GaC8H12O4), 1.80 (s, 1.5 H, GaC8H12O4), 1.74 (s, 1.5 H, GaC8H12O4), 1.53 (m, 18 H, CH(CH3)2, ArNCCH3), 1.12 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 1.04 (d, 3JHH = 6.9 Hz, 9 H, CH(CH3)2), 0.95 (s, 4.5 H, GaC8H12O14). 1H NMR (300 MHz, C6D6, 80 °C): δ 7.06 (s, 6 H, N-C6H5), 4.94 (s, 1 H, γ-CH), 3.31 (sept, 3JHH = 6.9 Hz, 4 H, CH(CH3)2), 1.86 (s(br), 6 H, GaC8H12O14), 1.67 (s(br), 1.5 H, GaC8H12O14), 1.63 (s,6 H, ArNCCH3), 1.49 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2), 1.14 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): many resonances were not observed due to line broadening (cp. 1H NMR spectrum) and low solubility in benzene.
4.3. Synthesis of LGa(C11H12O3) (3)
21.4 μL of benzaldehyde (22.2 mg, 209.3 μmol) was added to 100 mg of LGa(C4H6O2) (1) (174 μmol) dissolved in 1 mL of toluene. A rapid color change from orange to yellow was observed and a precipitate began to form. The suspension was stored in the freezer overnight, filtered, and washed with n-hexane, yielding 75 mg of LGa(C11H12O3). Yield: 70 mg (111 μmol, 64%).
Anal. Calcd. for C40H53GaN2O3: C, 70.69, H, 7.86; N, 4.12; Found: C, 70.6, H, 7.96; N, 3.95%. ATR-IR: υ 2960, 2927, 2868, 2814, 1702, 1537, 1438, 1394, 1367, 1359, 1320, 1268, 1182, 1142, 1096, 1051, 1028, 929, 877, 796, 757, 732, 699, 672, 604, 569, 492, 474 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.44, 7.23–7.00 (m, 11 H, C6H3-2,6iPr2 and C6H5), 4.88 (s, 1 H, γ-CH), 4.60 (s, 1 H, GaOCH), 3.61 (sept, 3JHH = 6.7 Hz, 1 H, CH(CH3)2), 3.47 (sept, 3JHH = 6.8 Hz, 1 H, CH(CH3)2), 3.15 (sept, 3JHH = 6.8 Hz, 1 H, CH(CH3)2), 3.04 (sept, 3JHH = 6.8 Hz, 1 H, CH(CH3)2), 1.74 (s, 3 H, GaOC(CO)CH3), 1.61 (s, 3 H, ArNCCH3), 1.60 (s, 3 H, ArNCCH3), 1.59 (d, 3JHH = 6.8 Hz, 3 H, CH(CH3)2), 1.51 (d, 3JHH = 6.8 Hz, 3 H, CH(CH3)2), 1.45 (d, 3JHH = 6.7 Hz, 3 H, CH(CH3)2), 1.26 (d, 3JHH = 6.8 Hz, 3 H, CH(CH3)2), 1.13 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 1.10 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 1.05 (d, 3JHH = 6.8 Hz, 3 H, CH(CH3)2), 0.92 (d, 3JHH = 6.8 Hz, 3 H, CH(CH3)2), 0.44 (s, 3 H, GaOCCH3). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 171.4 (GaOC(CO)CH3), 172.0 (ArNCCH3), 172.0, 144.4, 144.4, 143.9, 143.9, 143.5, 139.7, 139.0, 127.6, 127.1, 126.6, 124.7, 124.7, 124.7, 124.6 (ArC), 95.8 (γ-CH), 84.7 (GaOCCH3), 80.7 (GaOCH), 29.0, 28.9, 28.6, 28.5 (CH(CH3)2), 25.4, 25.0, 24.7, 24.7, 24.5, 24.2, 24.1, 23.7 (CH(CH3)2), 24.6 (GaOC(CO)CH3), 23.4, 23.4 (ArNCCH3), 17.9 (GaOCCH3).
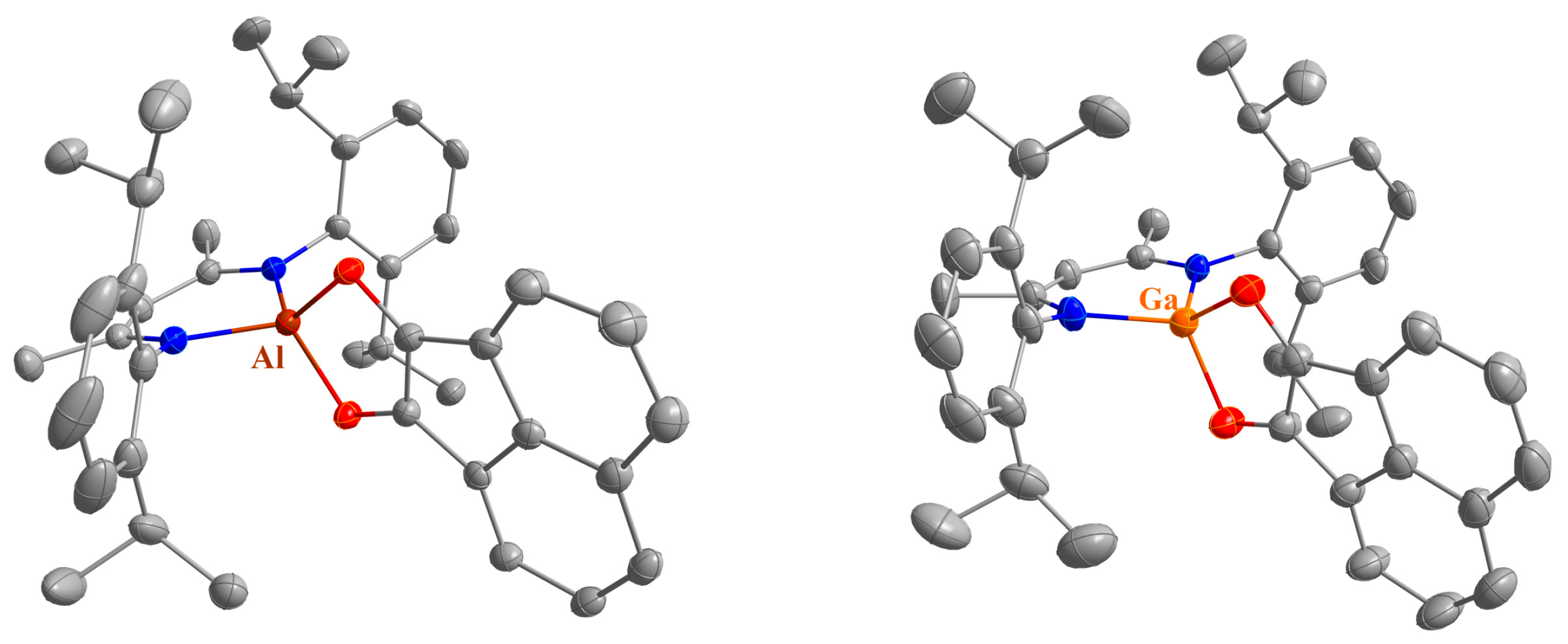
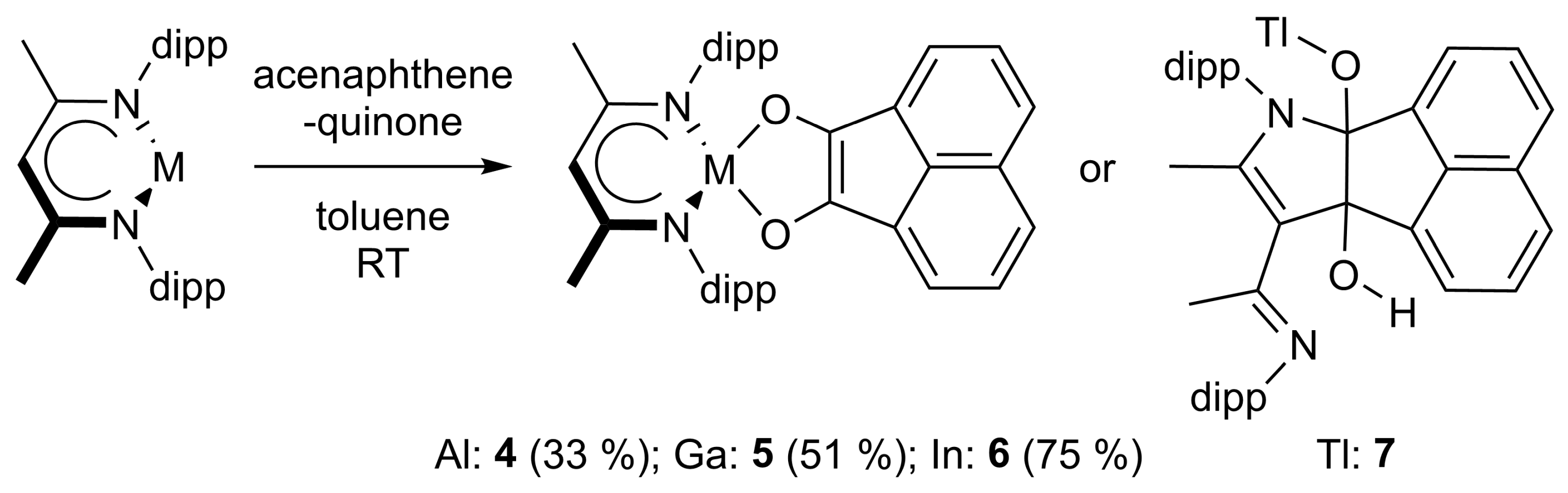
4.4. Synthesis of LAl(C12H6O2) (4)
50 mg of LAl (112 μmol) and 20.5 mg of acenaphthenequinone (112 μmol) were dissolved in 5 mL of toluene and the resulting dark purple solution was stirred overnight at ambient temperature. All volatiles were removed, and the dark purple residue was washed with small amounts of n-hexane. Yield: 23 mg (37 μmol, 33%).
Anal. Calcd. for C41H47AlN2O2: C, 78.56, H, 7.56; N, 4.47; Found: C, 78.1, H, 7.37; N, 4.28%. ATR-IR: υ 2964, 2927, 2868, 1531, 1462, 1444, 1382, 1313, 1249, 1196, 1133, 1105, 1024, 917, 898, 803, 772, 759, 738, 703, 596, 482, 472, 445, 416 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.18 (d, 3JHH = 6.8 Hz, 2 H, AlC12H6O2), 7.10 (d, 3JHH = 8.3 Hz, 2 H, AlC12H6O2), 6.98 (dd, 3JHH = 6.7, 8.3 Hz, 2 H, AlC12H6O2), 6.95-6.87 (m, 6 H, C6H3-2,6iPr2), 5.02 (s, 1 H, γ-CH), 3.35 (sept, 3JHH = 6.8 Hz, 4 H, CH(CH3)2), 1.54 (s, 6 H, ArNCCH3), 1.52 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.10 (d, 3JHH = 6.9 Hz, 12 H CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 172.5 (ArNCCH3), 144.4, 137.2, 128.4, 124.7 (ArC), 146.0, 133.5, 127.4, 127.1, 124.9, 123.4, 116.8 (AlC12H6O2), 98.1 (γ-CH), 28.9 (CH(CH3)2), 24.9, 24.8 (CH(CH3)2), 23.0 (ArNCCH3).
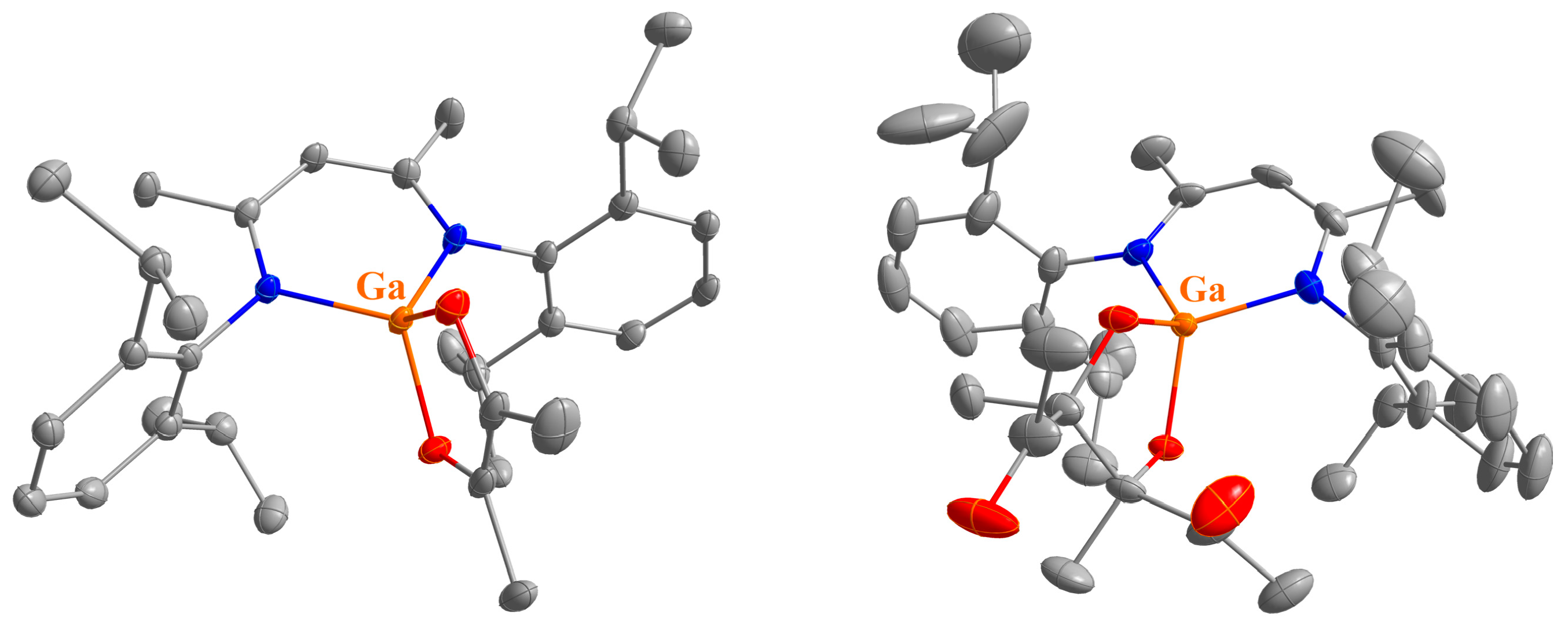
4.5. Synthesis of LGa(C12H6O2) (5)
A total of 50 mg of LGa (103 μmol) and 18.7 mg of acenaphthenequinone (103 μmol) were dissolved in 5 mL of toluene and the resulting dark purple solution was stirred overnight at room temperature. Crystals suitable for sc-XRD were obtained from a highly concentrated solution after storage at −30 °C. However, the product can be more conveniently isolated as a purple powder by removing all volatiles and washing the residue with n-hexane. Yield: 35 mg (52 μmol, 51%).
Anal. Calcd. for C41H47GaN2O2: C, 73.55, H, 7.08; N, 4.18; Found: C, 73.3, H, 7.09; N, 4.49%. ATR-IR: υ 3061, 3034, 2964, 2923, 2866, 1526, 1462, 1442, 1382, 1313, 1256, 1192, 1180, 1131, 1106, 1055, 1024, 921, 900, 883, 800, 769, 759, 627, 596, 581, 526, 472, 441, 420 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.22 (d, 3JHH = 6.6 Hz, 2 H, GaC12H6O2), 7.12 (d, 3JHH = 9.4 Hz, 2 H, GaC12H6O2), 7.22 (dd, 3JHH = 6.6, 8.3 Hz, 2 H, GaC12H6O2), 6.91 (s, 6 H, C6H3-2,6iPr2), 4.89 (s, 1 H, γ-CH), 3.35 (sept, 3JHH = 6.7 Hz, 4 H, CH(CH3)2), 1.54 (s, 6 H, ArNCCH3), 1.51 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2), 1.11 (d, 3JHH = 6.9 Hz, 12 H CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 172.1 (ArNCCH3), 144.0, 137.8, 128.5, 124.7 (ArC), 146.6, 134.1, 127.4, 127.1, 124.7, 123.2, 116.7 (GaC12H6O2), 96.5 (γ-CH), 28.9 (CH(CH3)2), 24.8, 24.7 (CH(CH3)2), 23.2 (ArNCCH3).
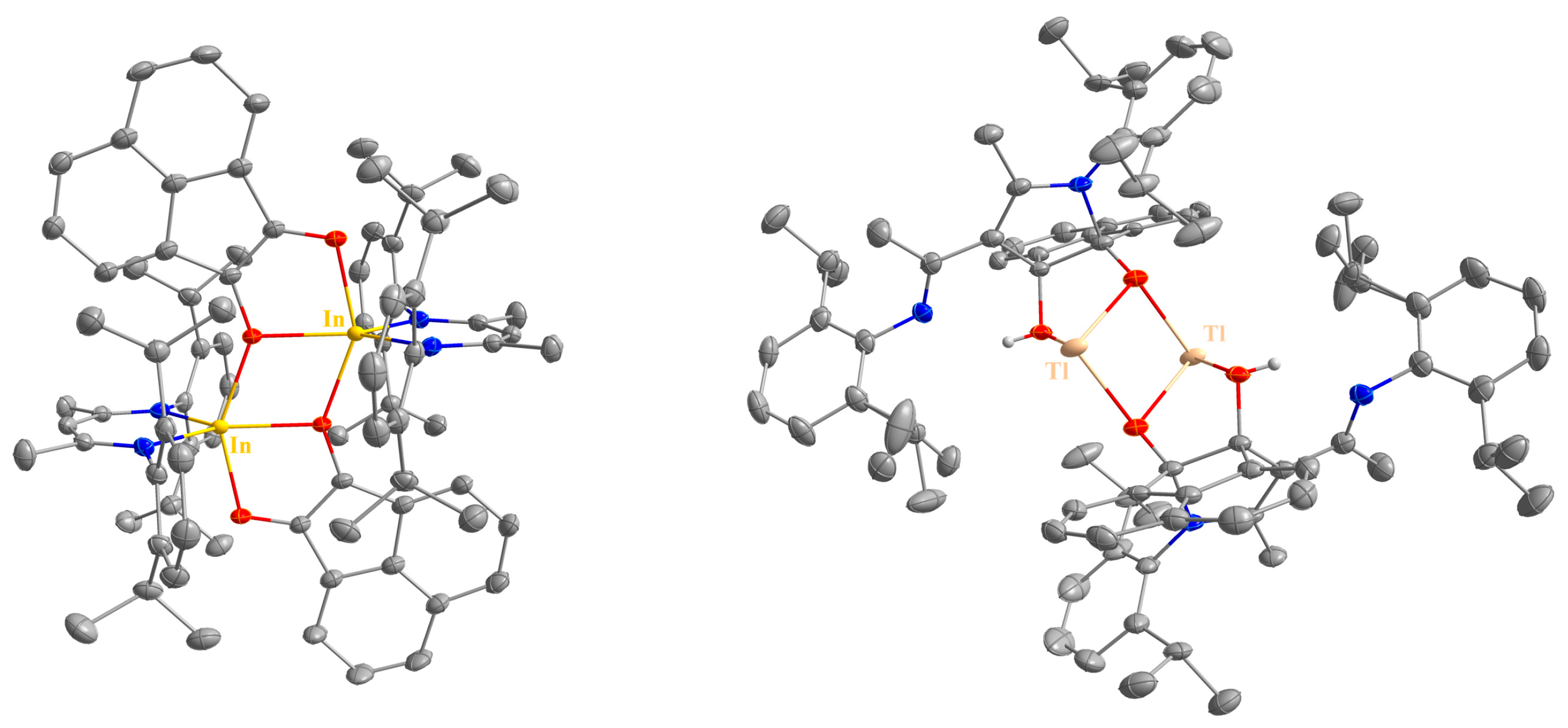
4.6. Synthesis of LIn(C12H6O2) (6)
200 mg of LIn (376 μmol) and 68.4 mg of acenaphthenequinone (376 μmol) were dissolved in 5 mL of toluene and the resulting dark purple suspension was stirred overnight at ambient temperature. The product was obtained as a purple powder by filtration. Yield: 201 mg (281 μmol, 75%).
Anal. Calcd. for C41H47InN2O2: C, 68.91, H, 6.63; N, 3.92; Found: C, 68.7, H, 6.54; N, 3.66%. ATR-IR: υ 2967, 2951, 2925, 2866, 1547, 1522, 1458, 1438, 1409, 1382, 1367, 1328, 1316, 1266, 1172, 1130, 1101, 1079, 1055, 1030, 935, 902, 854, 798, 761, 730, 592, 536, 481, 428 cm−1. 1H NMR (400 MHz, thf-d8, 25 °C): δ 7.52, 7.36, 7.24, 7.07 (m, 6 H, InC12H6O2), 7.18, 7.07, 6.63 (m, 6 H, C6H3-2,6iPr2), 5.40 (s, 1 H, γ-CH), 3.72 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.09 (sept, 3JHH = 6.8 Hz, 2 H, CH(CH3)2), 1.76 (s, 6 H, ArNCCH3), 1.27 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 1.19 (d, 3JHH = 6.7 Hz, 6 H CH(CH3)2), 0.20 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2), −0.28 (d, 3JHH = 6.8 Hz, 6 H CH(CH3)2). 13C NMR (100.6 MHz, thf-d8, 25 °C): δ 172.7 (ArNCCH3), 150.6, 145.2, 144.0, 143.5, 137.2, 135.7, 135.5, 127.6, 127.4, 127.1, 126.9, 126.1, 125.3, 125.2, 123.0, 121.3, 120.0, 116.9 (ArC + InC12H6O2), 95.6 (γ-CH), 28.5, 28.0 (CH(CH3)2), 26.4, 25.5, 24.6, 22.2 (CH(CH3)2), 25.7 (ArNCCH3).
4.7. Synthesis of LTl(C14H10O2) (7)
A total of 100 mg of LTl (161 μmol) and 27.8 mg of acenaphthenequinone (153 μmol) were cooled to −80 °C and 2 mL of n-hexane was added. The resulting mixture was warmed to ambient temperature within 8 h, and the resulting off-white solid (60 mg) was separated by filtration. The 1H NMR spectrum of this solid is essentially consistent with that obtained from isolated crystals from the reaction mixture, which were characterized by sc-XRD and are highly reproducible between different batches. All attempts to further purify compound 9 failed due to decomposition in the solution state, resulting in the formation of the protonated ligand LH.
1H NMR (400 MHz, C6D6, 25 °C): δ 7.62–6.84 (m, 22 H, C6H3-2,6iPr2 and TlC12H6O2), 4.04, 3.20, 2.57, 1.98 (broad, 4 H, CH(CH3)2), 1.91, 1.85 (s, 6 H, ArNCCH3), 1.24, 1.21, 1.20, 1.12, 1.11, 0.74, 0.49, −0.18 (d, 24 H, CH(CH3)2).
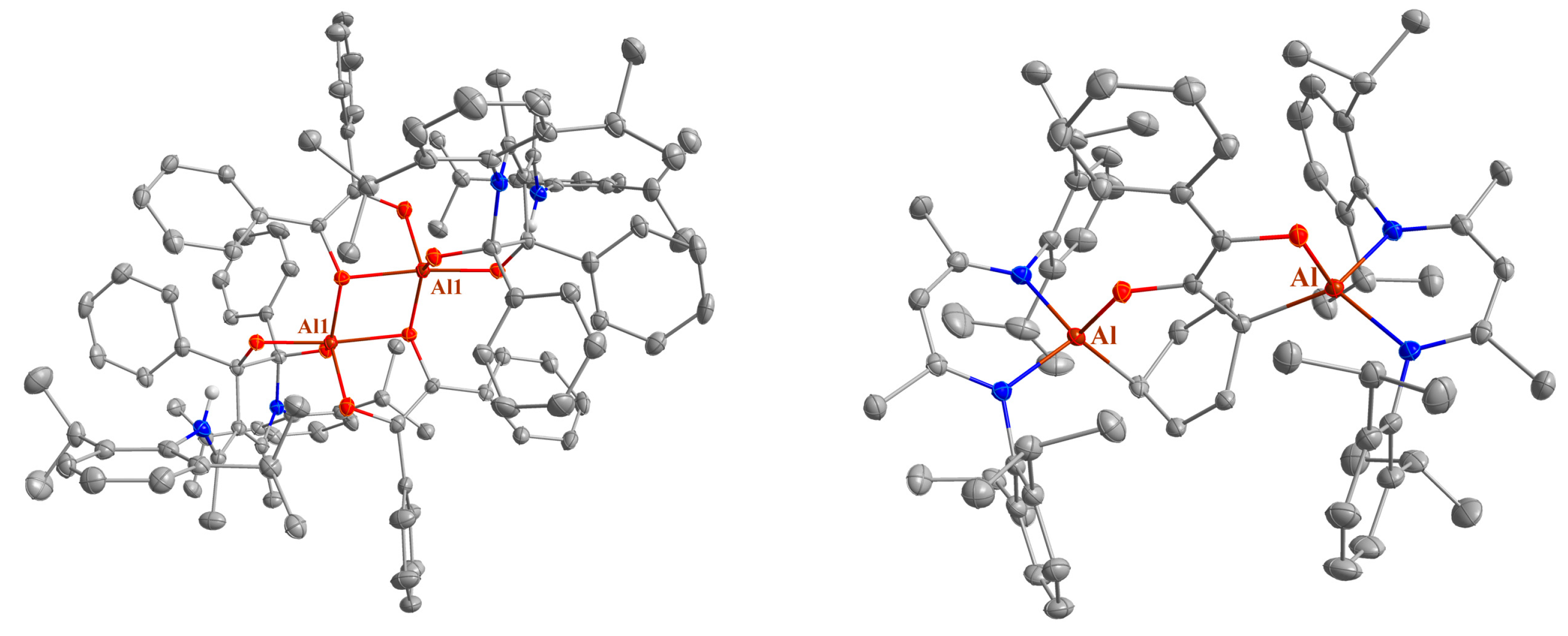
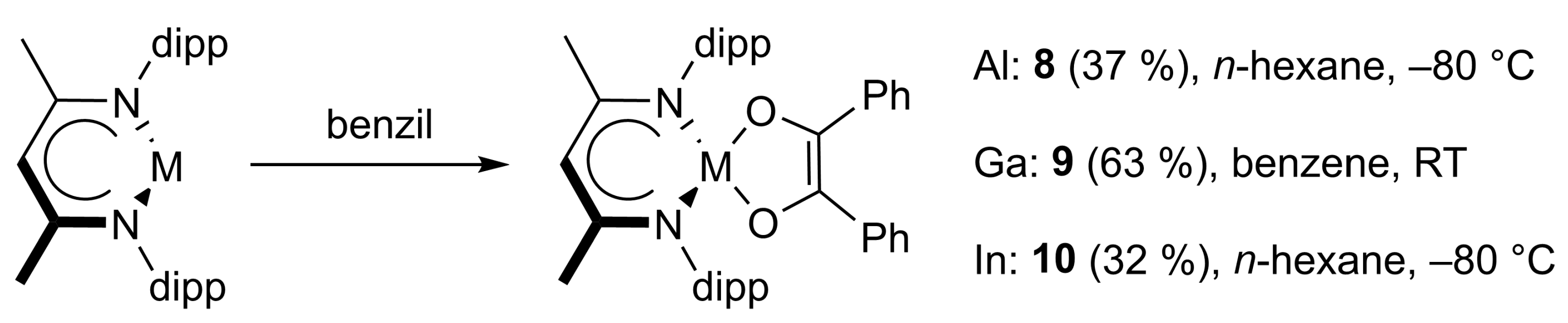
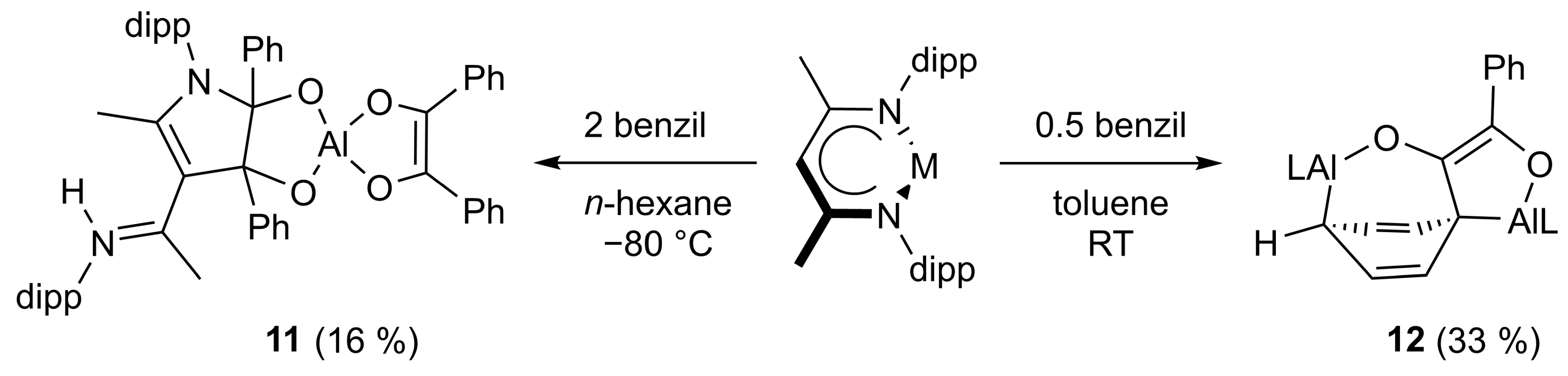
4.8. Synthesis of LAl(C14H10O2) (8) and LAl(C14H10O2)2 (11)
LAl(C14H10O2)2 (11): 100 mg of LAl (225 μmol) and 94.6 mg of benzil (450 μmol) were cooled to −80 °C and 5 mL of n-hexane was slowly added and the mixture was warmed to ambient temperature overnight with stirring. The resulting solid was filtered off and dissolved in toluene. Storage at −30 °C afforded 30 mg of LAl(C14H10O2)2 as a crystalline yellow solid (suitable for sc-XRD). Yield: 40 mg (35 μmol, 16%).
Anal. Calcd. for C57H61AlN2O4: C, 79.14, H, 7.11; N, 3.24; Found: C, 78.7, H, 7.17; N, 3.23%. ATR-IR: υ 2964, 2928, 2868, 1578, 1529, 1462, 1442, 1371, 1334, 1316, 1253, 1214, 1131, 1057, 1023, 933, 839, 803, 776, 759, 747, 732, 709, 695, 664, 613, 577, 528, 480 cm−1. 1H NMR (600 MHz, C6D6, 25 °C): δ 11.76 (s, 1 H, ArNHCCH3), δ 7.88 (d, 3JHH = 7.7 Hz, 2 H, C6H5), 7.82 (d, 3JHH = 7.7 Hz, 2 H, C6H5), 7.32 (d, 3JHH = 7.7 Hz, 1 H, C6H5), 7.27 (m, 3 H, C6H5), 7.22 (t, 3JHH = 7.6 Hz, 2 H, C6H5), 7.13-6.93 (m, 11 H, C6H3-2,6iPr2 and C6H5), 6.88 (t, 3JHH = 7.3 Hz, 1 H, C6H5), 6.83 (t, 3JHH = 7.6 Hz, 1 H, C6H5), 6.80 (d, 3JHH = 7.7 Hz, 1 H, C6H5), 6.72 (t, 3JHH = 7.6 Hz, 2 H, C6H5), 6.65 (t, 3JHH = 7.4 Hz, 1 H, C6H5), 6.62 (t, 3JHH = 7.7 Hz, 1 H, C6H5), 6.07 (d, 3JHH = 7.9 Hz, 1 H, C6H5), 4.69 (sept, 3JHH = 6.7 Hz, 1 H, CH(CH3)2), 4.45 (sept, 3JHH = 6.7 Hz, 1 H, CH(CH3)2), 3.35 (sept, 3JHH = 6.5 Hz, 1 H, CH(CH3)2), 2.20 (sept, 3JHH = 6.8 Hz, 1 H, CH(CH3)2), 2.11 (s, 3 H, C6H5CH3), 1.92 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 1.78 (s, 36 H, ArNCCH3), 1.69 (s, 3 H, ArNCCH3), 1.47 (d, 3JHH = 6.8 Hz, 3 H, CH(CH3)2), 1.41 (d, 3JHH = 6.4 Hz, 3 H, CH(CH3)2), 1.22 (d, 3JHH = 6.5 Hz, 3 H, CH(CH3)2), 1.01 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 0.92 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 0.90 (d, 3JHH = 7.0 Hz, 3 H, CH(CH3)2), 0.45 (d, 3JHH = 6.7 Hz, 3 H, CH(CH3)2). Due to the low solubility of the sample, no meaningfull 2D NMR spectrum could be recorded. Signals in the aromatic region could not be assigned. 13C NMR (150.9 MHz, C6D6, 25 °C): δ 168.8, 166.6 (ArNCCH3), 137.9, 129.3, 128.6, 125.7, 21,4 (toluene), 150.7, 148.0, 146.6, 146.6, 145.9, 145.6, 143.0, 139.5, 137.0, 134.7, 133.4, 133.1, 131.4, 130.3, 130.2, 129.2, 129.0, 128.9, 127.9, 127.8, 126.9, 126.5, 126.5, 126.3, 126.1, 126.0, 125.4, 124.7, 124.3, 124.1, 124.0, 123.6, 115.1, 110.6 (C6H3-2,6iPr2 and (COC6H5)2), 89.0 (γ-CH), 29.1, 28.2, 28.1, 28.1 (CH(CH3)2), 27.4, 26.4, 25.6, 25.5, 24.2, 24.1, 23.4, 23.1, (CH(CH3)2), 19.1, 17.8 (ArNCCH3).
LAl(C14H10O2) (8): The mother liquor from the synthesis of LAl(C14H10O2)2 (11) was dried in vacuo and the resulting residue was washed with 20 mL of n-hexane, yielding 40 mg of LAl(C14H10O2). The washing liquid was concentrated to 10 mL and stored at −30 °C to give a second fraction of LAl(C14H10O2). Yield: 55 mg (84 μmol, 37%).
Anal. Calcd. for C43H51AlN2O2: C, 78.87, H, 7.85; N, 4.28; Found: C, 78.5, H, 7.79; N, 4.25%. ATR-IR: υ 3056, 3018, 2962, 2926, 2868, 1585, 1535, 1462, 1442, 1384, 1316, 1253, 1176, 1105, 1055, 1022, 925, 912, 900, 803, 790, 769, 759, 736, 695, 643, 546, 507, 447, 416 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.48 (d, 3JHH = 7.8 Hz, 4 H, C6H5), 7.02 (m, 6 H, C6H3-2,6iPr2), 6.99 (m, 4 H, C6H5), 6.86 (tt, 3JHH = 7.2 Hz, 4JHH = 1.4 Hz, 2 H, C6H5), 5.03 (s, 1 H, γ-CH), 3.35 (sept, 3JHH = 6.7 Hz, 4 H, CH(CH3)2), 1.55 (s, 6 H, ArNCCH3), 1.44 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.08 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 172.5 (ArNCCH3), 139.7 (COAl), 138.8, 127.9, 127.6, 125.6 (C6H5), 144.4, 138.0, 128.2, 124.6 (ArC), 98.2 (γ-CH), 28.9 (CH(CH3)2), 25.0, 24.7 (CH(CH3)2), 23.2 (ArNCCH3).
4.9. Synthesis of LGa(C14H10O2) (9)
A total of 100 mg of LGa (205 μmol) and 43.1 mg of benzil (205 μmol) were combined with 2 mL of benzene and the mixture was stirred overnight, during which the product precipitated as an orange crystalline solid. Yield: 90 mg (129 μmol, 63%).
Anal. Calcd. for C43H51GaN2O2: C, 74.03, H, 7.37; N, 4.02; Found: C, 73.6, H, 7.37; N, 4.15%. ATR-IR: υ 2962, 2927, 2865, 1585, 1533, 1491, 1464, 1442, 1384, 1318, 1260, 1180, 1105, 1065, 1051, 1022, 929, 912, 803, 765, 724, 699, 683, 627, 535, 481, 445 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.47 (d, 3JHH = 7.8 Hz, 4 H, C6H5), 7.01 (m, 10 H, C6H5, and C6H3-2,6iPr2), 6.87 (tt, 3JHH = 7.1 Hz, 4JHH = 1.4 Hz, 2 H, C6H5), 4.90 (s, 1 H, γ-CH), 3.35 (sept, 3JHH = 6.8 Hz, 4 H, CH(CH3)2), 1.54 (s, 6 H, ArNCCH3), 1.46 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2), 1.09 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 172.1 (ArNCCH3), 139.6 (COGa), 139.3, 128.6, 127.6, 125.4 (C6H5), 144.1, 138.5, 128.6, 124.5 (ArC), 96.4 (γ-CH), 28.9 (CH(CH3)2), 24.8, 24.6 (CH(CH3)2), 23.4 (ArNCCH3).
4.10. Synthesis of LIn(C14H10O2)·MeCN (10)
A total of 100 mg of LIn (188 μmol) and 39.4 mg of benzil (188 μmol) were balanced in a Schlenk flask and cooled to −80 °C. 5 mL of n-hexane was added to the solids, and the mixture was stirred overnight and allowed to warm to room temperature. All volatiles were removed under reduced pressure, the residue was dissolved in a mixture of 1 mL benzene and 10 mL acetonitrile and concentrated to about 1 mL. 10 was obtained as an orange crystalline solid (suitable for sc-XRD) after storage at 6 °C. Yield: 45 mg (61 μmol, 32%).
ATR-IR: υ 2962, 2924, 2868, 2296, 2268, 1592, 1519, 1435, 1384, 1356, 321, 1270, 1178, 1133,1051, 1021, 924, 906, 863, 800, 757, 711, 693, 665, 609, 530, 439 cm−1. 1H NMR (400 MHz, thf-d8, 25 °C): δ 7.24-7.12 (m, 6 H, C6H3-2,6iPr2), 6.90 (d, 3JHH = 7.0 Hz, 4 H, C6H5), 6.74 (d, 3JHH = 7.4 Hz, 4 H, C6H5), 6.70 (t, 3JHH = 7.2 Hz, 2 H, C6H5), 5.19 (s, 1 H, γ-CH), 3.32 (sept, 3JHH = 6.9 Hz, 4 H, CH(CH3)2), 1.94 (s, 2.5 H, MeCN), 1.84 (s, 6 H, ArNCCH3), 1.25 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.23 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2). 13C NMR (100.6 MHz, thf-d8, 25 °C): δ 172.5 (ArNCCH3), 139.7 (COIn), 143.8, 143.3, 127.1, 124.4 (ArC), 142.9, 128.6, 126.9, 123.8 (C6H5), 96.9 (γ-CH), 28.8 (CH(CH3)2), 25.0, 24.5 (CH(CH3)2), 24.5 (ArNCCH3), 117.3, 0.4 (MeCN). 100 mg LGa
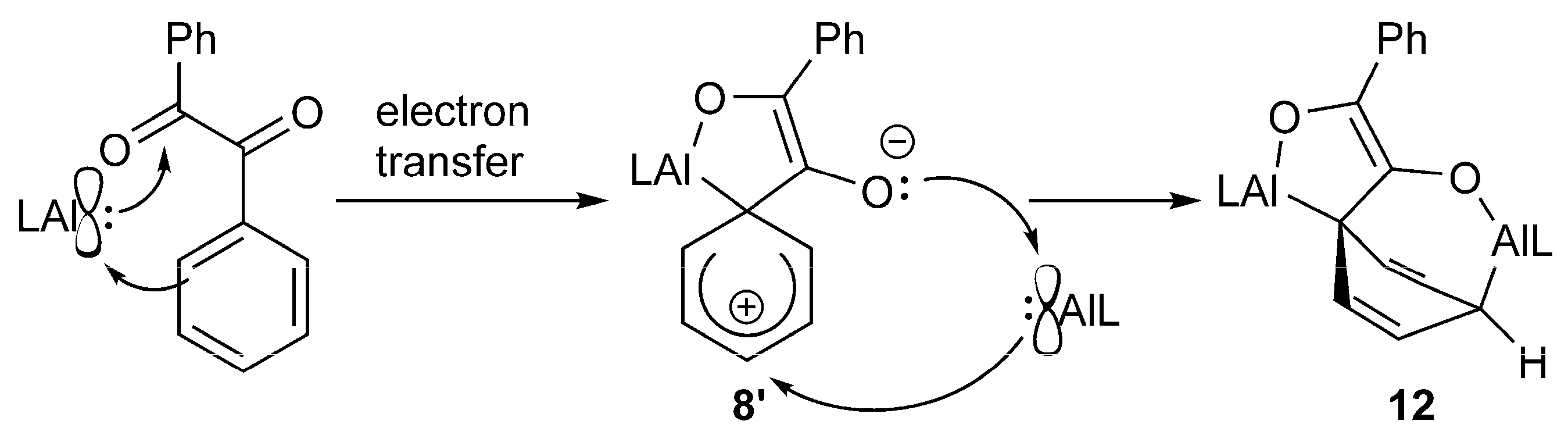
4.11. Synthesis of (LAl)2(C14H10O2) (12)
A total of 23.6 mg of benzil (113 μmol) dissolved in 1 mL of toluene was added dropwise to a solution of 100 mg of LAl (225 μmol) in 1 mL of toluene. The mixture was stirred for 2 days, after which an orange precipitate formed. The suspension was stored overnight at −6 °C. 40 mg of 12 was obtained as an orange powder by filtration. The filtrate consists mainly of LAl and (LAl)(C14H10O2) (8). Yield: 40 mg (36 μmol, 33%).
Anal. Calcd. for C72H92Al2N4O2: C, 78.65, H, 8.43; N, 5.10; Found: C, 78.9, H, 8.51; N, 4.90%. ATR-IR: υ 2964, 2928, 2865, 1529, 1462, 1440, 1384, 1316, 1253, 1179, 1156, 1102, 1061, 1022, 972, 937, 899, 873, 800, 786, 774, 759, 734, 693, 618, 532, 480, 445, 422 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 8.53 (d, 3JHH = 7.9 Hz, 2 H, C6H5), 7.42 (dd, 3JHH = 7.0, 8.4 Hz, 2 H, C6H5), 7.24–7.11 (m, 8 H, C6H3-2,6iPr2), 7.07 (t, 3JHH = 7.3 Hz, 1 H, C6H5), 7.00 (m, 4 H, C6H3-2,6iPr2), 4.96 (s, 1 H, γ-CH), 4.88 (dd, 3JHH = 6.0, 8.4 Hz, 2 H, AlC(CHCH)2CHAl), 4.84 (s, 1 H, γ-CH), 4.02 (d, 3JHH = 8.4 Hz, 2 H, AlC(CHCH)2CHAl), 3.45 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.33–3.20 (m, 4 H, CH(CH3)2), 3.07 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 1.92 (t, 3JHH = 6.0, 1 H, AlC(CHCH)2CHAl), 1.53 (s, 6 H, ArNCCH3), 1.52 (s, 6 H, ArNCCH3), 1.33 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.30 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2), 1.25 (d, 3JHH = 7.0 Hz, 6 H, CH(CH3)2), 1.16 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.12 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 0.96 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2), 0.92 (d, 3JHH = 6.78 Hz, 6 H, CH(CH3)2), 0.82 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 170.8, 170.3 (ArNCCH3), 148.5, 132.7 (COAl), 140.5, 126.9, 126.9, 123.0 (C6H5), 128.6, 123.3 (AlC(CHCH)2CHAl), 146.2, 145.3, 143.3, 142.5, 141.4, 141.3, 127.4, 127.1, 125.3, 124.7, 123.8, 123.2 (ArC), 98.1, 97.4 (γ-CH), 39.6, 32.7 (AlC(CHCH)2CHAl), 30.0, 28.9, 28.5, 28.2 (CH(CH3)2), 26.6, 26.5, 25.0, 24.9, 24.8, 24.6, 23.8, 23.6 (CH(CH3)2), 23.8 (ArNCCH3). Peaks in italic were only observed in HSQC or HMBC respectively.
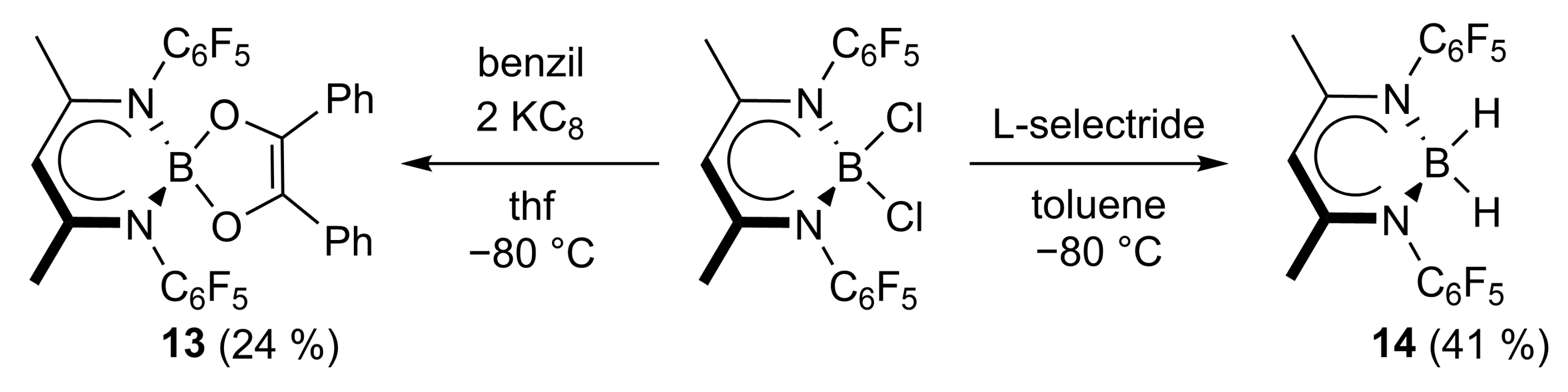
4.12. Synthesis of L’B(C14H10O2) (13)
A total of 56.9 mg of KC8 (421 μmol) and 41.1 mg of benzil (198 μmol) were cooled to −80 °C, 5 mL of thf was added and the resulting mixture was warmed to room temperature upon stirring. The liquid phase was transferred through a filter cannula into a cooled (−80 °C) Schlenk flask containing 100 mg of L’BCl2 (198 μmol). All volatiles were removed at room temperature in a vacuo, and the resulting residue was extracted with 5 mL of toluene. The extract was concentrated and stored at −30 °C, yielding 20 mg of an orange crystalline solid. The mother liquor was further concentrated to give a second fraction. Yield: 30 mg (46 μmol, 24%).
Anal. Calcd. for C31H17BF10N2O2: C, 57.26, H, 2.64; N, 4.31; Found: C, 57.4, H, 2.63; N, 4.31%. ATR-IR: υ 1564, 1508, 1469, 1450, 1392, 1290, 1264, 1130, 1084, 1065, 1042, 1022, 987, 925, 871, 761, 724, 693, 663, 619, 597, 523, 457 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.62–7.57 (m, 4 H, C6H5), 6.98 (t, 3JHH = 7.8 Hz, 4 H, C6H5), 6.86 (t, 3JHH = 7.41 Hz, 2 H, C6H5), 4.69 (s, 1 H, γ-CH), 1.26 (s, 6 H, ArNCCH3). 11B NMR (128.4 MHz, C6D6, 25 °C): δ 7.0. 13C NMR (100.6 MHz, C6D6, 25 °C): δ 166.2 (ArNCCH3), 136.7 (COB), 133.4, 128.4, 127.1, 126.1 (C6H5), 97.6 (γ-CH), 20.6 (ArNCCH3). 13C {19F} NMR (150.9 MHz, C6D6, 25 °C): δ 144.4, 141.0, 137.9, 116.4 (C6F5). 19F NMR (376.5 MHz, C6D6, 25 °C): δ −143.5 (d, 3JHH = 17.6 Hz, 4 F, C6F5), −153.8 (t, 3JHH = 22.5 Hz, 2 F, C6F5), −162.3 (m, 4 F, C6H5).
4.13. Synthesis of L’BH2 (14)
At –80 °C, 783 μL (783 μmol) of a 1 M (n-hexane) L-selectride solution was added to 200 mg of L’BCl2 (391 μmol) dissolved in 10 mL of toluene. The reaction mixture was allowed to reach ambient temperature overnight. All volatiles were then removed in vacuo, and the resulting residue was extracted with 10 mL of n-pentane. The extract was concentrated and stored at −30 °C to give 70 mg of 14 as a yellow crystalline solid. Yield: 70 mg (158 μmol, 41%).
Anal. Calcd. for C17H9BF10N2: C, 46.19, H, 2.05; N, 6.34; Found: C, 46.6, H, 2.32; N, 6.16%. ATR-IR: υ 2964, 2428, 2370, 2291, 2192, 1582, 1549, 1504, 1467, 1438, 1392, 1344, 1260, 1206, 1069, 1051, 1024, 1009, 989, 939, 817, 788, 782, 726, 647, 561, 472 cm−1. 1H NMR (400 MHz, C6D6, 25 °C): δ 4.77 (s, 1 H, γ-CH), 3.77 (s (br), 1 H, BH2), 3.46 (s (br), 1 H, BH2), 1.24 (s, 6 H, ArNCCH3). 11B NMR (192.5 MHz, C6D6, 25 °C): δ 7.0 (B). 13C NMR (100.6 MHz, C6D6, 25 °C): δ 167.6 (ArNCCH3), 143.7 (m, 2JHH = 250 Hz, C6F5), 140.3 (m, 2JHH = 252 Hz, C6F5), 138.2 (m, 2JHH = 252 Hz, C6F5), 120.1 (td, JHH = 17.8, 5.2 Hz, C6F5), 99.6 (γ-CH), 19.8 (ArNCCH3). 19F NMR (376.5 MHz, C6D6, 25 °C): δ −143.7 (d, 3JHH = 20.8 Hz, 4 F, C6F5), −156.2 (t, 3JHH = 22.3 Hz, 2 F, C6F5), −161.9 (td, JHH = 23.3, 6.1 Hz, 4 F, C6H5).
4.14. Crystallographic Details
Crystals were mounted on nylon loops in inert oil. Data of were collected on a Bruker AXS D8 Kappa diffractometer (
2, 5,
7,
9,
10,
11,
12b) with APEX2 detector (monochromated Mo
Ka radiation,
λ = 0.71073 Å) and on a Bruker AXS D8 Venture diffractometer (
1,
4,
6,
8, 12a,
13) with Photon II detector (monochromated Cu
Ka radiation,
λ = 1.54178 Å, microfocus source) at 100(2) K. Structures were solved by Direct Methods (SHELXS-2013) [
40] and refined anisotropically by full-matrix least-squares on
F2 (SHELXL-2014) [
41,
42,
43]. Absorption corrections were performed semi-empirically from equivalent reflections based on multi-scans (Bruker AXS APEX2). Hydrogen atoms were refined using a riding model or rigid methyl groups.
1: An isopropyl group is disordered over two positions. C24 and C24′ were refined with common positions and displacement parameters (EXYZ, EADP). RIGU restraints were applied to the displacement parameters of the disordered atoms.
2: Two isopropyl groups are disordered over two positions. Their bond lengths and angles were restrained to be equal (SADI) and RIGU restraints were applied to their displacement parameters. Additional SIMU restraints were applied to the group in residue 2. In residue 1, the diol ligand is disordered over two positions. All corresponding bond lengths were restrained to be equal (SADI) and RIGU restraints were used to refine the displacement parameters. Atoms in close proximity (O4, O4′ and C33, C33′) were refined with common displacement parameters (EADP). One solvent molecule is disordered over two positions. All bond lengths and angles of the solvent molecules were restrained to be equal (SADI) and the molecule was restrained to be planar (FLAT). RIGU restraints were applied to the displacement parameters of the solvent atoms. The quantitative results of the disordered moieties should be scrutinized, and especially the data for the diol ligand may be unreliable. In addition, ice formed during the measurement and combined with a scattering of the mount/oil used as glue, clearly visible background scattering was found in the frames. This leads to some distorted intensities. The resolution of (090) could be related to the (103) reflection of water and was therefore omitted. Three reflections shaded by the beamstop were also ignored in the refinement. The remaining most disagreeable reflections have a resolution of >2 Å with Fobs higher than Fcalc. 2 was refined as a 2-component inversion twin.
5: The crystal was a non-merohedral twin and the model was refined against de-twinned HKLF4 data. At low angles, some disagreeable reflections are found. These are either due to poor separation of overlaps in the integration or, more likely, to background scattering caused by the icing of the crystal. Three reflections shaded by the beam-stop were ignored in the refinement (OMIT).
6: The structure contains a 2-methyltetrahydrofuran molecule highly disordered over an inversion center. The final refinement was conducted with a solvent-free data set from a PLATON/SQUEEZE run [
44]. The molecule was included in the sum formula for completeness.
7: An isopropyl group is disordered over two positions. Its bond lengths and angles were restrained to be equal (SADI) and RIGU restraints were applied to its displacement parameters. The structure contains two highly disordered
n-hexane molecules. The final refinement was conducted with a solvent-free data set from a PLATON/SQUEEZE run [
44]. The molecules were included in the sum formula for completeness.
9: A diisopropylphenyl group is disordered over two positions. RIGU restraints were applied to the displacement parameters of the corresponding atoms.
12a: A benzene molecule is disordered over two positions. RIGU restraints were applied to the displacement parameters of the atoms of the solvent molecules.
12b: An n-hexane molecule is disordered over a center of inversion. The bond lengths and bond angles of all solvent molecules were restrained to be equal and RIGU restraints were applied to the displacement parameters of their atoms. An additional SIMU restraint was used for the disordered molecule on the center of inversion. Its displacement parameters suggested further disorder that could not be resolved any further.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}