
Metal–Organic Cages: Applications in Organic Reactions



1
Key Laboratory of Resource Chemistry of Ministry of Education, Shanghai Key Laboratory of Rare Earth Functional Materials, Department of Chemistry, Shanghai Normal University, Shanghai 200234, China
2
Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia
*
Author to whom correspondence should be addressed.
Chemistry 2022, 4(2), 494-519; https://doi.org/10.3390/chemistry4020036
Submission received: 30 April 2022
/
Revised: 15 May 2022
/
Accepted: 16 May 2022
/
Published: 18 May 2022
(This article belongs to the Special Issue Metal-Organic Cages and Their Applications)
Abstract
:Supramolecular metal–organic cages, a class of molecular containers formed via coordination-driven self-assembly, have attracted sustained attention for their applications in catalysis, due to their structural aesthetics and unique properties. Their inherent confined cavity is considered to be analogous to the binding pocket of enzymes, and the facile tunability of building blocks offers a diverse platform for enzyme mimics to promote organic reactions. This minireview covers the recent progress of supramolecular metal–organic coordination cages for boosting organic reactions as reaction vessels or catalysts. The developments in the utilizations of the metal–organic cages for accelerating the organic reactions, improving the selectivity of the reactions are summarized. In addition, recent developments and successes in tandem or cascade reactions promoted by supramolecular metal–organic cages are discussed.
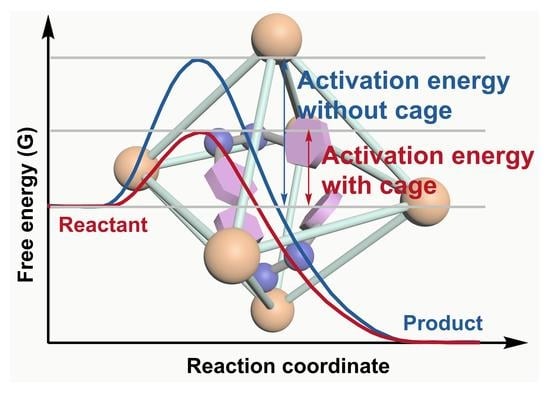
1. Introduction
Catalysis, one of the twelve principles of green chemistry [1], is considered to be a fundamental technology for saving energy and protecting the environment [2]. The ultimate aim of catalysis is to improve catalytic efficiency and reduce waste with environmentally friendly reagents [3]. Considerable and continued research attention has been attracted by supramolecular catalysis, which can control the chemical reactivity by exploiting the intermolecular interaction [4,5,6]. By mimicking the working principles of structurally much more complex enzymes, the relatively simple artificial supramolecular catalysts with discrete and confined cavities can accomplish specific and powerful catalysis, with rate enhancements of 105-fold and higher compared to classical homogeneous catalysis in bulk solvent [7,8,9]. A number of different artificial supramolecular host systems have been developed in the last several decades, constructed by covalent bonds [10], the hydrophobic effect [11], hydrogen bonds [12], and metal–ligand interactions [13,14]. Among them, metal–organic cages have shown several advantages in their application for boosting organic reactions [15,16]. (i) The self-assembly of metal–organic cages is driven by the coordination between the metal ions and ligands, which requires less synthetic effort than covalent versions [17,18,19,20]. Consequently, the space, shape, and hydrophobic environment of the cavity can be well controlled by simply modulating the properties of the metal centers and/or the organic ligands [21]. This means that specific cavities could be well constructed for particular reactions [22,23]. (ii) Catalytic sites or catalysts can be easily incorporated onto the cage framework for catalysis. The catalytic moieties can either be introduced onto the organic ligands or onto the metal centers. Thus, the number and type of the catalytic sites or catalysts integrated in the cavity can be well designed. In this context, a high local concentration of the catalytic active sites can be obtained, which could lead to the improvement of catalytic performance. Moreover, the synergistic interaction between the catalytic active sites and the confined cavity (such as the electron transfer between the active sites and the cavity, the stabilization of the active sites by the cavity, and so on) may also contribute to the catalytic activity or selectivity [24,25,26]. (iii) Different and even incompatible catalysts or active sites could be spatially segregated by the cavities of the metal–organic cages [27]. This property is extremely desirable for the development of high efficient tandem or cascade reactions, where two or more successive independent catalytic steps were performed in a one pot [28]. Consequently, complicated and high value products could be efficiently prepared from simple starting materials with limited reaction steps, energy consumption, and waste [29].
In this mini-review, we focus on describing the organic reactions boosted by metal–organic cages, which are used as reaction vessels and also catalysts. We highlight the structure–property relationship between the cage and the reaction and the principles to boost organic reactions. We first summarize the methods for accelerating the organic reactions, followed by methods to improve the selectivity of the reactions, and finally we target tandem or cascade reactions, which have been extremely rare in supramolecular catalysis. Since there are many wonderful reviews about supramolecular catalysis [30,31,32,33,34], only selected examples of the above three aspects in the past ten years will be included. The synthesis of the metal–organic cages is outside the scope of this review. Readers who are interested in the self-assembly of the supramolecular cages and other related topics are referred to the excellent reviews [35,36,37,38].
2. Acceleration of Catalytic Activity by Metal–Organic Cages
Metal–organic cages are not only able to modulate the reactivity of the molecules by encapsulating them within their cavities, but are also capable of influencing the reactive states of the catalysts or the active sites in the framework of the cage. These characterizations of the cage can be used to accelerate the reaction rate. The strategies to accelerate organic reactions include (i) fixing the reagents in a favored reactive conformation in the cavity, (ii) significantly promoting the local concentration of the catalysts within the cavity or on the surface of the cavity, and (iii) stabilizing the active sites by the cage.
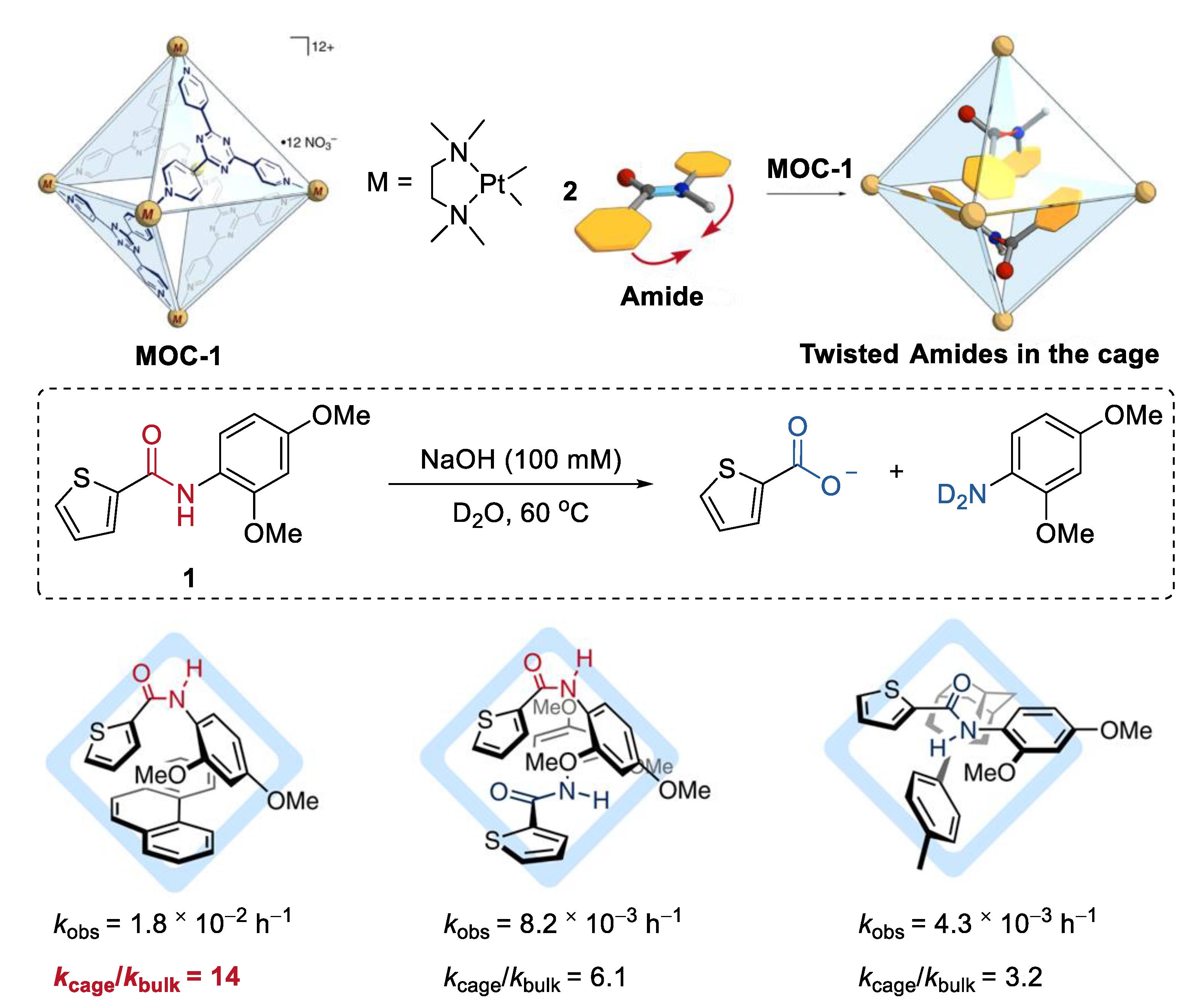
Improving the hydrolysis reactivity of amides by mechanically twisting them within the cavity of a molecular metal cage, without any chemical processes, was recently achieved by Fujita and co-workers [39]. A Td-symmetric platinum molecular metal cage (Pt6L4(NO3)12, MOC-1) containing electron-deficient panel ligands (2,4,6-tris(4-pyridyl)-1,3,5-triazine, L) was used as the host (Figure 1) [40]. When an aromatic amide (1) and a large modulator guest phenanthrene were co-encapsulated within the host with a large hydrophobic cavity under the basic solution at 60 °C [41], an outstanding rate enhancement with kcage/kbulk = 14 was observed (kcage and kbulk are rate constants with and without the cage). When two aromatic amides were co-included, rate enhancement with decreased kcage/kbulk value range from about 3 to 6 was also observed. However, when only one aromatic amide was located with its latent trans-planar conformation, negligible enhanced reactivity of the amide was observed. The crystal structures of the inclusion complex, aromatic amide, a large modulator guest, and the molecular metal cage (palladium instead of platinum [42]) reveal that the aromatic amide adopts a twisted cis conformation. A 1:1 cis–trans dimer formation from two aromatic amides was also found in the inclusion complex of two aromatic amides. The authors concluded that the enhanced hydrolysis reactivity of amides may result from the favored cis-twisted conformation over the trans-planar one due to the inclusion of them within the molecular metal cage [43].
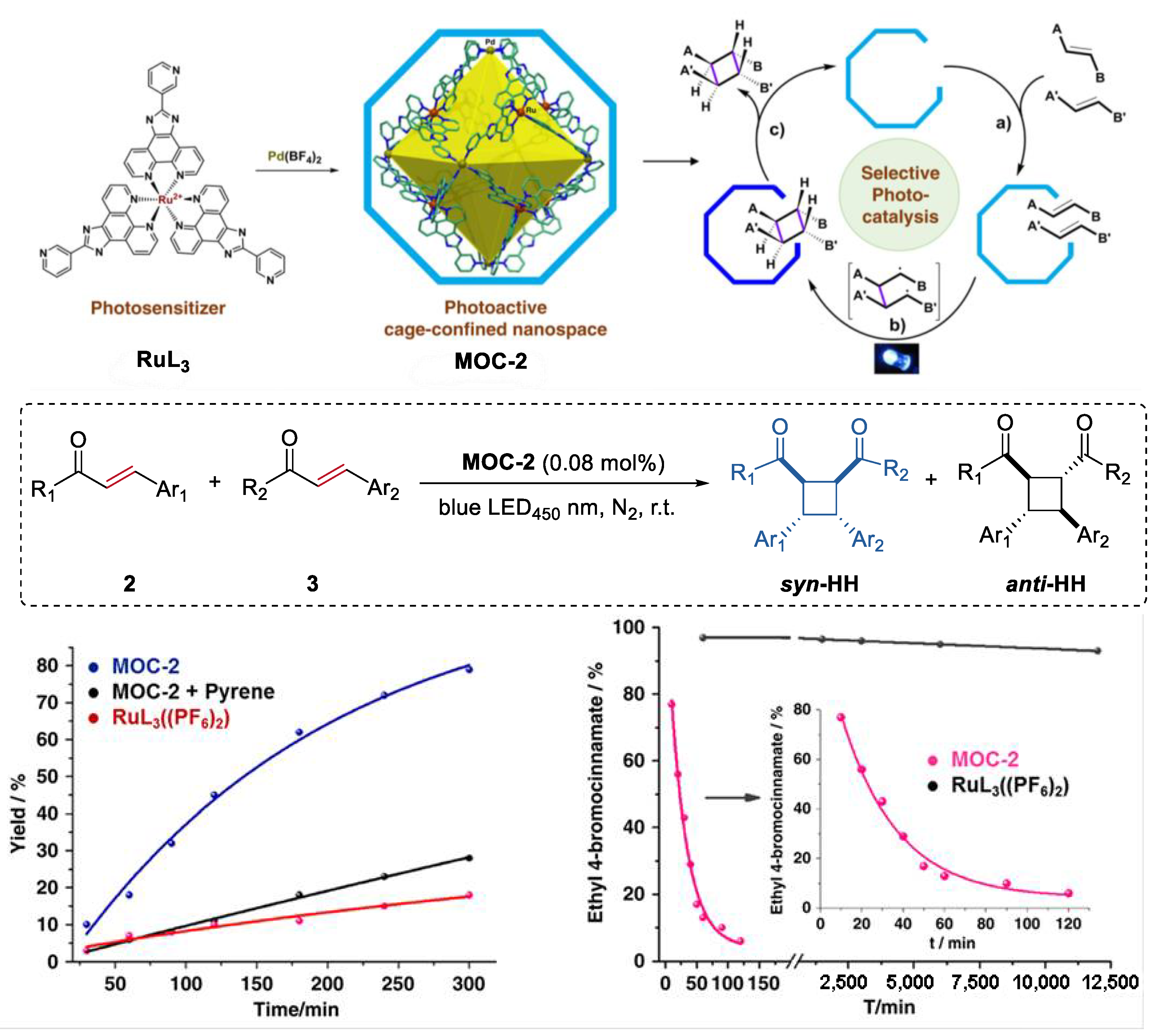
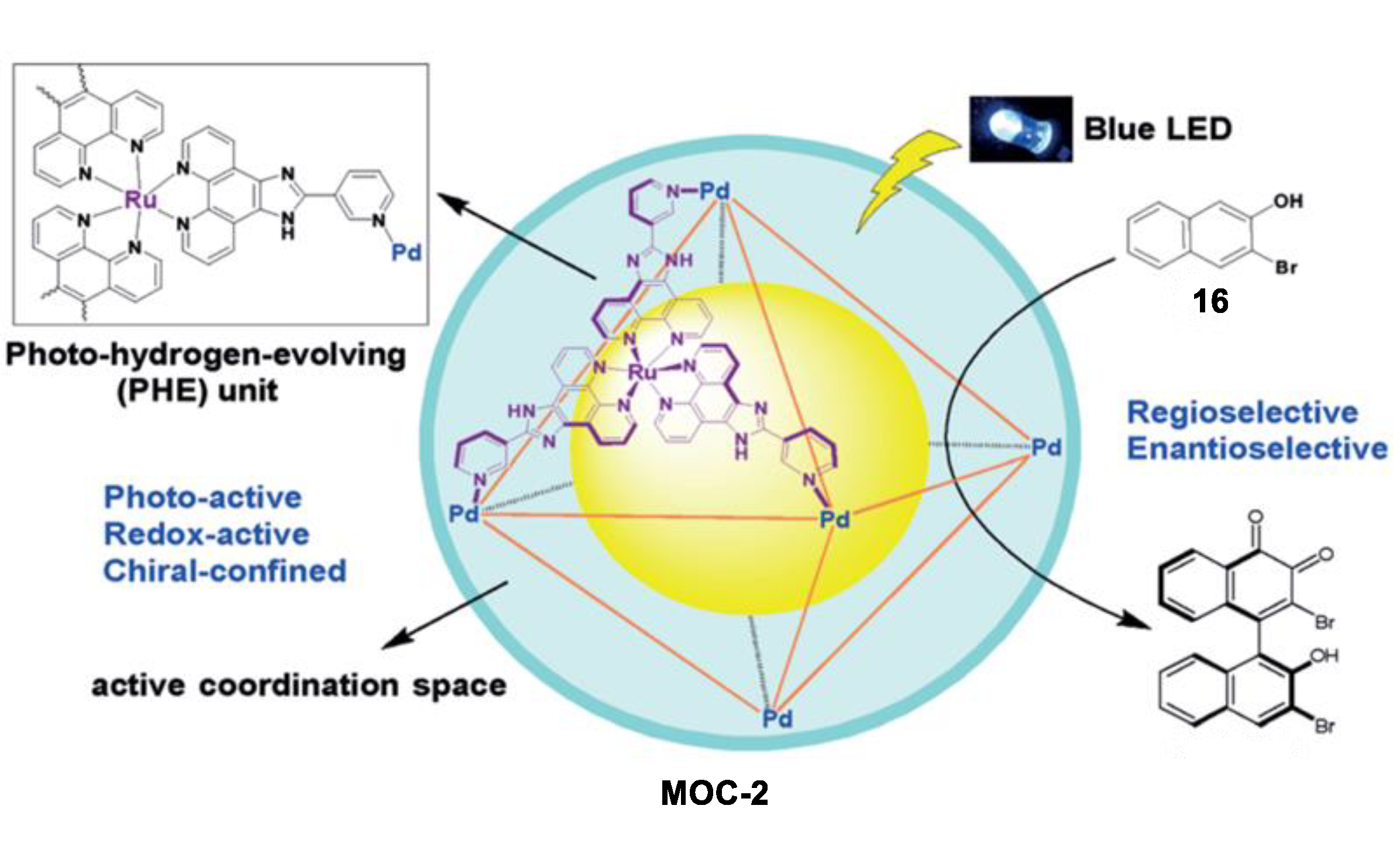
An unusual intermolecular [2 + 2] cycloaddition of acyclic alkenes such as chalcones, cinnamates, and benzylideneacetones to produce homo- and hetero- coupled syn-HH cyclobutanes with high diastereoselectivity and yields, which was catalyzed by a molecular metal cage-confined Ru photo-catalyst, was reported by Su and co-workers (Figure 2) [44]. The (Pd6RuL3)8(BF4)28 cage catalyst (MOC-2) was self-assembled from eight RuL3(PF6)2 (L = 2-(pyridin-3-yl)-1H-imidazo [4,5-f] [1,10]-phena-nthroline) visible-light photosensitizer and six Pd(BF4)2, resulting in a truncated octahedron photoactive nanospace for dynamic guests inclusion and exchange [45]. Mixing the cage catalyst with 6 eq of acyclic alkenes (2 and 3, for example chalcone and ethyl 4-bromocinnamate) in a DMSO-d6-D2O solution (1:3, v/v) resulted in a remarkable upfield shift of proton signals of substrates and splitting of host signals in the 1HNMR spectra, revealing the efficiently capture of the substrates in the cavity of the cage. Visible-light irradiation (450 nm, N2 atmosphere, 3 h, r.t.) leads to the formation of the cyclobutane products, as demonstrated by the disappearance of the resonances of substrate protons and the restoration of those of the cage catalyst. The cycloaddition reaction can even proceed very well in a 93% yield with at a very low photocatalyst loading of 0.08 mol%. To prove the catalytic efficiency of the cage catalyst, cycloaddition reactions between chalcone and methyl cinnamate as well as cycloaddition of methyl cinnamate were performed with the cage catalyst and the free photosensitizer RuL3(PF6)2. A ~12-fold and a surprising 1.044 × 104-fold rate enhancement was observed, respectively, in these two reactions. The box-like cavity of the cage as revealed by the crystal structure was believed to selectively arrange two substrates in one portal, and facilitated the following dimerization and dynamic processes of guest capture and delivery [46]. Moreover, the cage catalyst also shows nice water-solubility due to the multiple nitrogen atoms in the ligands of RuL3(PF6)2. The hydrophobic effect of the cage was considered to accelerate the transfer of the insoluble substrates and photoproducts out of the aqueous reaction media, thus helping the product to release from the host [47].
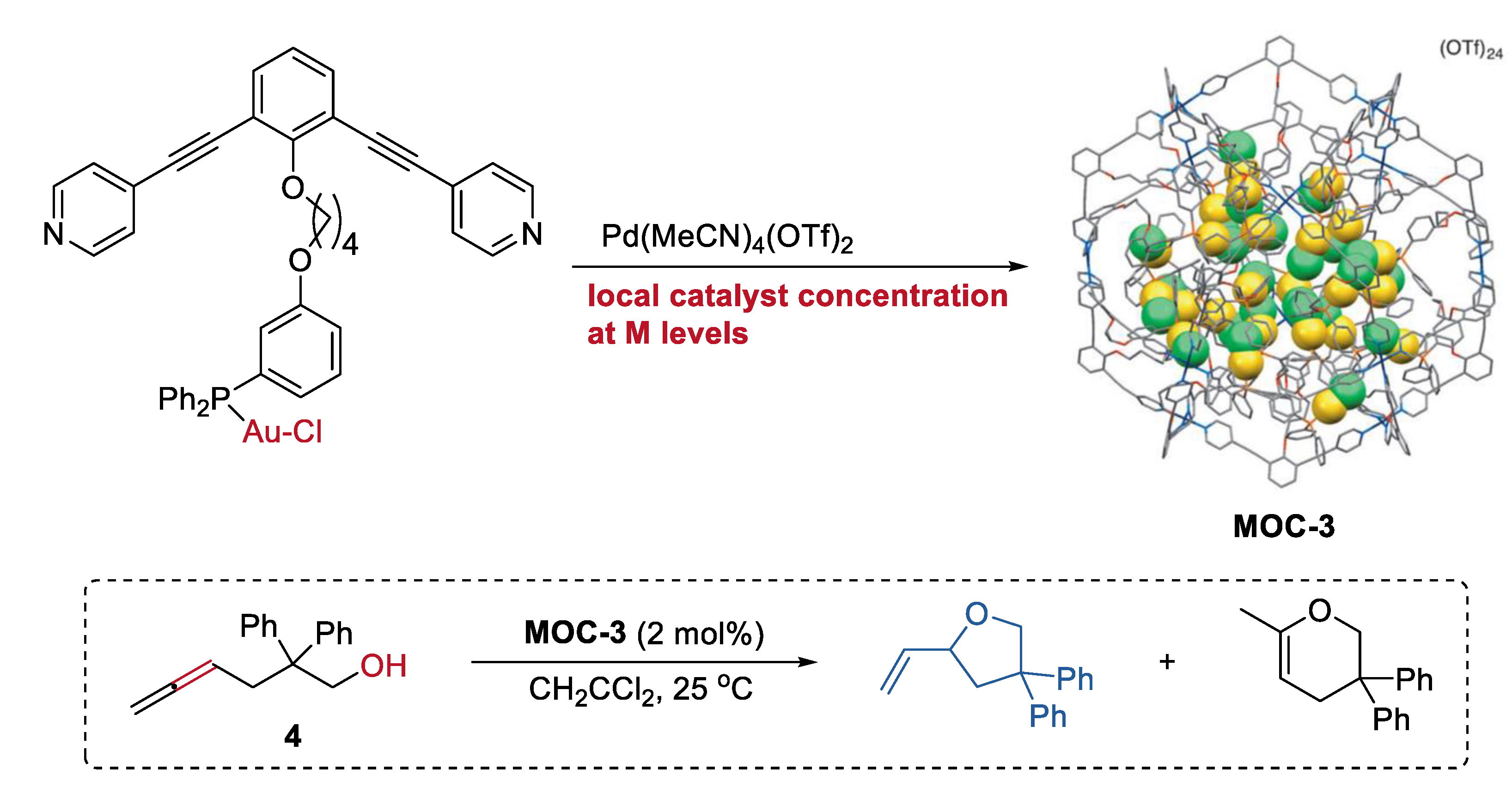
Increasing the local catalyst concentration to the mole level in a metal–organic cage, which is unfavorable for general homogenous transition-metal catalysts in solution, can significantly improve the activity and selectivity of catalyst [48]. A metal–organic cage (Pd12L24(OTf)24, MOC-3) with tunable high concentration (0.05 M to 1.1 M) of gold(I) catalyst inside (Figure 3) was prepared by Joost N. H. Reek and co-workers through a self-assembly strategy using ditopic nitrogen ligands (L) with Ph3PAuCl unit [49]. The hydroalkoxylation of the g-allenol (4) catalyzed by the metal–organic cage with a high local concentration of gold-chloride catalyst of 1.07 M selectively leads to the five-membered ring product with a high yield of 90% and a turnover numbers (TON) number of 1.86. The metal–organic cages with decreasing catalyst concentrations generally give lower yields of the product. In contrast, the free gold(I) complexes, including the AuCl, Ph3PAuCl, and Ph3PAuCl containing ditopic nitrogen ligands, are all inactive for the reaction under the same reaction conditions. Based on the UV/Vis spectra of the solution of the metal–organic cage, which show a broad shoulder for the d10–d10 aurophilic interactions of the metal complex [50], the author proposed that the unexpected activity and selectivity of the metal–organic cage may be a result of the new formed active gold species (active cationic gold species due to chloride dissociation or a multinuclear (P-Au-Cl)n complex) inside the cage. It should be mentioned that the cage collapsed when a lot of ring product was formed.
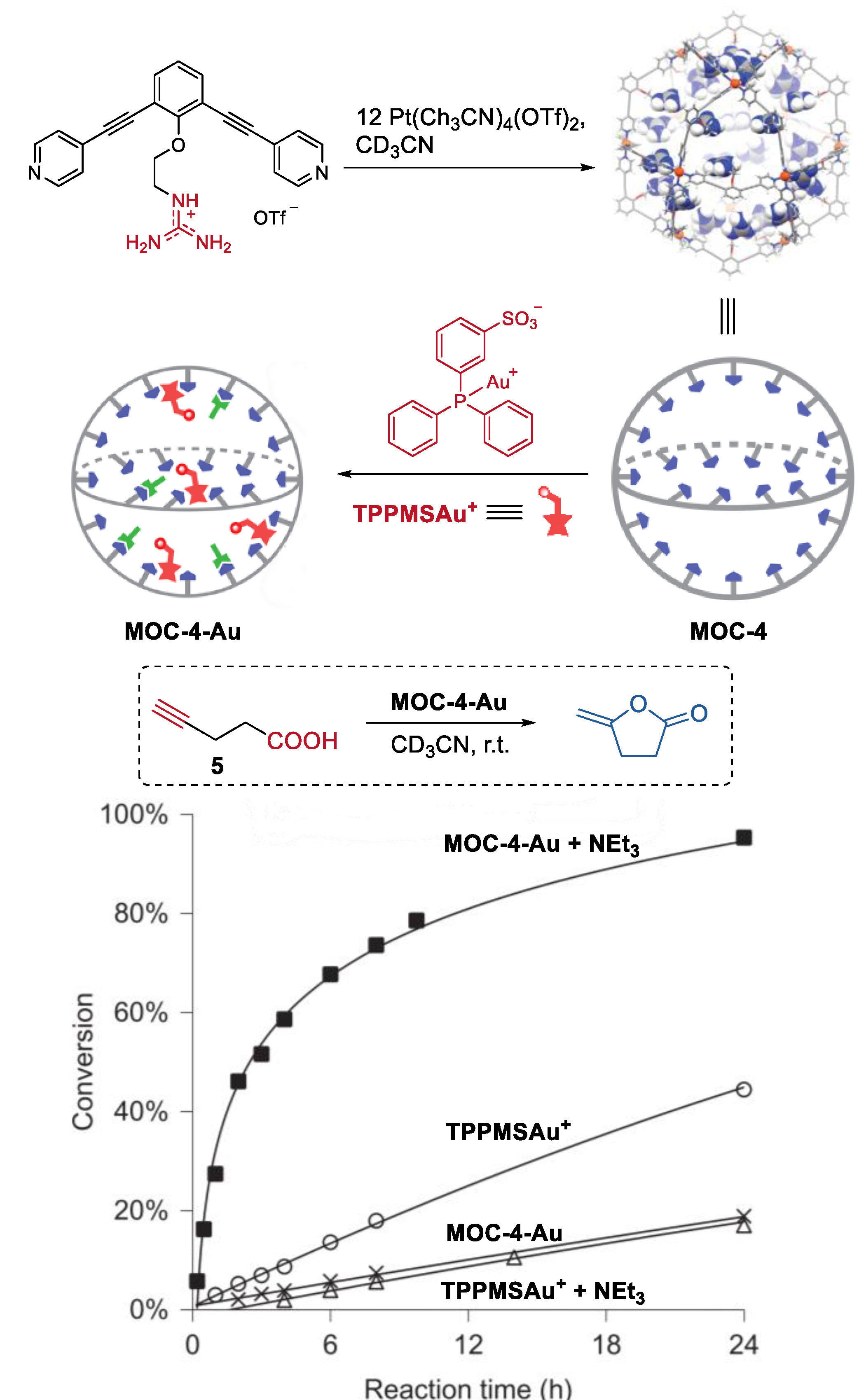
They also developed a non-covalent anchoring strategy to improve the catalyst concentration within the molecular metal cage (Figure 4) [51]. Guanidinium-binding motifs (L), which can form complexes with sulfonates and carboxylates via hydrogen bonding [52], were introduced into organic ligands of the cage (Pd12L24(OTf)48, MOC-4). The resulting molecular metal cage with 24 endohedral guanidinium-binding sites can strongly bind sulfonate-containing gold catalyst (TPPMSAuCl, TPPMS = triphenylphosphinomonosulfonate), leading to a high gold catalyst concentration in the cavity of the cage. Meanwhile, the guanidinium sites can also weakly bind carboxylate-functionalized substrates, thus locating them in the proximity of the gold catalysts. As a consequence, in the Au(I)-catalyzed cyclization reaction of acetylenic acid (5) to give enol lactone under basic conditions, the gold catalysts inside the molecular metal cage show a remarkable 40-fold rate enhancement compared to that of the free TPPMSAuCl. The authors believe that the cooperative binding, in which sulfonate-bearing catalyst bounding stronger than the carboxylates, and the pre-organization effect of the cage were the keys to the increased activity of the encapsulated sulfonate-containing Au(I) catalysts.
The same supramolecular encapsulation strategy was also applied to prepare high local concentrations of electrochemical ruthenium catalyst (up to 0.54 M) [53]. In water oxidation, the pre-organized Ru catalyst (Ru(bda)(PySO3TBA)2) inside the cage (MOC-4) shows a high TOF value of 125 s−1, which is two orders of magnitude (130 times) higher than that of the homogeneous ruthenium catalyst. The clear increase of kcat, which was determined by combining the Randles–Sevcik equation with kinetics plots, along with the increased catalyst concentration, indicated the reaction proceeded via a I2M mechanism, where the ruthenium–oxo intermediate was formed via a rate determining proton coupled electron transfer step. In the kinetic isotope effect (KIE, H2O/D2O) studies with different local catalyst concentrations (0–0.54 M), relatively small KIE value (<1.5) was observed at low catalyst concentrations (0–0.27 M), but the KIE increases up to 2.2 with higher catalyst concentration (0.27–0.54 M). These results further confirmed that the dinuclear radical coupling with typical diffusion limitations was facilitated in higher catalyst concentration and the rate-determining step (RDS) step was changed from the radical coupling step to the preceding proton coupled oxidation step by altering the local catalyst concentration [54].
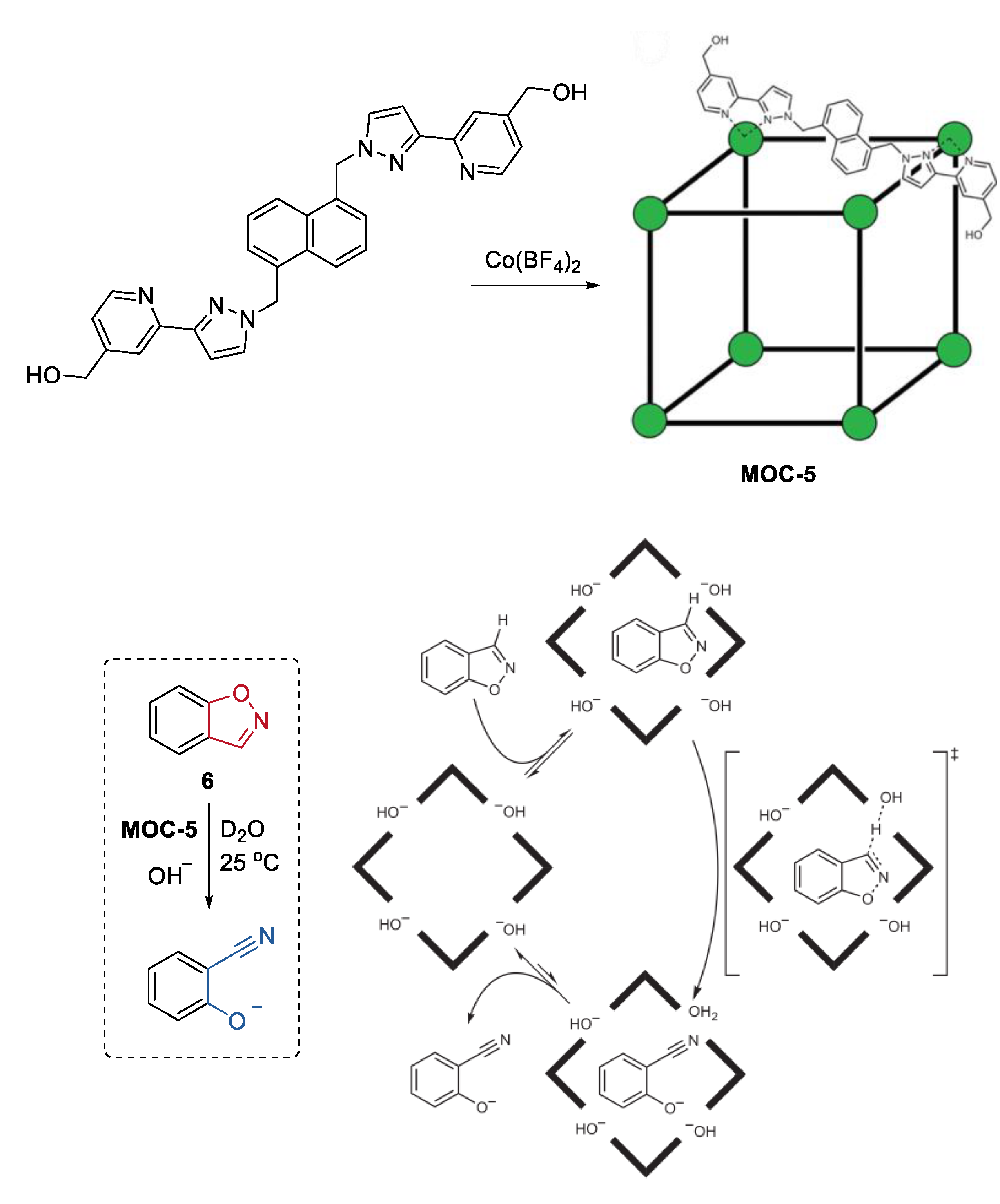
An impressively efficient catalysis of the Kemp elimination, the ring-opening reaction of benzisoxazole to give 2-cyanophenolate in the presence of a base, in the cavity of a metal coordination cage, was recently reported by Ward, Williams, and Hunter [7]. The cubic Co8L12(BF4)16 (MOC-5) coordination cage catalyst (Figure 5), which contains eight Co2+ ions at the vertices and sixteen hydroxyl groups on the external surface, features by its water solubility, hydrophobic interior cavity, and positive charge on the cage [55]. These characteristics of the cage promoted the binding of benzisoxazole (6) in the cavity, the accumulation of hydroxide ions around the cage surface, and the release of the hydrophilic product when the reaction was carried out in a basic aqueous media [56]. As a result, the cage catalyst showed pH independence of catalytic activity in the Kemp elimination of benzisoxazole, leading to a significant rate enhancement (2 × 105-fold) for the cage-catalyzed reaction compared with the un-catalyzed one at pD of 8.5. The concentrated partially desolvated hydroxide ions around the cage surface were considered to be the critical factor for the high catalytic activity of the cage. This was confirmed by competition experiments where the observed rate of reaction was reduced to that of the background rate by adding a large excess of chloride ions (47 mM) into the reaction solution. In their continued study of this catalytic system, they discovered a new autocatalytic pathway catalyzed by the accumulated basic product 2-cyanophenolate at the cage surface [57]. When chloride or fluoride inhibited the reaction to some extent, the product 2-cyanophenolate with a bigger affinity toward the cationic cage could displace them and acted as a base for the next reaction cycle. This autocatalytic reaction inspired them to develop an anion-controlled reaction for a cavity-bound guest. For instance, the Kemp elimination could optionally be accelerated by adding phenolates with different basicity.
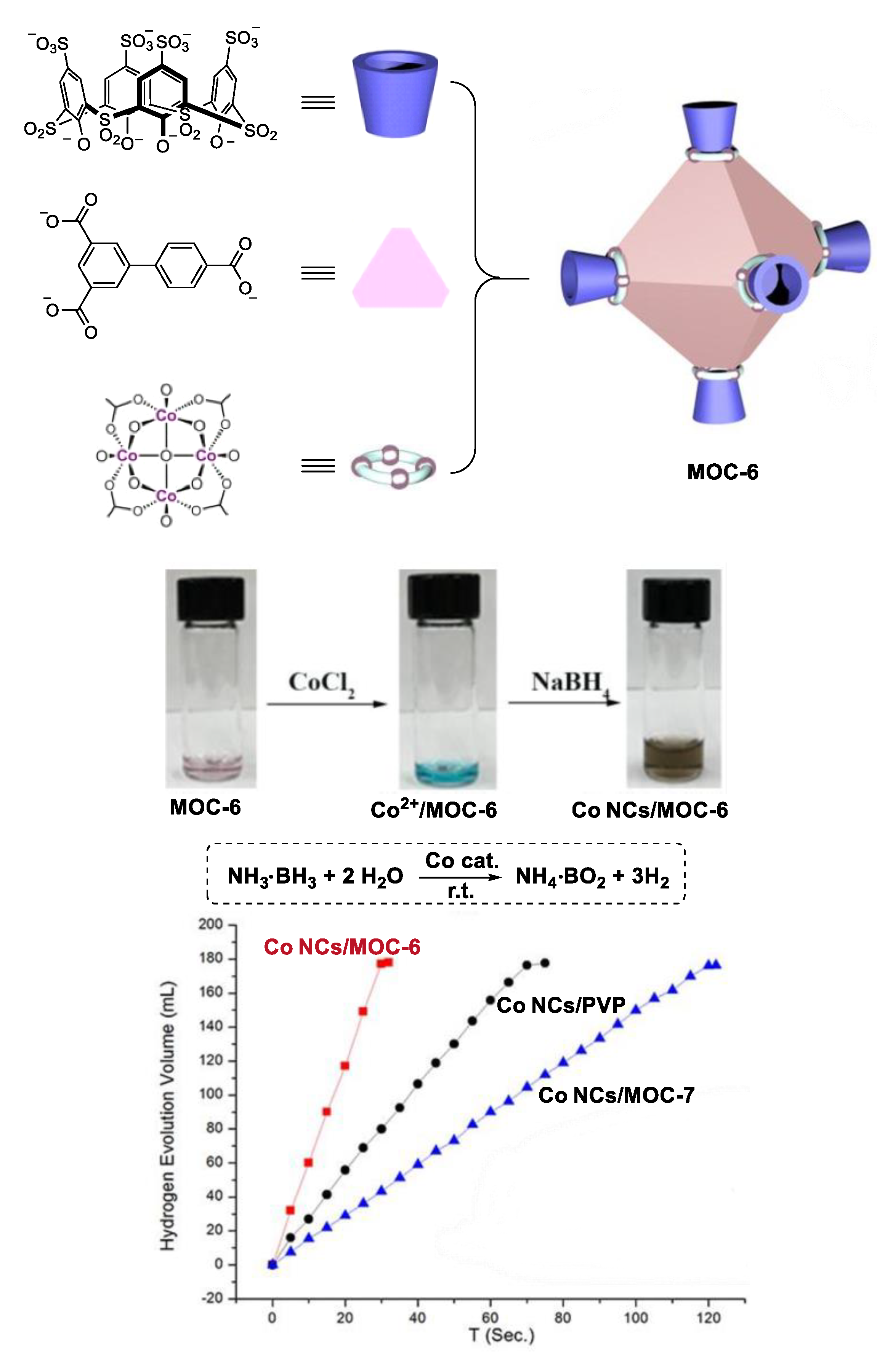
By encapsulating small Co nanoparticles in soluble porous coordination cages, Zhou and co-workers developed a highly catalytically active Co catalyst for hydrolysis of ammonium borane (Figure 6) [58]. Two metal coordination cages ([Co24V6L8][(Et3NH) 6Na24]) MOC-6 and ([Co24V6L’8][(Et3NH)6Na24]) MOC-7 with similar structure (same cavity in shape and size) but different charge properties (MOC-6 has five times more net charge than MOC-7) were synthesized as supports of the Co catalysts. The cage was mixed with CoCl2 in N,N-dimethylformamide (DMF) at room temperature for 30 min. The encapsulated Co2+ ions within the cage were then reduced by NaBH4. Disappearance of the adsorption band of Co2+ from 600 to 700 nm accompanied by an immediate color change from green to dark brown were observed upon the addition of NaBH4. These findings clearly indicate the formation of Co nanoparticles (Co Ps@MOC-6 and Co NPs@MOC-7), which were characterized by Transmission Electron Microscopy (TEM), Energy Dispersive X-ray Spectroscopic (EDS), and X-ray Photoelectron Spectroscopic (XPS). The accumulated Co2+ can be reduced into small Co nanoparticles (2.5 nm) without aggregation by NaBH4 due to the stronger interaction between MOC-6 and Co ions. In contrast, large Co nanoparticles (100 nm) were obtained in the case of MOC-7 with fewer net charges. As a result, better catalytic performances in the hydrolysis of ammonium borane were observed for the catalyst Co Ps@MOC-6 (TOF, 90.1 min−1) than Co NPs@MOC-7 (TOF, 22.5 min−1). They also used the same strategy to synthesize an encapsulated small Ru nanoparticle catalyst (2.5 nm), which showed high activity in the methanolysis of ammonia borane (TOF, 304.4 min−1) [59].
In most of the reported metal–organic cages involving catalysis, the reactions were generally considered to take place inside the cavity. However, Jonathan R. Nitschke and co-workers reported an unusual mode of catalytic action where the reaction occurs on the surface of the cage [60]. In their case, the cage was used to mediate electron transfer rather than to encapsulate the substrate (Figure 7). To achieve this goal, a redox-switchable ZnII4L6 metal–organic cage (MOC-8) decorated with NDI naphthalenediimide (NDI) moieties at its edges was synthesized. C60, a co-catalyst that can stabilize radicals, was then included inside the cage to enhance the catalytic activity [61]. The resulting catalyst system (10 mol% cage, 20 mol% C60) showed moderate activity in the oxidative coupling reaction of tetraaryl borates (37%−81% yield). EPR experiments of the mixed cage, C60 and a tetraaryl borate(potassium tetrakis(4-chlorophenyl)borate, 8) showed sharp signals at 2.0016 and 2.0013, which was attributed to C60* radical species. Based on this, the authors concluded that C60 may serve as a stabilizing agent by taking up some radical spin density from the cage, which was reduced by the borates.
3. Improvement of Selectivity by Metal–Organic Cages
Improving the selectivity toward the desired product is one of the most important goals for the design of a catalyst. Common synthetic catalysts usually require complex ligands and specific reaction conditions to render selectivity. In contrast, metal–organic cage catalysts readily self-assemble from simple components, offering tailored confined spaces to enable remarkable product selectivity. Strategies to improve the selectivity of a supramolecular cage catalyst include (i) taking advantage of the constriction imposed by the size and shape of the cavity to limit the type of the products, (ii) using the cage as a stoichiometric supramolecular “protecting group” to selectively hide functional groups at multiple sites of the substrate, and (iii) encapsulating a catalyst inside the cage to provide a secondary environment for direct chemo and regioselectivity.
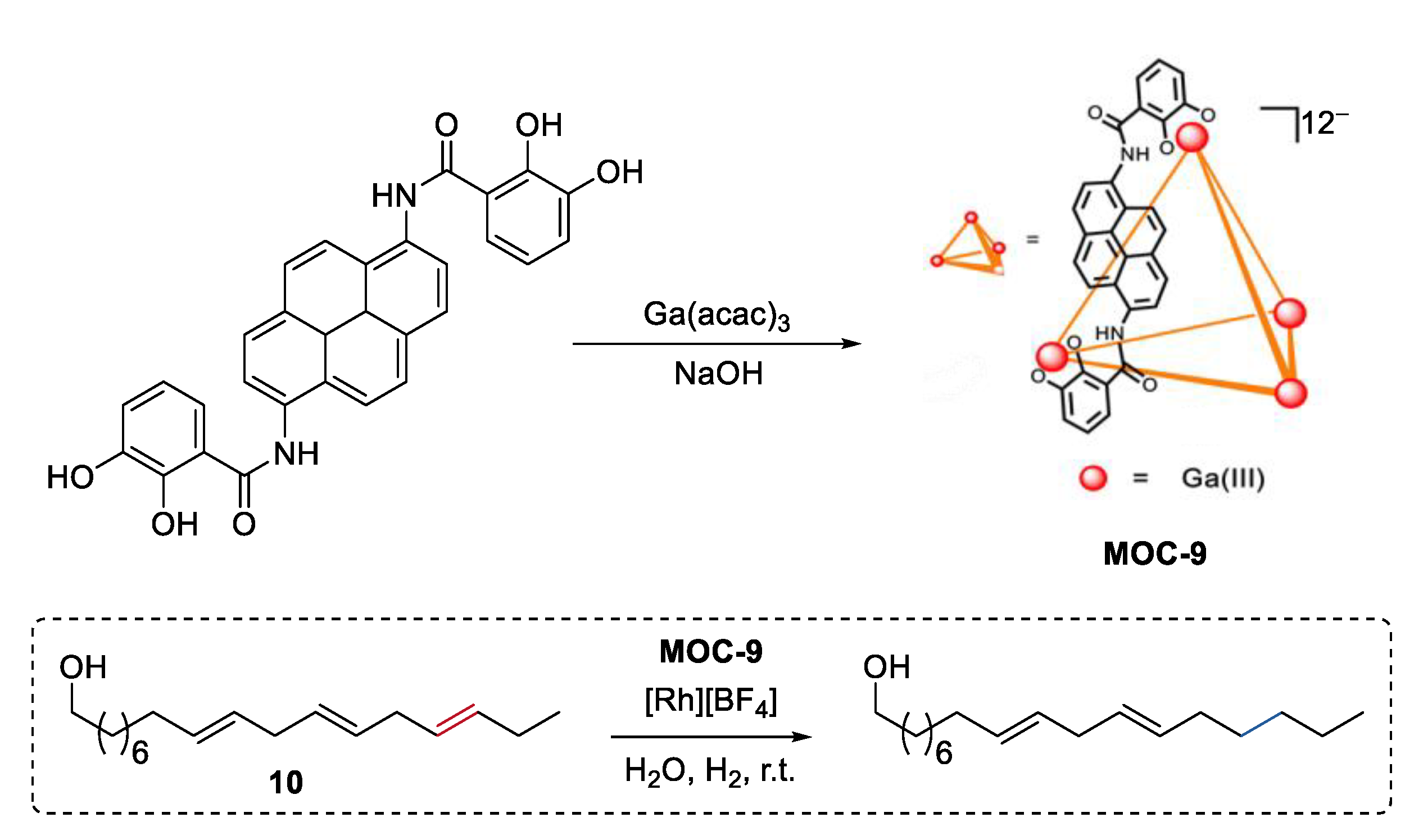
Instead of size discrimination between substrates [62], partial confinement of a single molecule can selectively protect functional groups at multiple sites of the molecule, thus leading to size-selective reactions. Very recently, Raymond, Bergman, and Toste reported a selective catalytic hydrogenation of a polyene group over other unsaturation bonds in the same substrate by encapsulating a rhodium complex hydrogenation catalyst [Rh][BH4] ([Rh] = [(DMPE)Rh(COD)], DMPE = 1,2-bis(dimethylphosphino)ethane) into a tetrahedron metal–organic cage (Na12Ga4L6, MOC-9, Figure 8) [63,64]. Polyenol containing three points of unsaturation (10) was selectively hydrogenated into a monohydrogenated product with a yield of 74% in the presence of the supramolecular catalyst, while was converted into the fully hydrogenated alcohol with the free rhodium complex. It is proposed that only a portion of the substrate (outermost unsaturation bonds) can enter into the cavity of the cage and then be catalyzed by the encapsulated rhodium complex. This partial confinement strategy provides an unusual and beautiful supramolecular catalyst-directed regioselectivity as opposed to common directing-group selectivity.
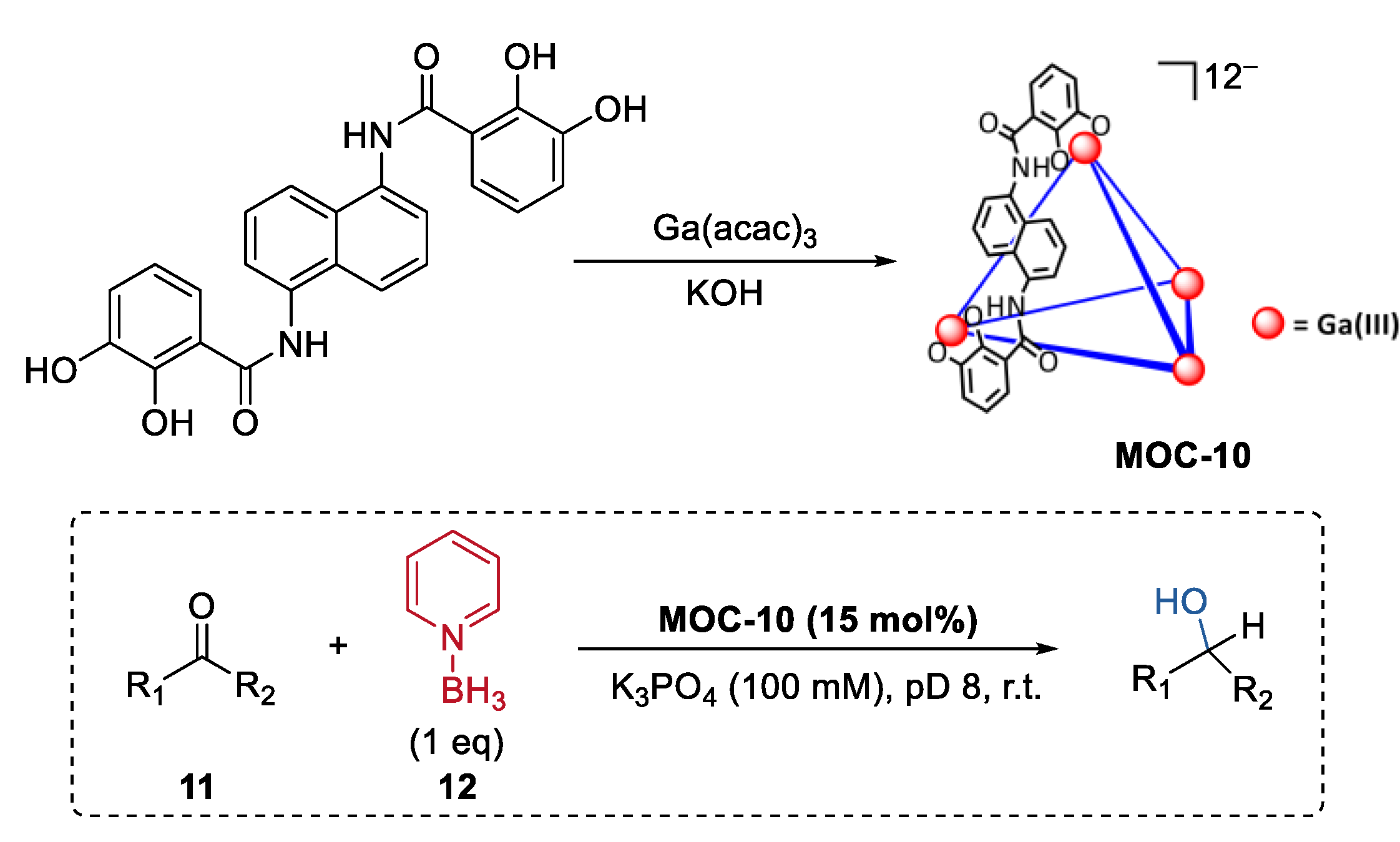
In contrast to small molecule synthesis, metal–organic cage catalysts having a wide breadth of reactivity ranging from small molecules to complex biomolecules such as proteins remain rare. By emulating the catalytic action of the pyridine-based cofactor of ketoreductase enzymes (KREDs) [65], Raymond, Bergman, and Toste reported a metal–organic cage host-mediated pyridine-borane reduction with a wide substrate (11) scope including enones, ketones, aldehydes, oximes, hydrazones, and imines (Figure 9) [66]. Pyridine-borane (12), a mild reducing agent, was encapsulated into a highly anionic [K2Ga4L6] (MOC-10) metal–organic cage [67]. The host systems showed selectivity towards ketones. In the reduction of α, β-unsaturated ketones with reducible C=C and C=O bonds, only the C=O bond was selectively reduced under the metal–organic cage-mediated reduction conditions. In contrast, when the reaction was run in the presence of a strongly binding inhibitor of the catalysts (NEt4+), almost only the corresponding saturated ketone products were consistently observed as the products. It was proposed by the authors that the co-encapsulation of the enone substrate with pyridine−borane and the stabilization of the protonated intermediate of the carbonyl group may accelerate the 1,2-reduction pathway over the 1,4-pathway [68]. Partial encapsulation was then employed for the host-catalyzed reduction to more complex substrates. Hydrophilic substrates like Lysine, amino acid peptide, and human insulin, which contain both anionic carboxylate groups and amine groups, were expected to be partially encapsulated by the catalyst due to the unfavorable coulombic interactions between the carboxylate group and the host. In turn, selectively reductive alkylation of the ε-terminus of the substrates with fluorobenzaldehydes was achieved in the presence of the supramolecular catalysts.
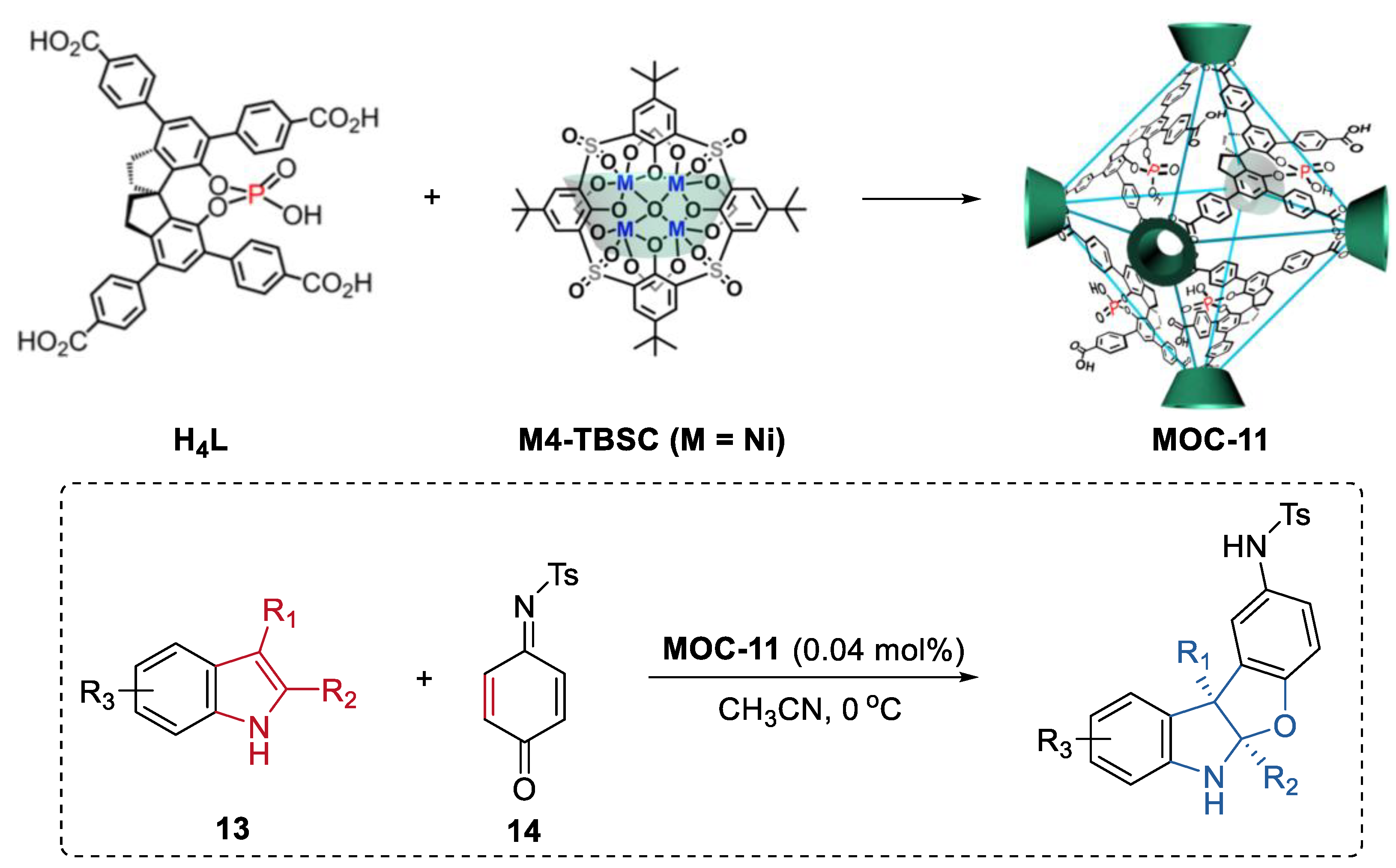
Cui, Liu, and co-workers reported a chiral cage featuring chiral dihydroxyl groups by installing a chiral molecular catalyst into a supramolecular metal–organic cage [69]. The chiral cage catalyst exhibited improved enantioselectivity in the asymmetric conjugate addition of styrylboronic acids to α,β-enones and the asymmetric addition diethylzinc to aldehydes compared to the free chiral molecular catalyst. Different from the free chiral molecular one, the cage catalyst showed substituent size-dependent activity and a much higher binding capacity towards the substrate in the asymmetric conjugate addition reaction between 4-hydroxychalcone and styrylboronic acids. The authors, therefore, proposed that the enhancement of stereoselectivity of the cage catalyst was due to the steric hindrance and confinement effect of the supramolecular porous cage. Moreover, the same group further developed a cage extension strategy to construct heterogeneous chiral porous catalysts through intracage hydrogen-bonds and hydrophobic interactions (Figure 10) [70]. A spinol-based metal–organic cage (MOC-11) with phosphoric and carboxylic groups capable of forming strong hydrogen bonds to another was first prepared using enantiopure 1,1′-spirobiindane-7,7′-phosphoric acid and Ni4-p-tertbutylsulfonylcalix[4]arene clusters as the ligands. The hierarchical assembly of the cage leaded to the formation of a porous hydrogen bonded organic framework (HOF) [71]. The resulting chiral HOF acted as a recyclable Brønsted acid catalyst in the asymmetric [3+2] coupling of indoles (13) with quinone monoimine (14) and Friedel-Crafts alkylations of indole with aryl aldimines. Up to a 92% isolated yield and a 99.9% ee for the benzofuroindoline derivatives and a 92% isolated yield and a 91% ee for 3-indolylmethanamine derivatives can be obtained with extremely low loading of the HOF catalyst (0.04 mol%). These results surpassed those of the homogeneous counterparts including the acidic ligand or the chiral free metal–organic cage. Moreover, the solid porous catalyst can maintain its efficiency and enantioselectivity after 10 runs.
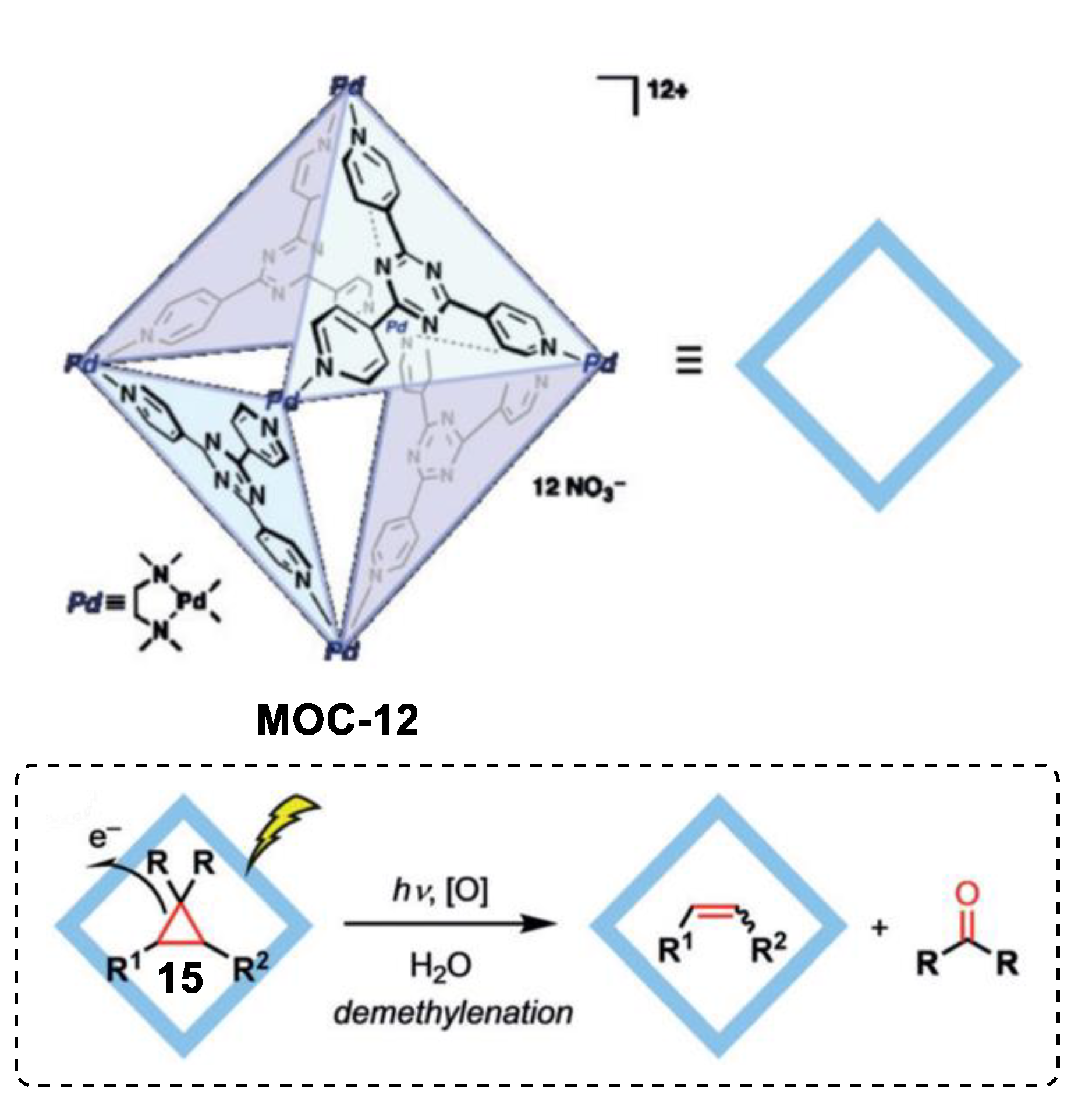
Very recently, Fujita and coworkers reported an unusual demethylenation reaction of cyclopropane with high chemo-selectivity towards the alkene product boosted by a Pd6L4 metal–organic cage (MOC-12) [72], which showed improvement in photo-oxidation reactions of inert alkanes and photohydrations of alkynes [73,74]. Cyclopropane compounds such as drospirenone, trans-2-phenyl-1- cyclopropanecarboxylic acid, and chrysanthemic acid underwent demethylenation reaction to give corresponding alkene products and formaldehyde or acetone inside the photoactive coordination cage under UV light irradiation (Figure 11). In contrast, the reference cage without the triazine-cored ligand was inactive in the demethylenation reaction of drospirenone. After the photoreaction, nitrite anion (NO2−) was detected by the Griess method. A photoinduced electron transfer from the guest to the cage was proposed for the demethylenation reaction. The electron-deficient triazine-based ligand acted as an electron acceptor and accepted an electron from the bound guest under UV irradiation, generating a radical anion centered on the cage and a radical cation centered on the guest. Nucleophilic attack of a nitrate anion of the radical cation and successive fragmentation leaded to the demethylenated product and a fragment nitrite radical. The catalytic cycle was completed by the one electron back to the nitrate radical from the cage. The same cage also showed site-selective electrophilic addition of linear diterpenoids with nitrate anions [75].
Oxidative coupling of 2-naphthol in bulk solution usually leads to 1,1-bis(2-naphthol), while the 1,4-coupling product was only detected as a minor byproduct. In contrast to these results in bulk solution, Su and co-workers recently reported an unusual photoinduced 1,4-coupling of naphthols and derivatives rather than the normal 1,1-coupling to form asymmetric biaryl compounds with moderate regio- and enantioselectivity in the above-mentioned MOC-2 [76]. The robust enantiopure metal coordination cage containing photoredox-active and stereogenic RuII metalloligands not only provided a confined chiral coordination space, but also photoactive active sites, thus serving as dual-functional photoredox- and stereochemically active nanoreactors. Under light irradiation, the oxidative coupling of 3-bromo-2-naphthol (16) in the presence of 10 mol% of the chiral metal coordination cage exclusively give the 1,4-coupling biaryl product with a yield of 32% and an ee value of 34% (Figure 12). On the contrary, a yield of only 8% and an ee value of 10% of the product were obtained by 80 mol% of the chiral RuL3 metalloligand complexes. In the control experiments using a racemic metal coordination cage and FeCl3 or CuCl2 as the coupling agent, both the 1,4-coupling and the normal 1,1-coupling reaction were prevented. The authors proposed that the confined chiral space of the metal coordination cage, which preferentially encapsulated the substrate between the two parallel walls of the homochiral configuration of the Ru centers, was attributed to the enhanced regio- and enantioselectivity of the naphthol coupling [77].
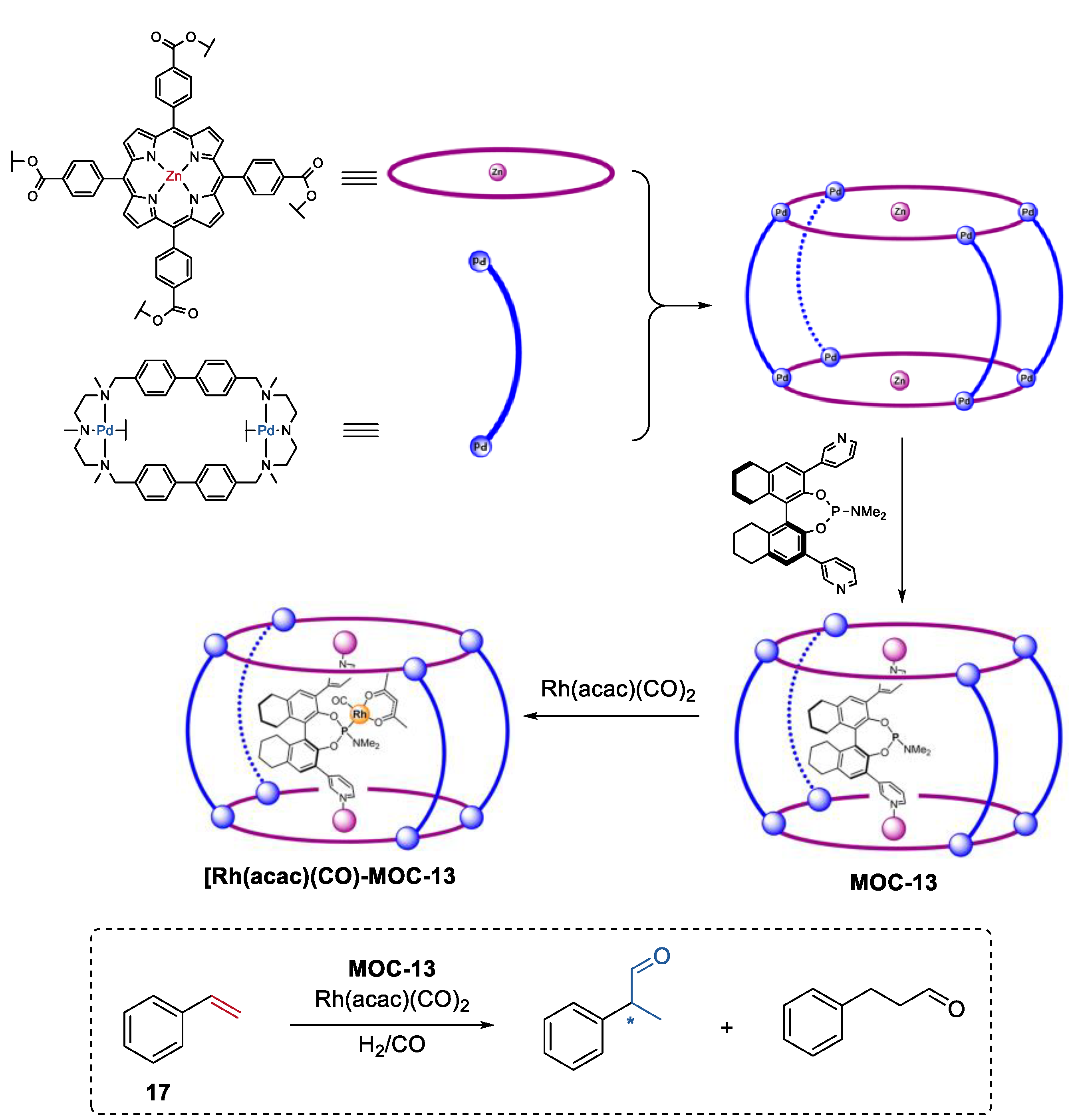
A second coordination-sphere tuning strategy to control the selectivity of the metal coordination cage catalysts was developed by Reek and co-workers [78]. One of the examples was the regio- and enantioselective hydroformylation of styrenes (17) catalyzed by the encapsulated chiral Rh catalyst in a metal coordination cage (Figure 13, MOC-13) [79]. The self-assembly of the macrocyclic dipalladium complexes and tetracarboxylate zinc porphyrins [80], and further inclusion of a chiral phosphoramidite led to the formation of the metal coordination cage with a chiral ligand inside its cavity. In the presence of this special supramolecular ligand, Rh(acac)(CO)2 provided high chemo- and stereoselectivities towards R-2-phenylpropanal, with an ee value of 74% and a TON of 797. In contrast, phosphoramidite ligand ((S)-α) (1% ee, TON of 342) alone or the Rh-complex of α(Zn-TPP)2 (9% ee, TON of 363) exhibited extremely low selectivity and activity. Molecular modeling studies indicated that the substrate could approach the Rh center with multiple orientations for the Rh-complex of phosphoramidite ligand, while most of the coordination modes of styrene to the Rh center were prevented in the cage catalyst. This was an elegant example of using space control of the chirality around the catalyst center to achieve enantioselectivity that cannot be attained in bulk.
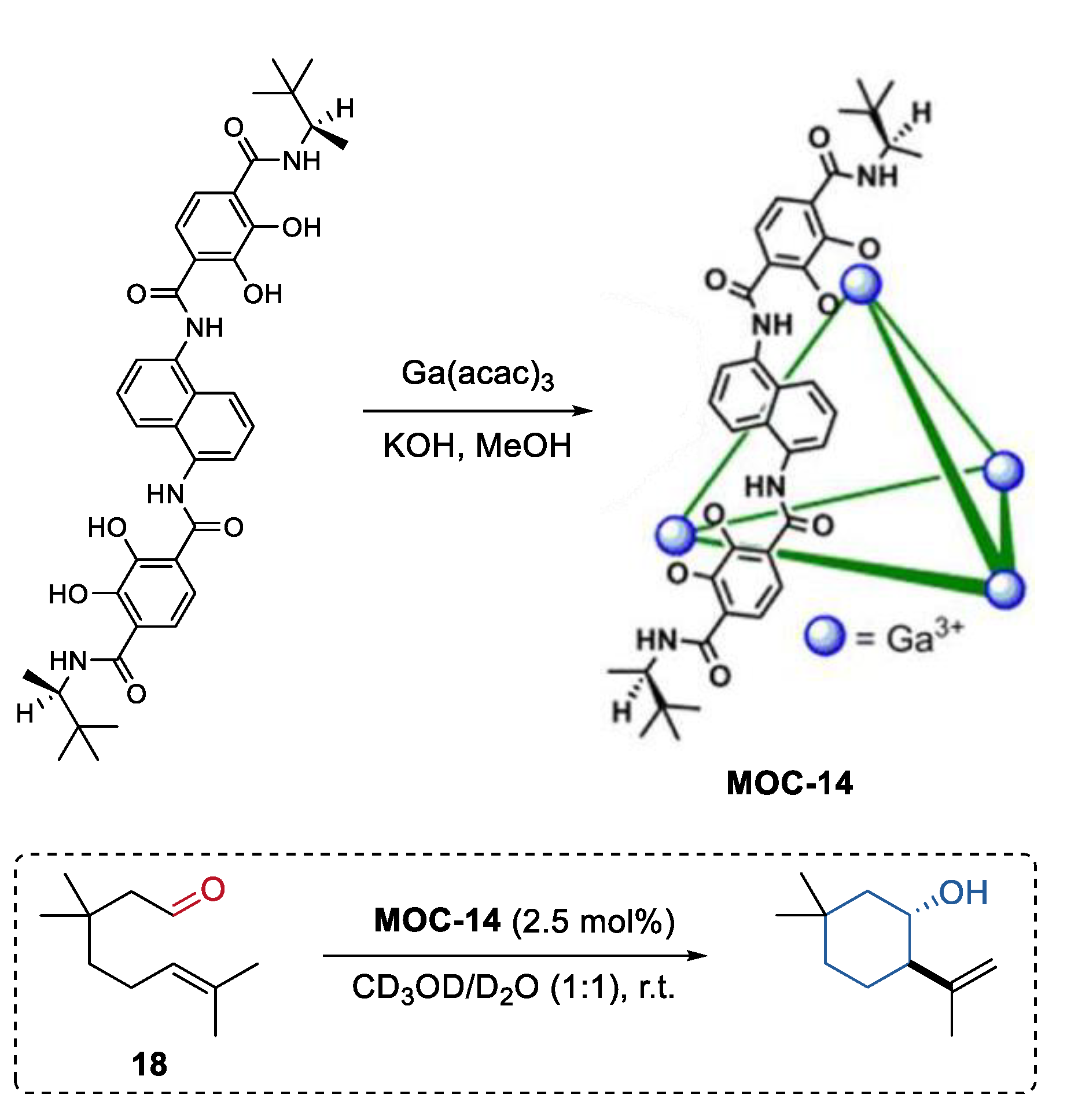
An enantiopure metal coordination cage was synthesized by Raymond, Bergman, and Toste using a terephthalamide-based ligand with chiral amide groups (Figure 14) [81]. Since this supramolecular cage (K4Ga4L6, MOC-14) was prepared without the use of any templates or cationic species, it was applied as a chiral catalyst for the transformation of neutral compounds, for example, the Prins cyclizations of monoterpene derivatives. The chiral metal coordination cage showed high conversion in the Prins cyclizations of citronellal (18) and its homologues (yield up to 94%), and gave an enantiomer cycle product with an ee value up to 65%. The effects of the host spacer on product selectivity were investigated by using an analogous pyrene-core metal coordination cage with a large cavity [64]. Product enantioselectivity was significantly decreased (33% ee) with the pyrene-core host. These results clearly demonstrated that the host-mediated enantioinduction decreases with host cavity size.
4. Tandem or Cascade Reactions Catalyzed by Metal–Organic Cages
Tandem or cascade reactions, which combine two or more incompatible successive catalytic steps in one pot, are highly attractive in terms of improving catalytic efficiencies, and reducing energy consumption and waste, as well as rendering more complex materials from a simple set of starting materials. However, such tandem or cascade reactions have been less developed due to the necessary compatibility of multiple catalysts or catalytic active sites, reagents, and intermediates generated during the reaction. The unique structure of metal–organic cages makes them desirable platforms for tandem or cascade reactions by (i) perfectly spatially segregating incompatible catalysts inside or outside their cavities, or (ii) integrating different catalytically active sites in the framework of a single cage, or (iii) constructing suitable reaction space to avoid interference between the different catalytic cycles. However, the construction of such distinctive metal–organic cages has proven to be challenging, and only small steps have been taken in this field.
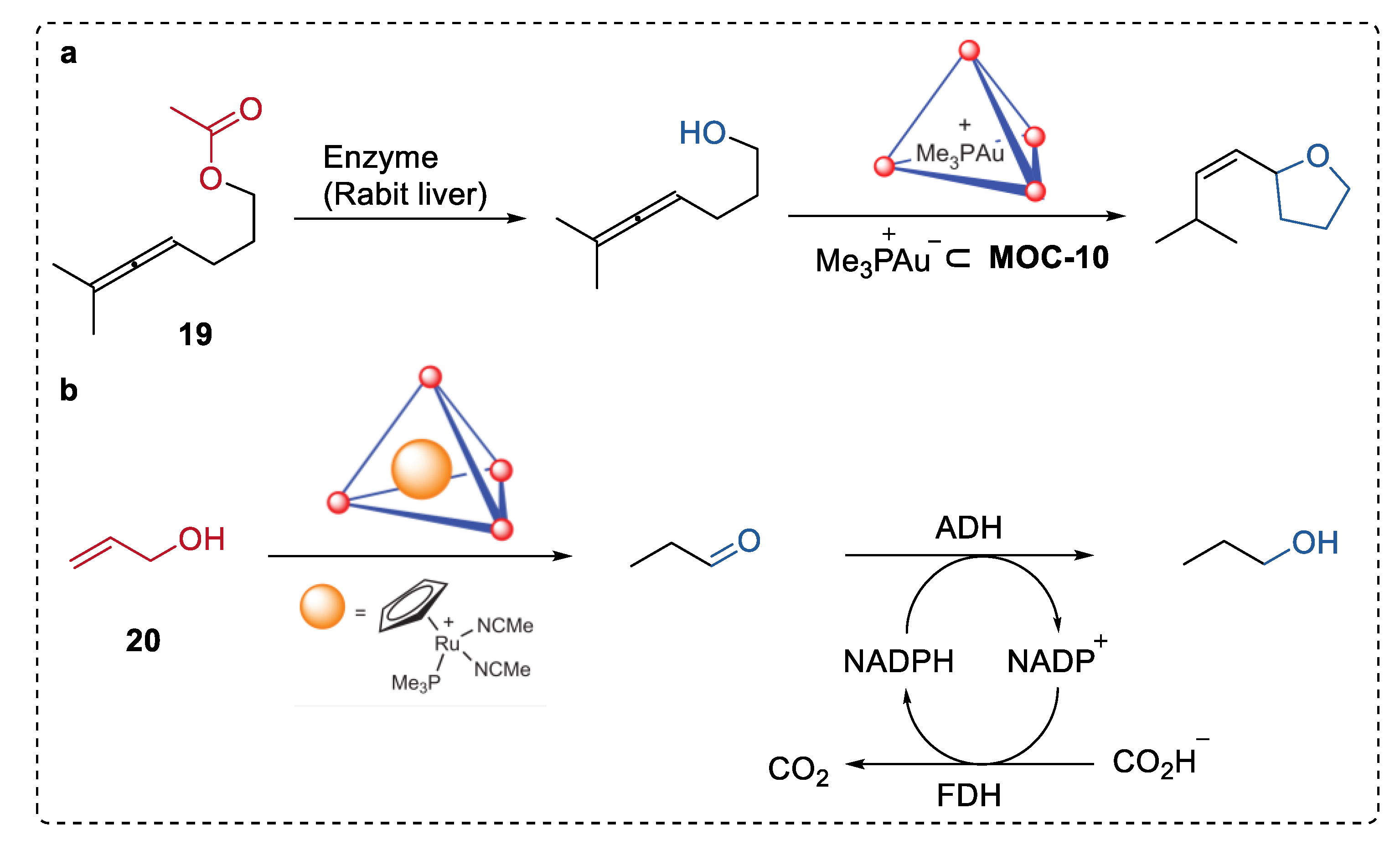
In 2013, Raymond, Bergman, and Toste illustrated an intriguing example of hydrolysis–cyclization tandem reactions catalyzed by a spatially separated natural enzyme (esterases, or lipases) and gold(I) complexes (Me3P+Au−) encapsulated in MOC-10 (Figure 15a) [82]. The supramolecular encapsulation of organometallic complexes prevented the detrimental interactions between the complexes and the enzyme in the solution, therefore preserving the activity of both. Thus, the sequential transformation was performed smoothly starting from the enzymatical hydrolysis of allenic acetate (19) by esterase to form alcohol, which was then cyclized into substituted tetrahydrofuran by (Me3P+Au−) encapsulated in MOC-10 with excellent selectivity (100%) and yield (100%). Only 62% yield of the product was formed when the free Au catalyst was used instead of the bound one, and no such tetrahydrofuran product was obtained in the absence of the esterase or the encapsulated gold catalysts. This strategy was also employed in the encapsulated ruthenium ((Me3P)CpRu(NCMe)2)− catalyzed isomerization of 1-propenol (20) to give propanal and the sequential reduction of propanal to propanol via ADH (Figure 15b).
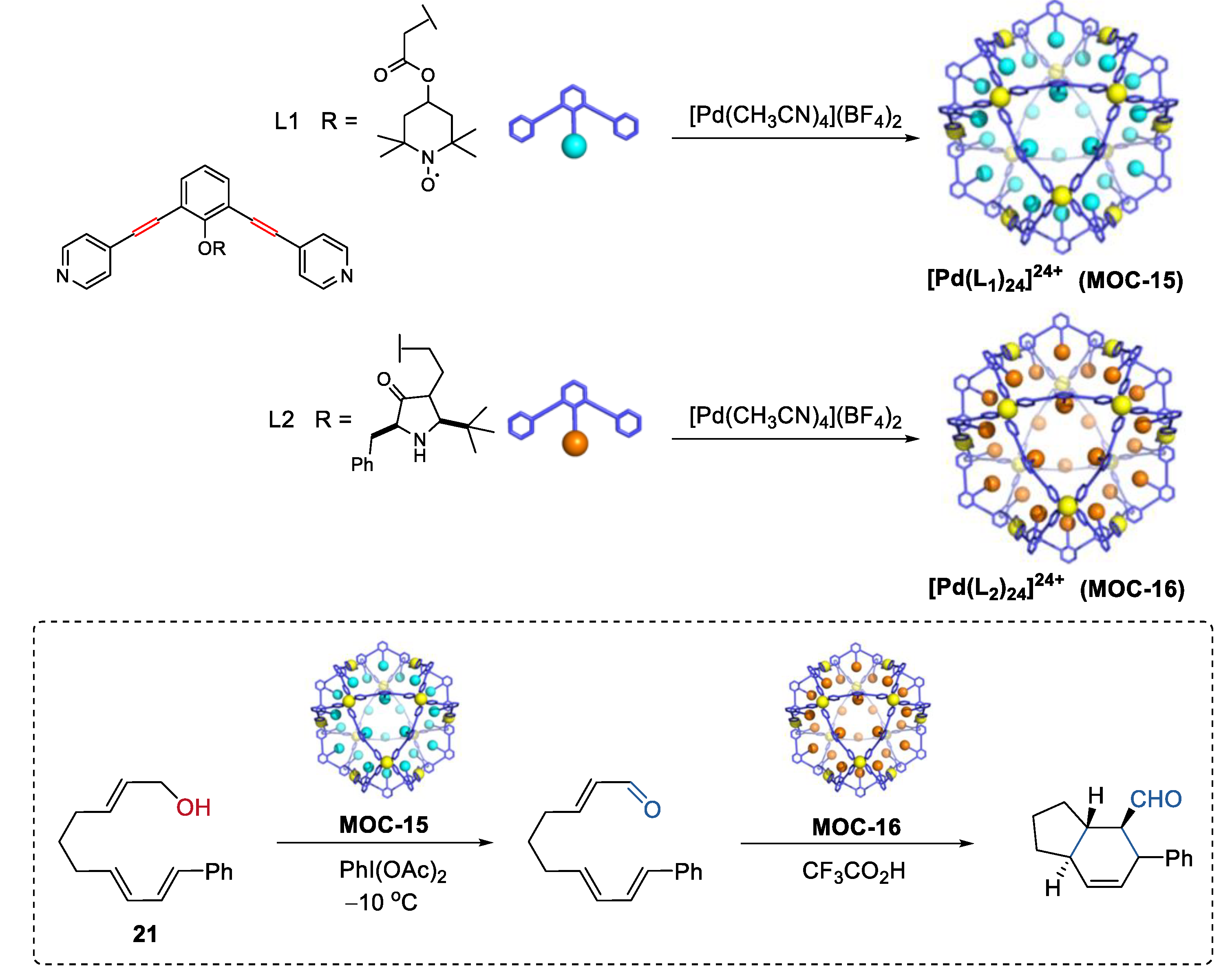
Very recently, Fujita and co-workers demonstrated an interesting one-pot tandem oxidation-Diels−Alder reaction using a site-isolation strategy (Figure 16) [83]. The incompatible oxidation catalyst TEMPO [84] and the Diels−Alder catalyst chiral amine [85] were physically separated within the cavity of two self-assembled metal–organic cages M12L24 (MOC-15, MOC-16), thus affording allylic oxidation of alcohol (21) followed by a stereoselective Diels−Alder cyclization. The cyclization product with four adjacent stereogenic centers was obtained in a moderate yield of 56% and a high ee value of 93% only in the presence of both two metal–organic cages, whereas in the absence of any of the metal–organic cages only the oxidation product formed. The M12L24 metal–organic cages, which have an inner space sterically insulated by a rigid self-assembled shell with numerous apertures allowing for small molecular permeability [86], was the key to this site-isolation strategy.
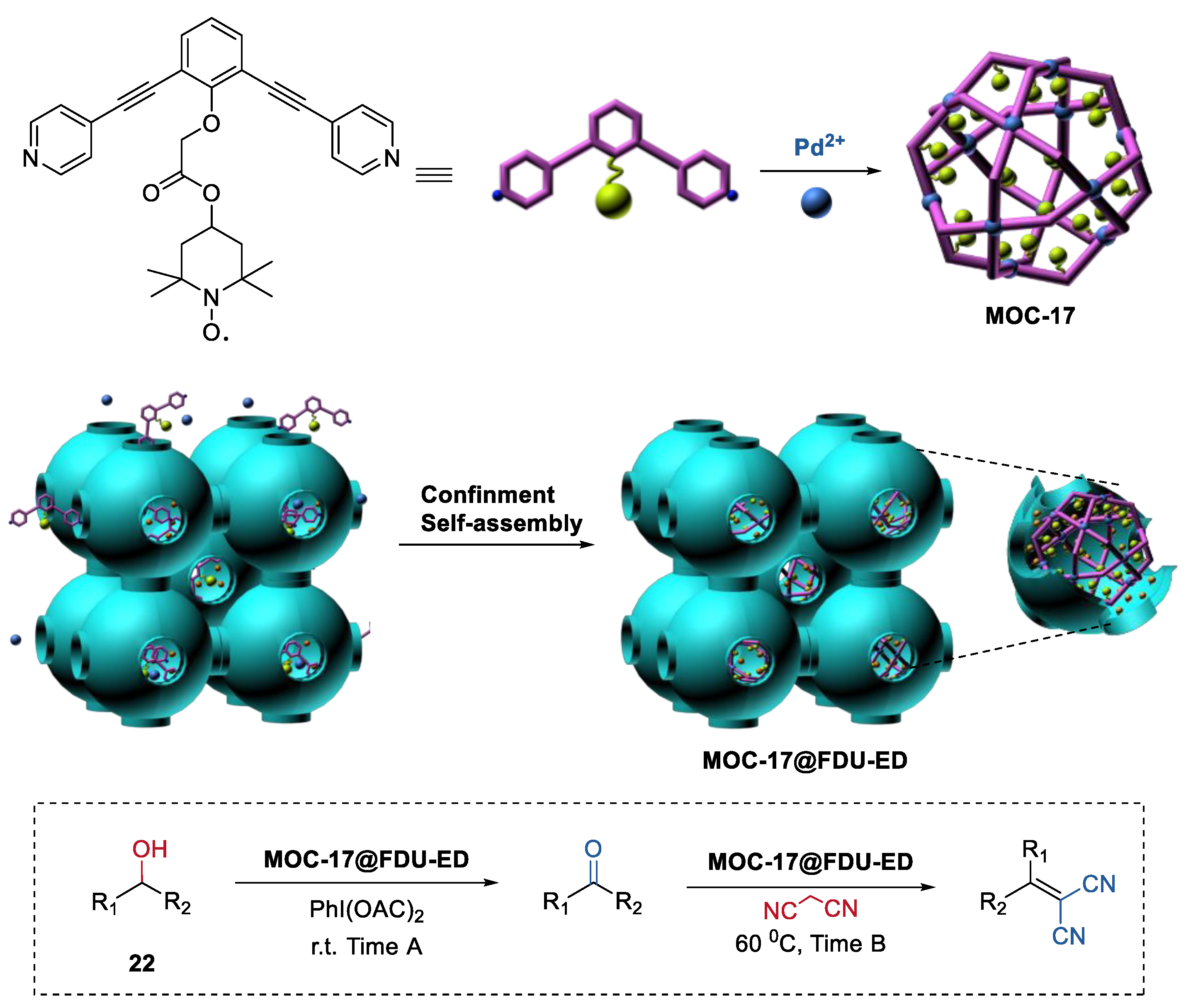
Isolating active sites within the metal–organic cages and the confined porous materials to protect the metal–organic cages is not only a novel way to improve the stability of metal–organic assemblies, but also a powerful strategy for constructing heterogeneous catalysts for one-pot sequential reactions. Very recently, we demonstrated the first confinement self-assembly of TEMPO-containing metal–organic cages (MOC-17, Figure 17) within amino-functionalized mesoporous carbon (FDU-ED) [87,88]. The resulting hybrid materials served as a recyclable heterogeneous catalyst (MOC-17@FDU-ED) for the one-pot alcohol oxidation (22) and Knoevenagel condensation sequential reactions.
The bifunctional catalyst MOC-17@FDU-ED exhibited enhanced catalytic activities (TOF, 261.18 min−1) compared to those of the simple mixing metal–organic cage and mesoporous matrix catalysts (TOF, 54.54 min−1). Moreover, MOC-17@FDU-ED maintained its catalytic performances after 5 runs. However, due to the leaching problem, the activities of the mixed catalysts (ligand L+FDU-ED and cage MOC-17+FDU-ED) dramatically decreased from the second run. The improved activities and recyclability of MOC-17@FDU-ED could be attributed to the high dispersion catalytically active sites inside the composites MOC-17@FDU-ED.
A single coordination cage containing multiple catalytically active sites can also promote one-pot sequential reactions [89]. A novel tetrahedral coordination cage (MOC-18, Figure 18) bearing three Mn(salen) and three Cr(salen) active species was constructed by Cui and coworkers using six dicarboxylate ligands derived-from enantiopure Mn(salen) and Cr(salen) as linear linkers and four Cp3Zr3 clusters as three-connected vertices. The resulting supramolecular catalyst shows high activity (62–82%, yield) and selectivity (91–99.9% ee) for the sequential asymmetric 2,2-dimethyl-2H-chromene (DMCH, 23) epoxidation/epoxide ring-opening reactions with TMSN3 or anilines. Reaction kinetics studies indicated that the cage catalyst remained active at a very low loading (0.005–0.01 mol%); however, the mixture of Mn(salen) and Cr(salen) catalysts were totally inactive [90]. It was proposed that the improved activity and enantioselectivity of the cage catalyst over the free catalysts was due to the stabilization of catalytically active metallosalen units and concentration of reactants within the cavity. It is also worth noting that this homogeneous cage catalyst can be easily recovered as precipitate by adding diethyl ether into the solution after the reaction. Its structure and the catalytic activity were maintained after at least five runs with slight deterioration in enantioselectivity.
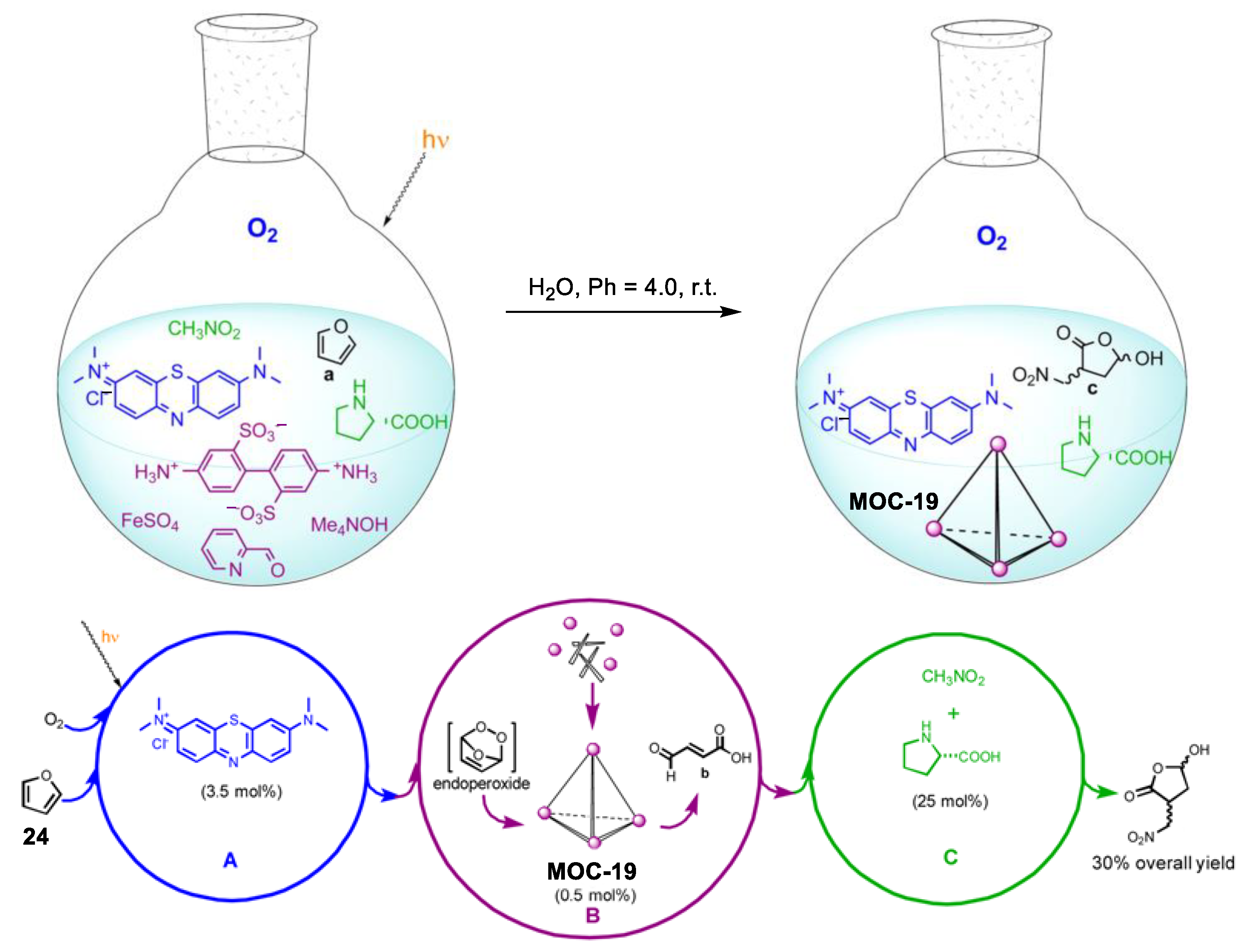
Relay multicatalysis is capable of transforming simple molecules into more complex and high-value products [91]. The key to this system is to avoid the possible interference between the different catalytic cycles as well as the tolerance between different substrates and intermediates. Nitschke and co-workers demonstrated a novel interference-free “assembly line” (Figure 19) capable of converting small molecules furan (24) into a chiral hydroxylactone, in which an in situ assembled metal−organic cage plays a vital role [92]. The precursors (i.e., 2-formylpyridine, iron(II) ions, and 4,4′-diaminobiphenyl-2,2′-disulfonic acid) of the metal−organic cage (MOC-19) were totally compatible with the substrates and other catalysts (nitromethane, dioxygen, methylene blue, and L-proline) in the present systems. The in situ assembled metal−organic cage selectively converted the high-energy endoperoxide intermediate generated from the hetero-Diels−Alder cycloaddition of oxygen with furan into fumaraldehydic acid, which is then catalyzed by the L-proline into the final hydroxyfuranone product. Control experiments in the absence of the metal−organic cage led to nonselective oxidation products and trace of the chiral hydroxylactone product, indicating the key role of the metal−organic cage.
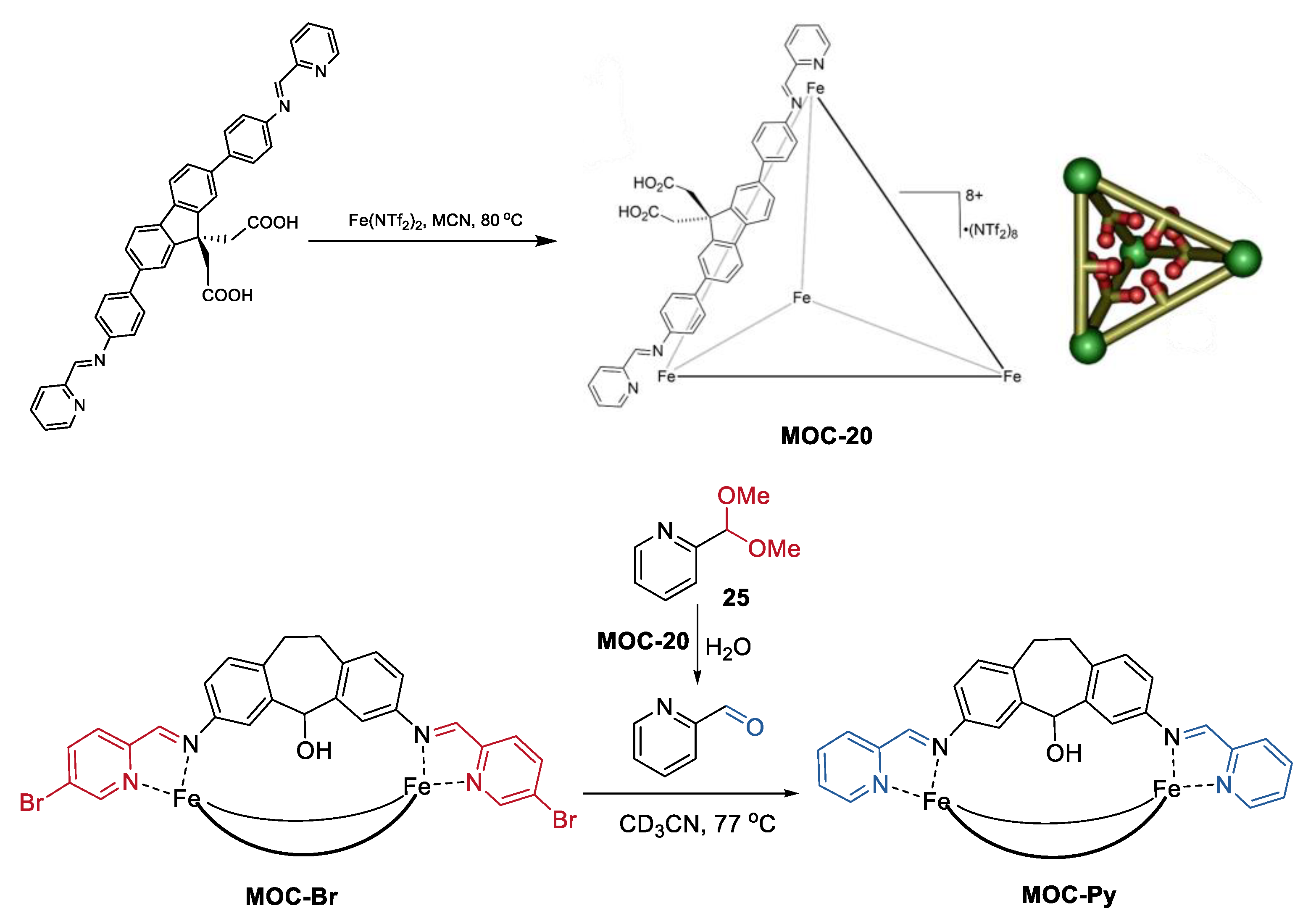
Besides the sequential multiple-steps transformation, an interesting tandem acetal hydrolysis/cage-to-cage interconversion was recently reported by Hooley and co-workers (Figure 20) [93]. Self-assembly of a carefully designed carboxylic acid-containing ligand into a Fe4L6 iminopyridine cage leaded to a tetrahedral metal−organic cage with internalized acid groups (MOC-20), which worked as an effective tandem catalyst for the hydrolysis of aromatic acetals (25) and cage-to-cage interconversion of self-assembled M2L3 helicates (MOC-Br, MOC-Py) [94]. The cage catalyst showed up to a 1000-fold rate enhancement of acetal solvolysis compared with the free carboxylic acid ligand. The rate enhancement was demonstrated by NMR analysis. There was a strong host–guest affinity between the cage catalyst and the aromatic acetals, while no binding was observed between the free acid ligand and the aromatic acetals. Moreover, the acid cage catalyst was tolerant of the M2L3 helicates, which are sensitive to acid, as well as carboxylate or chloride ions. Therefore, the deprotection of the 2-formylpyridine acetal and the followed helicate aldehyde exchange can proceed smoothly in the presence of the acid cage catalyst, while it cannot be performed using a stronger acid such as CF3CO2H.
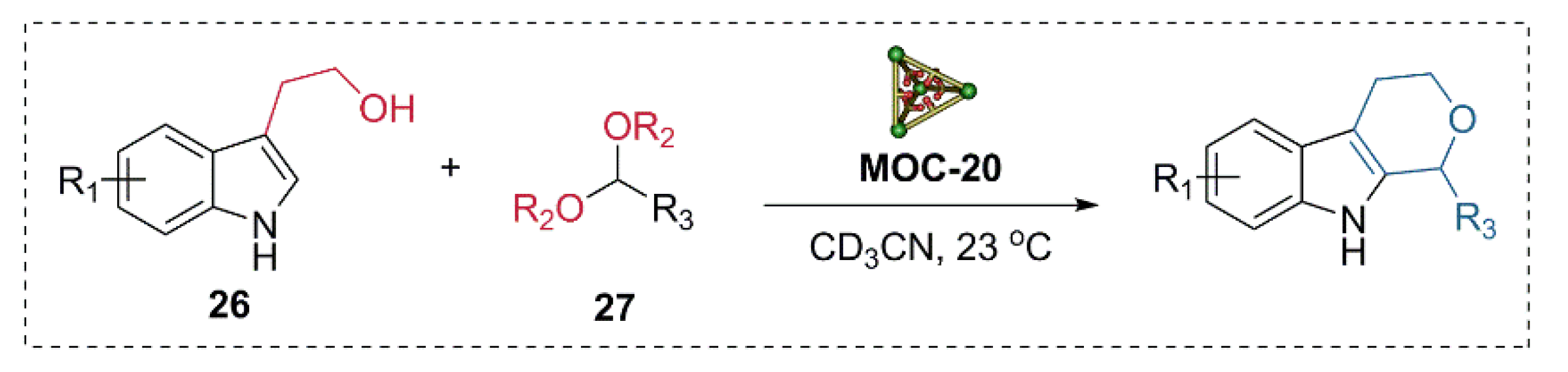
Very recently, the same catalyst was applied to the multi-step Pictet-Spengler cyclization of tryptophols and various aldehyde derivatives (Figure 21) [95]. The flexible endohedrally acid-functionalized metal−organic cage showed good activities in the Pictet-Spengler cyclization of tryptophol (26) and serials of benzaldehyde dimethyl acetal (27) with a good yield of cycle product (80–87%), while the unfunctionalized cage and the carboxylic acid ligand were both not capable of catalyzing the reaction. UV/vis absorbance titration experiments indicated that the acid functionalized metal−organic cage MOC-20 with a large cavity can bind the products, intermediates, and both substrates with suitable size at the same time. Further in situ monitoring of the reaction by 1H NMR spectra provided evidence for the formation of an acetal intermediate and the final cycle product. The binding and activation of two or more different substrates in a single active site of the same cage were proposed for the high activity of the acid functionalized supramolecular catalyst.
5. Conclusions
This paper provides and overview of several recent examples illustrating the breadth and attractive characteristics of metal–organic cages in boosting organic reactions. Different metal–organic cages that offer tailored confined spaces have been shown to be able to accelerate reactions and enhance or alter product selectivity, as well as promote tandem or cascade reactions. The unique cavity structures of metal–organic cages and the various functional groups provide many advantages for catalysis. Regarding rate accelerations, metal–organic cages work by fixing the reagents in a favored reactive conformation, or promoting the local concentration of the catalysts within the cavity, or stabilizing the active sites by the cage. For enhancement or alteration of selectivity, metal–organic cages offer confinement effects to limit the type of the products, or selectively hide functional groups at multiple sites of the substrate, or encapsulating a catalyst inside the cage to provide a secondary environment for directing chemo and regioselectivity. Chiral metal–organic cages have also been developed as catalysts in asymmetric synthesis for achieving enantioselectivity. Metal–organic cages have also emerged as a powerful platform for engineering multifunctional catalysts for tandem or cascade reactions by perfectly spatially segregating incompatible catalysts inside or outside their cavities, or integrating different catalytically active sites in the framework of a single cage, or constructing suitable reaction space to avoid interference between the different catalytic cycles. It should be noted that the use of MOCs to achieve synergistic catalysis and promote tandem reactions is relatively rare compared with other reactions and should be explored more in the future.
Although impressive progress has been made in the application of metal–organic cages for catalysis, tuning reactivity within well-defined metal–organic cages is still in its infancy, and some challenges and limitations still need to be surmounted. For example, the range of reaction types is narrow, limiting their use in practical organic synthesis. Moreover, accurate and selective recognition and anchoring of specific molecules remains challenging. To address this, there is much to explore for the further development of novel classes of metal–organic cages with targeted functions. Furthermore, the balance between stability and activity should be emphasized and investigated, due to the relatively low stabilities of supramolecular metal–organic cages systems during reaction processes. There is no doubt that metal–organic cages have an extremely bright future in catalysis applications, and we hope that this minireview offer researchers general guidance and insight into the rational design of sophisticated metal–organic cages containers to manipulate chemical reactivity within confined nanospaces.
Author Contributions
S.C. writing—original draft preparation, writing—review and editing; L.-J.C. writing—original draft preparation, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the International Joint Laboratory on Resource Chemistry of China (IJLRC), Shanghai Key Laboratory of Rare Earth Functional Materials, and Shanghai Frontiers Science Center of Biomimetic Catalysis. We also acknowledge technical assistance from the Australian National Fabrication Facility, the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. The e factor 25 years on: The rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Song, J.; Han, B. Green chemistry: A tool for the sustainable development of the chemical industry. Natl. Sci. Rev. 2014, 2, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef]
- Morimoto, M.; Bierschenk, S.M.; Xia, K.T.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Advances in supramolecular host-mediated reactivity. Nat. Catal. 2020, 3, 969–984. [Google Scholar] [CrossRef]
- Cullen, W.; Misuraca, M.C.; Hunter, C.A.; Williams, N.H.; Ward, M.D. Highly efficient catalysis of the kemp elimination in the cavity of a cubic coordination cage. Nat. Chem. 2016, 8, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Grommet, A.B.; Feller, M.; Klajn, R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 2020, 15, 256–271. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Reek, J.N. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. [Google Scholar] [CrossRef]
- Zhang, G.; Mastalerz, M. Organic cage compounds—From shape-persistency to function. Chem. Soc. Rev. 2014, 43, 1934–1947. [Google Scholar] [CrossRef]
- Jordan, J.H.; Gibb, B.C. Molecular containers assembled through the hydrophobic effect. Chem. Soc. Rev. 2015, 44, 547–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajami, D.; Liu, L.; Rebek, J., Jr. Soft templates in encapsulation complexes. Chem. Soc. Rev. 2015, 44, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Ueda, Y.; Sato, S.; Mizuno, N.; Kumasaka, T.; Fujita, M. Self-assembly of tetravalent goldberg polyhedra from 144 small components. Nature 2016, 540, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.J.; Rowland, C.A.; Bloch, E.D. Permanently microporous metal–Organic polyhedra. Chem. Rev. 2020, 120, 8987–9014. [Google Scholar] [CrossRef]
- Brown, C.J.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. Supramolecular catalysis in metal–Ligand cluster hosts. Chem. Rev. 2015, 115, 3012–3035. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, H.; Nam, D.; Lah, M.S.; Choe, W. The rise of metal–Organic polyhedra. Chem. Soc. Rev. 2021, 50, 528–555. [Google Scholar] [CrossRef]
- Chen, L.-J.; Yang, H.-B. Construction of stimuli-responsive functional materials via hierarchical self-assembly involving coordination interactions. Acc. Chem. Res. 2018, 51, 2699–2710. [Google Scholar] [CrossRef]
- Pullen, S.; Tessarolo, J.; Clever, G.H. Increasing structural and functional complexity in self-assembled coordination cages. Chem. Sci. 2021, 12, 7269–7293. [Google Scholar] [CrossRef]
- Chen, L.; Chen, Q.H.; Wu, M.Y.; Jiang, F.L.; Hong, M.C. Controllable coordination-driven self-assembly: From discrete metallocages to infinite cage-based frameworks. Acc. Chem. Res. 2015, 48, 201–210. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.-X.; Chen, L.-J.; Yang, H.-B. Construction of multiferrocenyl metallacycles and metallacages via coordination-driven self-assembly: From structure to functions. Chem. Soc. Rev. 2015, 44, 2148–2167. [Google Scholar] [CrossRef]
- Mouarrawis, V.; Plessius, R.; van der Vlugt, J.I.; Reek, J.N.H. Confinement effects in catalysis using well-defined materials and cages. Front. Chem. 2018, 6, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardhan, H.; Verpoort, F. Metal-organic polyhedra: Catalysis and reactive intermediates. Adv. Synth. Catal. 2015, 357, 1351–1368. [Google Scholar] [CrossRef]
- Xue, Y.; Hang, X.; Ding, J.; Li, B.; Zhu, R.; Pang, H.; Xu, Q. Catalysis within coordination cages. Coord. Chem. Rev. 2021, 430, 213656. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Q.; Zhang, Y.; Duan, C. Electron transfer in the confined environments of metal-organic coordination supramolecular systems. Chem. Soc. Rev. 2020, 49, 5561–5600. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.M.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Self-assembled tetrahedral hosts as supramolecular catalysts. Acc. Chem. Res. 2018, 51, 2447–2455. [Google Scholar] [CrossRef]
- Zhao, L.; Jing, X.; Li, X.; Guo, X.; Zeng, L.; He, C.; Duan, C. Catalytic properties of chemical transformation within the confined pockets of werner-type capsules. Coord. Chem. Rev. 2019, 378, 151–187. [Google Scholar] [CrossRef]
- Vazquez-Gonzalez, M.; Wang, C.; Willner, I. Biocatalytic cascades operating on macromolecular scaffolds and in confined environments. Nat. Catal. 2020, 3, 256–273. [Google Scholar] [CrossRef]
- Lohr, T.L.; Marks, T.J. Orthogonal tandem catalysis. Nat. Chem. 2015, 7, 477–482. [Google Scholar] [CrossRef]
- Catti, L.; Zhang, Q.; Tiefenbacher, K. Advantages of catalysis in self-assembled molecular capsules. Chem. Eur. J. 2016, 22, 9060–9066. [Google Scholar] [CrossRef]
- Gaeta, C.; La Manna, P.; De Rosa, M.; Soriente, A.; Talotta, C.; Neri, P. Supramolecular catalysis with self-assembled capsules and cages: What happens in confined spaces. ChemCatChem 2021, 13, 1638–1658. [Google Scholar] [CrossRef]
- Fang, Y.; Powell, J.A.; Li, E.; Wang, Q.; Perry, Z.; Kirchon, A.; Yang, X.; Xiao, Z.; Zhu, C.; Zhang, L.; et al. Catalytic reactions within the cavity of coordination cages. Chem. Soc. Rev. 2019, 48, 4707–4730. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jordan, J.H.; Hu, X.Y.; Wang, L. Supramolecular strategies for controlling reactivity within confined nanospaces. Angew. Chem. Int. Ed. 2020, 59, 13712–13721. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Q.; Liu, Y.; Cui, Y. Supramolecular chirality in metal-organic complexes. Acc. Chem. Res. 2021, 54, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Koblenz, T.S.; Wassenaar, J.; Reek, J.N.H. Reactivity within a confined self-assembled nanospace. Chem. Soc. Rev. 2008, 37, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Stang, P.J. Recent developments in the preparation and chemistry of metallacycles and metallacages via coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef]
- Percastegui, E.G.; Ronson, T.K.; Nitschke, J.R. Design and applications of water-soluble coordination cages. Chem. Rev. 2020, 120, 13480–13544. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Gao, W.X.; Lin, L.; Jin, G.X. Recent advances in the construction and applications of heterometallic macrocycles and cages. Coord. Chem. Rev. 2017, 344, 323–344. [Google Scholar] [CrossRef]
- McConnell, A.J. Metallosupramolecular cages: From design principles and characterisation techniques to applications. Chem. Soc. Rev. 2022, 51, 2957–2971. [Google Scholar] [CrossRef]
- Takezawa, H.; Shitozawa, K.; Fujita, M. Enhanced reactivity of twisted amides inside a molecular cage. Nat. Chem. 2020, 12, 574–578. [Google Scholar] [CrossRef]
- Ibukuro, F.; Kusukawa, T.; Fujita, M. A thermally switchable molecular lock. Guest-templated synthesis of a kinetically stable nanosized cage. J. Am. Chem. Soc. 1998, 120, 8561–8562. [Google Scholar] [CrossRef]
- Takezawa, H.; Akiba, S.; Murase, T.; Fujita, M. Cavity-directed chromism of phthalein dyes. J. Am. Chem. Soc. 2015, 137, 7043–7046. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Oguro, D.; Miyazawa, M.; Oka, H.; Yamaguchi, K.; Ogura, K. Self-assembly of ten molecules into nanometre-sized organic host frameworks. Nature 1995, 378, 469–471. [Google Scholar] [CrossRef]
- Takezawa, H.; Murase, T.; Fujita, M. Temporary and permanent trapping of the metastable twisted conformer of an overcrowded chromic alkene via encapsulation. J. Am. Chem. Soc. 2012, 134, 17420–17423. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Wu, K.; Yin, C.; Li, K.; Huang, Y.; Ruan, J.; Feng, X.; Hu, P.; Su, C.Y. Cage-confined photocatalysis for wide-scope unusually selective [2 + 2] cycloaddition through visible-light triplet sensitization. Nat. Commun. 2020, 11, 4675. [Google Scholar] [CrossRef]
- Li, K.; Zhang, L.-Y.; Yan, C.; Wei, S.-C.; Pan, M.; Zhang, L.; Su, C.-Y. Stepwise assembly of pd6(rul3)8 nanoscale rhombododecahedral metal–organic cages via metalloligand strategy for guest trapping and protection. J. Am. Chem. Soc. 2014, 136, 4456–4459. [Google Scholar] [CrossRef]
- Chen, S.; Li, K.; Zhao, F.; Zhang, L.; Pan, M.; Fan, Y.-Z.; Guo, J.; Shi, J.; Su, C.-Y. A metal-organic cage incorporating multiple light harvesting and catalytic centres for photochemical hydrogen production. Nat. Commun. 2016, 7, 13169. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, M.; Tamura, M.; Fujita, M. Diels-alder in aqueous molecular hosts: Unusual regioselectivity and efficient catalysis. Science 2006, 312, 251–254. [Google Scholar] [CrossRef]
- Gonell, S.; Reek, J.N.H. Gold-catalyzed cycloisomerization reactions within guanidinium M12L24 nanospheres: The effect of local concentrations. ChemCatChem 2019, 11, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Gramage-Doria, R.; Hessels, J.; Leenders, S.H.; Troppner, O.; Durr, M.; Ivanovic-Burmazovic, I.; Reek, J.N. Gold(I) catalysis at extreme concentrations inside self-assembled nanospheres. Angew. Chem. Int. Ed. 2014, 53, 13380–13384. [Google Scholar] [CrossRef]
- Cauteruccio, S.; Loos, A.; Bossi, A.; Blanco Jaimes, M.C.; Dova, D.; Rominger, F.; Prager, S.; Dreuw, A.; Licandro, E.; Hashmi, A.S.K. Gold(I) complexes of tetrathiaheterohelicene phosphanes. Inorg. Chem. 2013, 52, 7995–8004. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Gonell, S.; Leenders, S.H.; Durr, M.; Ivanovic-Burmazovic, I.; Reek, J.N. Self-assembled nanospheres with multiple endohedral binding sites pre-organize catalysts and substrates for highly efficient reactions. Nat. Chem. 2016, 8, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Schug, K.A.; Lindner, W. Noncovalent binding between guanidinium and anionic groups: Focus on biological- and synthetic-based arginine/guanidinium interactions with phosph[on]ate and sulf[on]ate residues. Chem. Rev. 2005, 105, 67–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Poole, D., 3rd; Mathew, S.; Yan, N.; Hessels, J.; Orth, N.; Ivanovic-Burmazovic, I.; Reek, J.N.H. Control over electrochemical water oxidation catalysis by preorganization of molecular ruthenium catalysts in self-assembled nanospheres. Angew. Chem. Int. Ed. 2018, 57, 11247–11251. [Google Scholar] [CrossRef] [PubMed]
- Daubignard, J.; Lutz, M.; Detz, R.J.; de Bruin, B.; Reek, J.N.H. Origin of the selectivity and activity in the Rhodium-catalyzed asymmetric hydrogenation using supramolecular ligands. ACS Catal. 2019, 9, 7535–7547. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, M.; Turega, S.; Stephenson, A.; Hunter, C.A.; Ward, M.D. Quantification of solvent effects on molecular recognition in polyhedral coordination cage hosts. Chem. Sci. 2013, 4, 2744–2751. [Google Scholar] [CrossRef] [Green Version]
- Turega, S.; Cullen, W.; Whitehead, M.; Hunter, C.A.; Ward, M.D. Mapping the internal recognition surface of an octanuclear coordination cage using guest libraries. J. Am. Chem. Soc. 2014, 136, 8475–8483. [Google Scholar] [CrossRef]
- Cullen, W.; Metherell, A.J.; Wragg, A.B.; Taylor, C.G.P.; Williams, N.H.; Ward, M.D. Catalysis in a cationic coordination cage using a cavity-bound guest and surface-bound anions: Inhibition, activation, and autocatalysis. J. Am. Chem. Soc. 2018, 140, 2821–2828. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Xiao, Z.; Li, J.; Lollar, C.; Liu, L.; Lian, X.; Yuan, S.; Banerjee, S.; Zhang, P.; Zhou, H.C. Formation of a highly reactive cobalt nanocluster crystal within a highly negatively charged porous coordination cage. Angew. Chem. Int. Ed. 2018, 57, 5283–5287. [Google Scholar] [CrossRef]
- Fang, Y.; Li, J.; Togo, T.; Jin, F.; Xiao, Z.; Liu, L.; Drake, H.; Lian, X.; Zhou, H.-C. Ultra-small face-centered-cubic ru nanoparticles confined within a porous coordination cage for dehydrogenation. Chem 2018, 4, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Lavendomme, R.; Burghaus, O.; Nitschke, J.R. A Zn4L6 capsule with enhanced catalytic C-C bond formation activity upon c60 binding. Angew. Chem. Int. Ed. 2019, 58, 9073–9077. [Google Scholar] [CrossRef]
- Lu, Z.; Ronson, T.K.; Nitschke, J.R. Reversible reduction drives anion ejection and C60 binding within an FeII4L6 cage. Chem. Sci. 2020, 11, 1097–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.F. Catalyst-controlled site-selective bond activation. Acc. Chem. Res. 2017, 50, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Bender, T.A.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A supramolecular strategy for selective catalytic hydrogenation independent of remote chain length. J. Am. Chem. Soc. 2019, 141, 11806–11810. [Google Scholar] [CrossRef] [PubMed]
- Hart-Cooper, W.M.; Zhao, C.; Triano, R.M.; Yaghoubi, P.; Ozores, H.L.; Burford, K.N.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. The effect of host structure on the selectivity and mechanism of supramolecular catalysis of prins cyclizations. Chem. Sci. 2015, 6, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.L. Biocatalysis in drug development—Highlights of the recent patent literature. Org. Process Res. Dev. 2018, 22, 1063–1080. [Google Scholar] [CrossRef]
- Morimoto, M.; Cao, W.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Chemoselective and site-selective reductions catalyzed by a supramolecular host and a pyridine-borane cofactor. J. Am. Chem. Soc. 2021, 143, 2108–2114. [Google Scholar] [CrossRef]
- Bierschenk, S.M.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A nanovessel-catalyzed three-component aza-darzens reaction. J. Am. Chem. Soc. 2020, 142, 733–737. [Google Scholar] [CrossRef]
- Xu, G.; Leloux, S.; Zhang, P.; Meijide Suárez, J.; Zhang, Y.; Derat, E.; Ménand, M.; Bistri-Aslanoff, O.; Roland, S.; Leyssens, T.; et al. Capturing the monomeric (L)CuH in nhc-capped cyclodextrin: Cavity-controlled chemoselective hydrosilylation of α,β-unsaturated ketones. Angew. Chem. Int. Ed. 2020, 59, 7591–7597. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Gong, W.; Jiang, H.; Tang, X.; Cui, Y.; Liu, Y. Boosting enantioselectivity of chiral molecular catalysts with supramolecular metal–organic cages. CCS Chem. 2021, 4, 1692–1700. [Google Scholar] [CrossRef]
- Gong, W.; Chu, D.; Jiang, H.; Chen, X.; Cui, Y.; Liu, Y. Permanent porous hydrogen-bonded frameworks with two types of bronsted acid sites for heterogeneous asymmetric catalysis. Nat. Commun. 2019, 10, 600. [Google Scholar] [CrossRef]
- Hu, F.; Liu, C.; Wu, M.; Pang, J.; Jiang, F.; Yuan, D.; Hong, M. An ultrastable and easily regenerated hydrogen-bonded organic molecular framework with permanent porosity. Angew. Chem. Int. Ed. 2017, 56, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Cullen, W.; Takezawa, H.; Fujita, M. Demethylenation of cyclopropanes via photoinduced guest-to-host electron transfer in an m6 l4 cage. Angew. Chem. Int. Ed. 2019, 58, 9171–9173. [Google Scholar] [CrossRef] [PubMed]
- Furutani, Y.; Kandori, H.; Kawano, M.; Nakabayashi, K.; Yoshizawa, M.; Fujita, M. In situ spectroscopic, electrochemical, and theoretical studies of the photoinduced host−guest electron transfer that precedes unusual host-mediated alkane photooxidation. J. Am. Chem. Soc. 2009, 131, 4764–4768. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Nishijima, Y.; Fujita, M. Unusual photoreaction of triquinacene within self-assembled hosts. Chem. Asian J. 2012, 7, 826–829. [Google Scholar] [CrossRef]
- Takezawa, H.; Kanda, T.; Nanjo, H.; Fujita, M. Site-selective functionalization of linear diterpenoids through u-shaped folding in a confined artificial cavity. J. Am. Chem. Soc. 2019, 141, 5112–5115. [Google Scholar] [CrossRef]
- Guo, J.; Xu, Y.W.; Li, K.; Xiao, L.M.; Chen, S.; Wu, K.; Chen, X.D.; Fan, Y.Z.; Liu, J.M.; Su, C.Y. Regio- and enantioselective photodimerization within the confined space of a homochiral ruthenium/palladium heterometallic coordination cage. Angew. Chem. Int. Ed. 2017, 56, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, K.; Hou, Y.-J.; Pan, M.; Zhang, L.-Y.; Chen, L.; Su, C.-Y. Homochiral d4-symmetric metal–Organic cages from stereogenic Ru(II) metalloligands for effective enantioseparation of atropisomeric molecules. Nat. Commun. 2016, 7, 10487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongkind, L.J.; Caumes, X.; Hartendorp, A.P.T.; Reek, J.N.H. Ligand template strategies for catalyst encapsulation. Acc. Chem. Res. 2018, 51, 2115–2128. [Google Scholar] [CrossRef]
- Garcia-Simon, C.; Gramage-Doria, R.; Raoufmoghaddam, S.; Parella, T.; Costas, M.; Ribas, X.; Reek, J.N. Enantioselective hydroformylation by a rh-catalyst entrapped in a supramolecular metallocage. J. Am. Chem. Soc. 2015, 137, 2680–2687. [Google Scholar] [CrossRef]
- García-Simón, C.; Garcia-Borràs, M.; Gómez, L.; Parella, T.; Osuna, S.; Juanhuix, J.; Imaz, I.; Maspoch, D.; Costas, M.; Ribas, X. Sponge-like molecular cage for purification of fullerenes. Nat. Commun. 2014, 5, 5557. [Google Scholar] [CrossRef]
- Zhao, C.; Sun, Q.-F.; Hart-Cooper, W.M.; DiPasquale, A.G.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. Chiral amide directed assembly of a diastereo- and enantiopure supramolecular host and its application to enantioselective catalysis of neutral substrates. J. Am. Chem. Soc. 2013, 135, 18802–18805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.J.; Clary, K.N.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. A supramolecular approach to combining enzymatic and transition metal catalysis. Nat. Chem. 2013, 5, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Ito, H.; Fujita, D.; Fujita, M. Permeable self-assembled molecular containers for catalyst isolation enabling two-step cascade reactions. J. Am. Chem. Soc. 2017, 139, 6090–6093. [Google Scholar] [CrossRef] [PubMed]
- Vogler, T.; Studer, A. Applications of tempo in synthesis. Synthesis 2008, 2008, 1979–1993. [Google Scholar]
- Jones, S.B.; Simmons, B.; Mastracchio, A.; MacMillan, D.W.C. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Iida, J.; Suzuki, K.; Kawano, M.; Ozeki, T.; Fujita, M. Fluorous nanodroplets structurally confined in an organopalladium sphere. Science 2006, 313, 1273–1276. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.-F.; Chen, L.-J.; Chen, S.; Wu, G.-Y.; Jiang, W.-L.; Shen, J.-C.; Qin, Y.; Xu, L.; Yang, H.-B. Confinement self-assembly of metal-organic cages within mesoporous carbon for one-pot sequential reactions. Chem 2020, 6, 2395–2406. [Google Scholar] [CrossRef]
- Chen, L.-J.; Chen, S.; Qin, Y.; Xu, L.; Yin, G.-Q.; Zhu, J.-L.; Zhu, F.-F.; Zheng, W.; Li, X.; Yang, H.-B. Construction of porphyrin-containing metallacycle with improved stability and activity within mesoporous carbon. J. Am. Chem. Soc. 2018, 140, 5049–5052. [Google Scholar] [CrossRef]
- Jiao, J.; Tan, C.; Li, Z.; Liu, Y.; Han, X.; Cui, Y. Design and assembly of chiral coordination cages for asymmetric sequential reactions. J. Am. Chem. Soc. 2018, 140, 2251–2259. [Google Scholar] [CrossRef]
- Srinivasan, K.; Michaud, P.; Kochi, J.K. Epoxidation of olefins with cationic (salen)manganese(III) complexes. The modulation of catalytic activity by substituents. J. Am. Chem. Soc. 1986, 108, 2309–2320. [Google Scholar] [CrossRef]
- Wende, R.C.; Schreiner, P.R. Evolution of asymmetric organocatalysis: Multi- and retrocatalysis. Green Chem. 2012, 14, 1821–1849. [Google Scholar] [CrossRef] [Green Version]
- Salles, A.G., Jr.; Zarra, S.; Turner, R.M.; Nitschke, J.R. A self-organizing chemical assembly line. J. Am. Chem. Soc. 2013, 135, 19143–19146. [Google Scholar] [CrossRef] [PubMed]
- Holloway, L.R.; Bogie, P.M.; Lyon, Y.; Ngai, C.; Miller, T.F.; Julian, R.R.; Hooley, R.J. Tandem reactivity of a self-assembled cage catalyst with endohedral acid groups. J. Am. Chem. Soc. 2018, 140, 8078–8081. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.A.; Holloway, L.R.; Miller, T.F.; Lyon, Y.; Julian, R.R.; Hooley, R.J. Electronic effects on narcissistic self-sorting in multicomponent self-assembly of fe-iminopyridine meso-helicates. Inorg. Chem. 2016, 55, 9805–9815. [Google Scholar] [CrossRef]
- Ngai, C.; Sanchez-Marsetti, C.M.; Harman, W.H.; Hooley, R.J. Supramolecular catalysis of the oxa-pictet-spengler reaction with an endohedrally functionalized self-assembled cage complex. Angew. Chem. Int. Ed. 2020, 59, 23505–23509. [Google Scholar] [CrossRef]
Figure 1.
Hydrolysis of amides catalyzed by MOC-1 by mechanically twisting them within the cavity of the cage [39]. Reaction additions: 1 (2.4 mM), co-encapsulated guest agent (1.05 eq.), MOC-1 (2.4 mM), NaOH (100 mM), 60 °C, D2O. Reprinted/adapted with permission from Ref. [39]. Copyright 2020 Springer Nature Ltd.
Figure 1.
Hydrolysis of amides catalyzed by MOC-1 by mechanically twisting them within the cavity of the cage [39]. Reaction additions: 1 (2.4 mM), co-encapsulated guest agent (1.05 eq.), MOC-1 (2.4 mM), NaOH (100 mM), 60 °C, D2O. Reprinted/adapted with permission from Ref. [39]. Copyright 2020 Springer Nature Ltd.

Figure 2.
Intermolecular [2 + 2] cycloaddition of acyclic alkenes catalyzed by MOC-2 through visible-light triplet sensitization [45]. Reaction additions: 2 (0.10 mmol), 3 (0.10 mmol), MOC-2 (0.08 mol%) or 0.64 mol% RuL3(PF6)2, pyrene (10 eq.), DMSO and H2O, r.t. Reprinted/adapted with permission from Ref. [45]. Copyright 2014 American Chemical Society.
Figure 2.
Intermolecular [2 + 2] cycloaddition of acyclic alkenes catalyzed by MOC-2 through visible-light triplet sensitization [45]. Reaction additions: 2 (0.10 mmol), 3 (0.10 mmol), MOC-2 (0.08 mol%) or 0.64 mol% RuL3(PF6)2, pyrene (10 eq.), DMSO and H2O, r.t. Reprinted/adapted with permission from Ref. [45]. Copyright 2014 American Chemical Society.

Figure 3.
Hydroalkoxylation of the g-allenol catalyzed by MOC-3 with a high local Au catalyst concentration [49]. Reaction additions: 4 (10.4 mM), MOC-3 (2 mol%), CH2CCl2, 25 °C. Reprinted/adapted with permission from Ref. [49]. Copyright 2014 Wiley VCH.

Figure 4.
Cyclization reaction of acetylenic acid catalyzed by MOC-4 binding with Au(I) catalysts [51]. Reaction additions: 5 (10.0 mM), Au+ (0.5 mM), MOC-4 (0.125 mM), NEt3 (0.5 mM), CD3CN, r.t. Reprinted/adapted with permission from Ref. [51]. Copyright 2016 Springer Nature Ltd.

Figure 5.
Kemp elimination of benzisoxazole catalyzed by MOC-5 in the presence of base [7]. Reaction additions: 6 (0.85 mM), MOC-5 (1.0 mM), D2O, 25 °C. Reprinted/adapted with permission from Ref. [7]. Copyright 2016 Springer Nature Ltd.

Figure 6.
Hydrolysis of ammonium borane catalyzed by Co nanoparticle catalysts [58]. Reaction additions: ammonia borane (1 mmol), H2O (1 mL), (Co 0.16 mmol), r.t. Reprinted/adapted with permission from Ref. [58]. Copyright 2018 Wiley VCH.

Figure 7.
Oxidative coupling reaction of tetraaryl borates catalyzed by MOC-8 with encapsulated C60 [60]. Reaction additions: 8 (0.014 mmol), MOC-8 (10 mol%), 9 (200 mol%), C60 (20 mol%), MeCN, 90 °C. Reprinted/adapted with permission from Ref. [60]. Copyright 2019 Wiley VCH.

Figure 8.
Selective hydrogenation of polyenol catalyzed by rhodium complex encapsulated within MOC-9 [63]. Reaction additions: 10 (0.05 mmol), MOC-9 (10 mol%), [Rh][BF4] (10 mol%), H2O (1.0 mL), H2, r.t. Reprinted/adapted with permission from Ref. [63]. Copyright 2019 American Chemical Society.

Figure 9.
Selective reduction of ketones catalyzed by pyridine-borane encapsulated within MOC-10 [66]. Reaction additions: 11 (0.023 mmol), MOC-10 (15 mol%), 12 (100 mol%), K3PO4 (100 mM), D2O, pD 8, r.t. Reprinted/adapted with permission from Ref. [66]. Copyright 2021 American Chemical Society.

Figure 10.
Asymmetric [3 + 2] coupling of indoles with quinone monoimine catalyzed by MOC-11 [70]. Reaction additions: 13 (0.10 mmol), MOC-11 (0.04 mol%), 14 (0.15 mmol), CH3CN, 0 °C. Reprinted/adapted with permission from Ref. [70]. Copyright 2020 Springer Nature Ltd.

Figure 11.
Demethylenation of cyclopropane catalyzed by MOC-12 [72]. Reaction additions: 15 (0.136 mmol), MOC-12 (100 mol%), UV light, H2O, r.t. Reprinted/adapted with permission from Ref. [72]. Copyright 2019 Wiley VCH.

Figure 12.
Oxidative coupling of 2-naphthol catalyzed by MOC-2 [76]. Reaction additions: 16 (2.5 × 10−3 mmol), MOC-2 (10 mol%), 8 W blue LEDs (λmax = 453 nm), CH3CN/H2O, r.t. Reprinted/adapted with permission from Ref. [76]. Copyright 2017 Wiley VCH.

Figure 13.
Regio- and enantioselective hydroformylation of styrenes catalyzed by the encapsulated chiral Rh catalyst in MOC-13 [79]. Reaction additions: 17 (8.3 × 10−2 mmol), MOC-13 (10 mol%), [Rh(acac)(CO)2] (0.019 mol%), CH3CN/Toluene, H2/CO (1:1, 10 bar), r.t. Reprinted/adapted with permission from Ref. [79]. Copyright 2015 American Chemical Society. “*” in the figure indicates the chirality of the carbon.
Figure 13.
Regio- and enantioselective hydroformylation of styrenes catalyzed by the encapsulated chiral Rh catalyst in MOC-13 [79]. Reaction additions: 17 (8.3 × 10−2 mmol), MOC-13 (10 mol%), [Rh(acac)(CO)2] (0.019 mol%), CH3CN/Toluene, H2/CO (1:1, 10 bar), r.t. Reprinted/adapted with permission from Ref. [79]. Copyright 2015 American Chemical Society. “*” in the figure indicates the chirality of the carbon.

Figure 14.
Prins cyclizations of monoterpene catalyzed by MOC-14 [81]. Reaction additions: 18 (0.04 mmol), MOC-14 (2.5 mol%), CD3OD/D2O (0.5 mL), pD = 8, r.t. Reprinted/adapted with permission from Ref. [81]. Copyright 2013 American Chemical Society.

Figure 15.
(a) Hydrolysis–cyclization tandem reactions of allenic acetate catalyzed by spatially separated natural enzyme (rabit liver) and Me3P+Au-encapsulated in MOC-10 [82]. Reaction additions: 19 (1.2 × 10−2 mmol), MOC-10 (10 mol%), Me3P+Au− (10 mol%), enzyme (Rabit liver, 6 unit), MeOH/DMSO (0.5 mL), pH = 8, 37 °C. (b) Isomerization of 1-propenol the sequential reduction of propanal catalysed by spatially separated (Me3P)CpRu(NCMe)2+ encapsulated in MOC-10 and alcohol dehydrogenase (ADH). Reprinted/adapted with permission from Ref. [82]. Copyright 2013 Springer Nature Ltd.
Figure 15.
(a) Hydrolysis–cyclization tandem reactions of allenic acetate catalyzed by spatially separated natural enzyme (rabit liver) and Me3P+Au-encapsulated in MOC-10 [82]. Reaction additions: 19 (1.2 × 10−2 mmol), MOC-10 (10 mol%), Me3P+Au− (10 mol%), enzyme (Rabit liver, 6 unit), MeOH/DMSO (0.5 mL), pH = 8, 37 °C. (b) Isomerization of 1-propenol the sequential reduction of propanal catalysed by spatially separated (Me3P)CpRu(NCMe)2+ encapsulated in MOC-10 and alcohol dehydrogenase (ADH). Reprinted/adapted with permission from Ref. [82]. Copyright 2013 Springer Nature Ltd.

Figure 16.
Oxidation of an alcohol followed by a stereoselective Diels−Alder cyclization catalyzed by MOC-15 and MOC-16 [83]. Reaction conditions: 21 (8.0 × 10−2 mmol), MOC-15 (10 mol%), MOC-16 (10 mol%), PhI(OAc)2 (115 mol%), CF3COOH (10 mol%), CD3NO2/D2O, −10 °C. Reprinted/adapted with permission from Ref. [83]. Copyright 2017 American Chemical Society.
Figure 16.
Oxidation of an alcohol followed by a stereoselective Diels−Alder cyclization catalyzed by MOC-15 and MOC-16 [83]. Reaction conditions: 21 (8.0 × 10−2 mmol), MOC-15 (10 mol%), MOC-16 (10 mol%), PhI(OAc)2 (115 mol%), CF3COOH (10 mol%), CD3NO2/D2O, −10 °C. Reprinted/adapted with permission from Ref. [83]. Copyright 2017 American Chemical Society.

Figure 17.
One-pot alcohol oxidation and Knoevenagel condensation sequential reactions catalyzed by MOC-17@FDU-ED [87]. Reaction conditions: 22 (5.0 × 10−2 mmol), MOC-17@FDU-ED (10 mol% based on TEMPO units), PhI(OAc)2 (120 mol%), Malononitrile (120 mol%), CH3CN, r.t., and then 60 °C. Reprinted/adapted with permission from Ref. [87]. Copyright 2020 Elsevier Inc.
Figure 17.
One-pot alcohol oxidation and Knoevenagel condensation sequential reactions catalyzed by MOC-17@FDU-ED [87]. Reaction conditions: 22 (5.0 × 10−2 mmol), MOC-17@FDU-ED (10 mol% based on TEMPO units), PhI(OAc)2 (120 mol%), Malononitrile (120 mol%), CH3CN, r.t., and then 60 °C. Reprinted/adapted with permission from Ref. [87]. Copyright 2020 Elsevier Inc.

Figure 18.
Asymmetric epoxidation/epoxide ring-opening reactions of 2,2-dimethyl-2H-chromene (DMCH) with TMSN3 catalyzed by MOC-18 [89]. Reaction conditions: 23 (0.1 mmol), MOC-18 (0.1 mol%), sPhIO (100 mol%), CH2Cl2, 0 °C. Reprinted/adapted with permission from Ref. [89]. Copyright 2018 American Chemical Society.
Figure 18.
Asymmetric epoxidation/epoxide ring-opening reactions of 2,2-dimethyl-2H-chromene (DMCH) with TMSN3 catalyzed by MOC-18 [89]. Reaction conditions: 23 (0.1 mmol), MOC-18 (0.1 mol%), sPhIO (100 mol%), CH2Cl2, 0 °C. Reprinted/adapted with permission from Ref. [89]. Copyright 2018 American Chemical Society.

Figure 19.
One-pot sequential transformation from into a chiral hydroxylactone catalyzed by singlet oxygen (photogenerated by methylene blue), in situ assembled metal−organic cage MOC-19, and L-proline [92]. Reaction conditions: 24 (2.6 mmol), MOC-19 (0.5 mol%), methylene blue (3.5 mol%), L-proline (25 mol%) H2O, r.t., O2 atmosphere. Reprinted/adapted with permission from Ref. [92]. Copyright 2013 American Chemical Society.
Figure 19.
One-pot sequential transformation from into a chiral hydroxylactone catalyzed by singlet oxygen (photogenerated by methylene blue), in situ assembled metal−organic cage MOC-19, and L-proline [92]. Reaction conditions: 24 (2.6 mmol), MOC-19 (0.5 mol%), methylene blue (3.5 mol%), L-proline (25 mol%) H2O, r.t., O2 atmosphere. Reprinted/adapted with permission from Ref. [92]. Copyright 2013 American Chemical Society.

Figure 20.
Tandem acetal hydrolysis/cage-to-cage interconversion catalyzed MOC-20 [93]. Reaction conditions: 25 (12.3 mM), MOC-20 (34 mol%), MOC-Br (1.5 mM) H2O, CD3CN. Reprinted/adapted with permission from Ref. [93]. Copyright 2018 American Chemical Society.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, S.; Chen, L.-J. Metal–Organic Cages: Applications in Organic Reactions. Chemistry 2022, 4, 494-519. https://doi.org/10.3390/chemistry4020036
AMA Style
Chen S, Chen L-J. Metal–Organic Cages: Applications in Organic Reactions. Chemistry. 2022; 4(2):494-519. https://doi.org/10.3390/chemistry4020036
Chicago/Turabian StyleChen, Shangjun, and Li-Jun Chen. 2022. "Metal–Organic Cages: Applications in Organic Reactions" Chemistry 4, no. 2: 494-519. https://doi.org/10.3390/chemistry4020036