
Recent Advances in Influenza, HIV and SARS-CoV-2 Infection Prevention and Drug Treatment—The Need for Precision Medicine


Abstract
:
1. Introduction
- ▪
- The toll of past and present pandemics;
- ▪
- RNA viruses: structures of IV, HIV, and SARS-CoV-2;
- ▪
- Host immune response against viruses;
- ▪
- Prevention of viral infection by vaccination;
- ▪
- Anti-influenza, anti-HIV, and anti-SARS-CoV-2 agents;
- ▪
- Summary and conclusion.
2. The Toll of Past and Present Pandemics
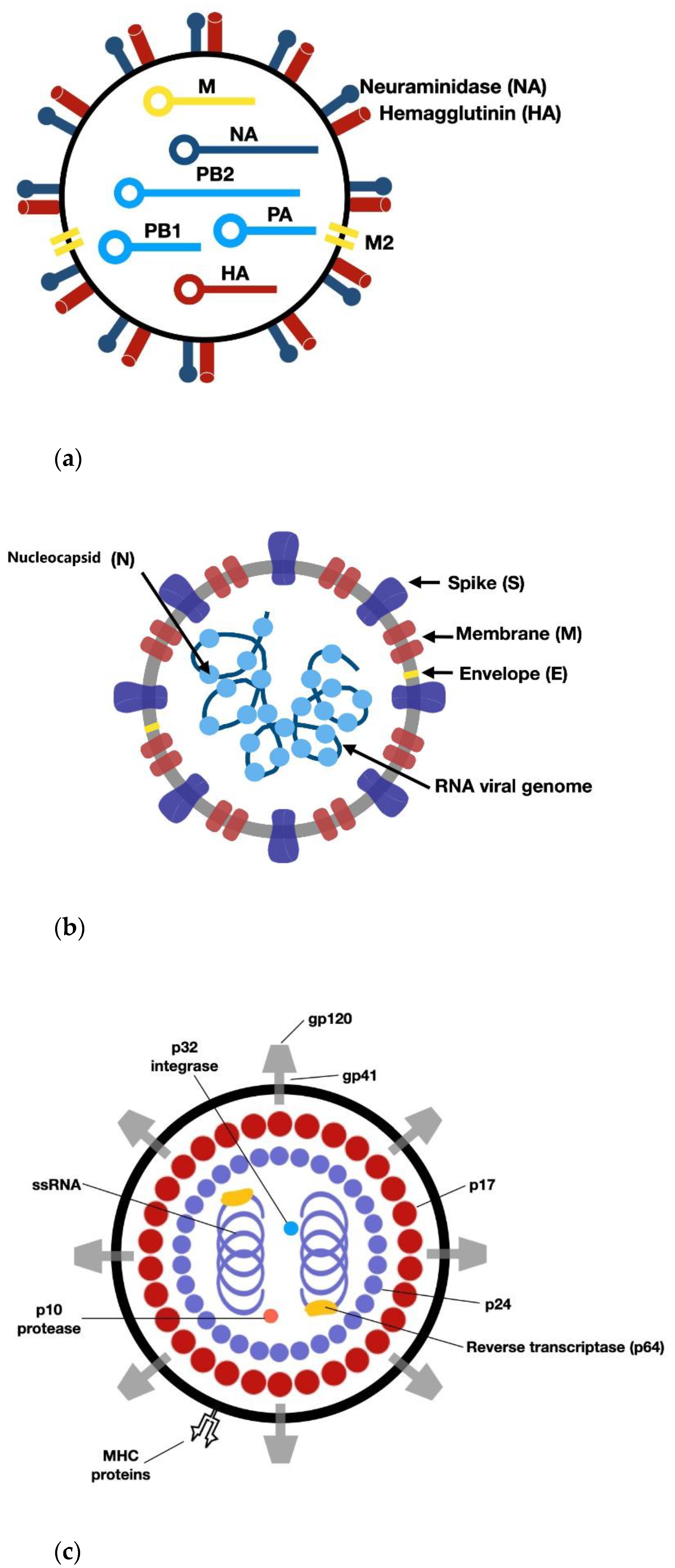
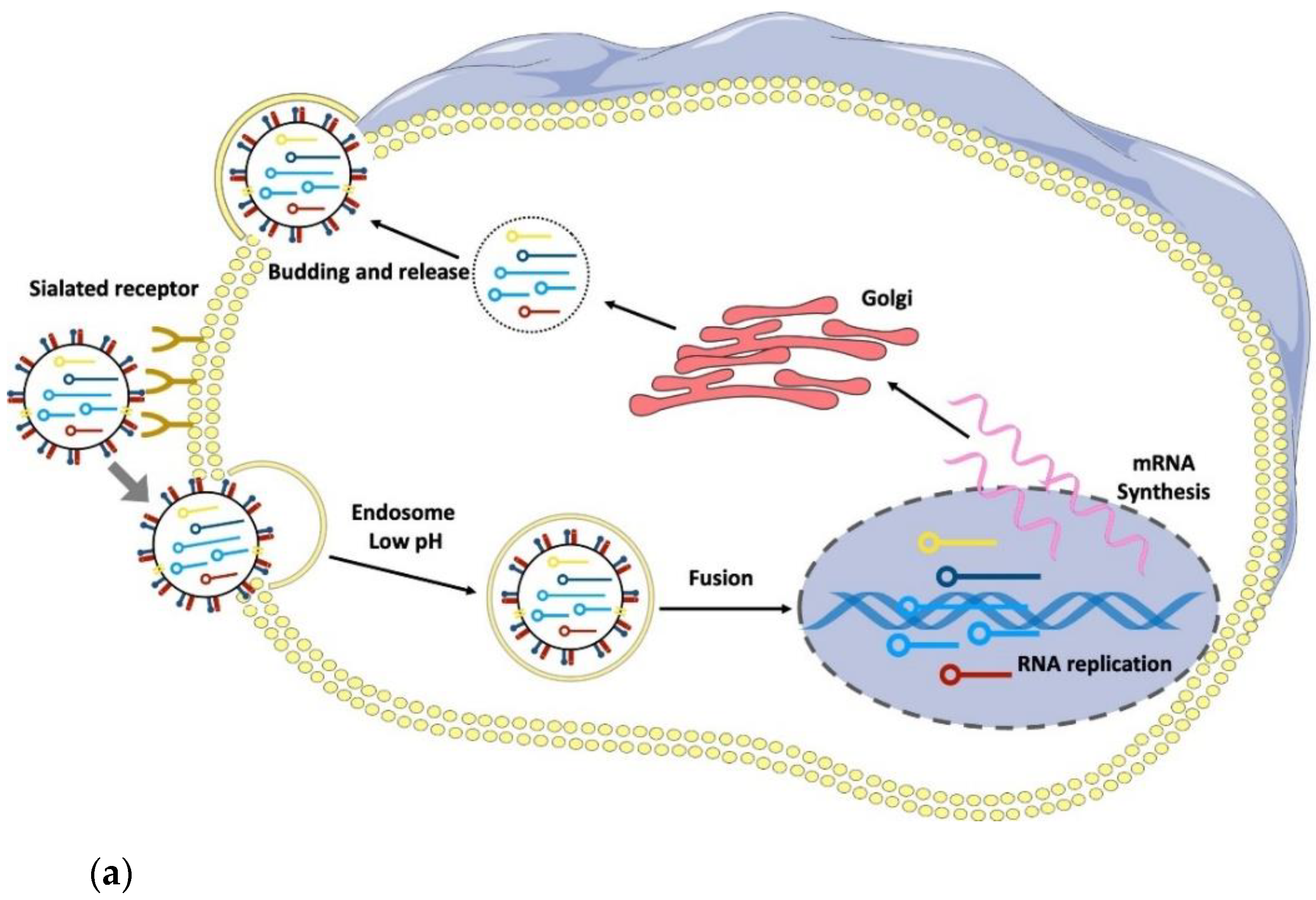
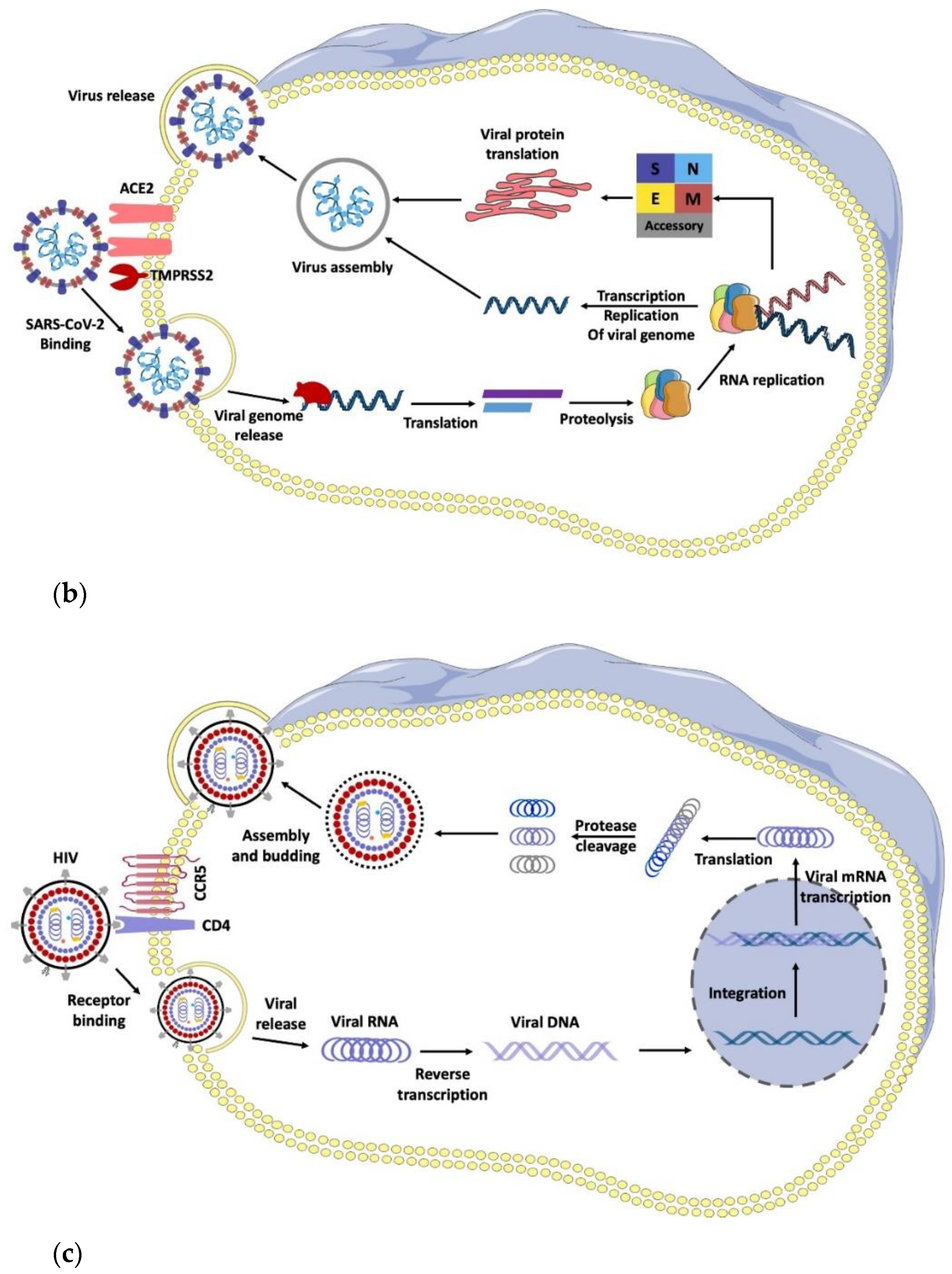
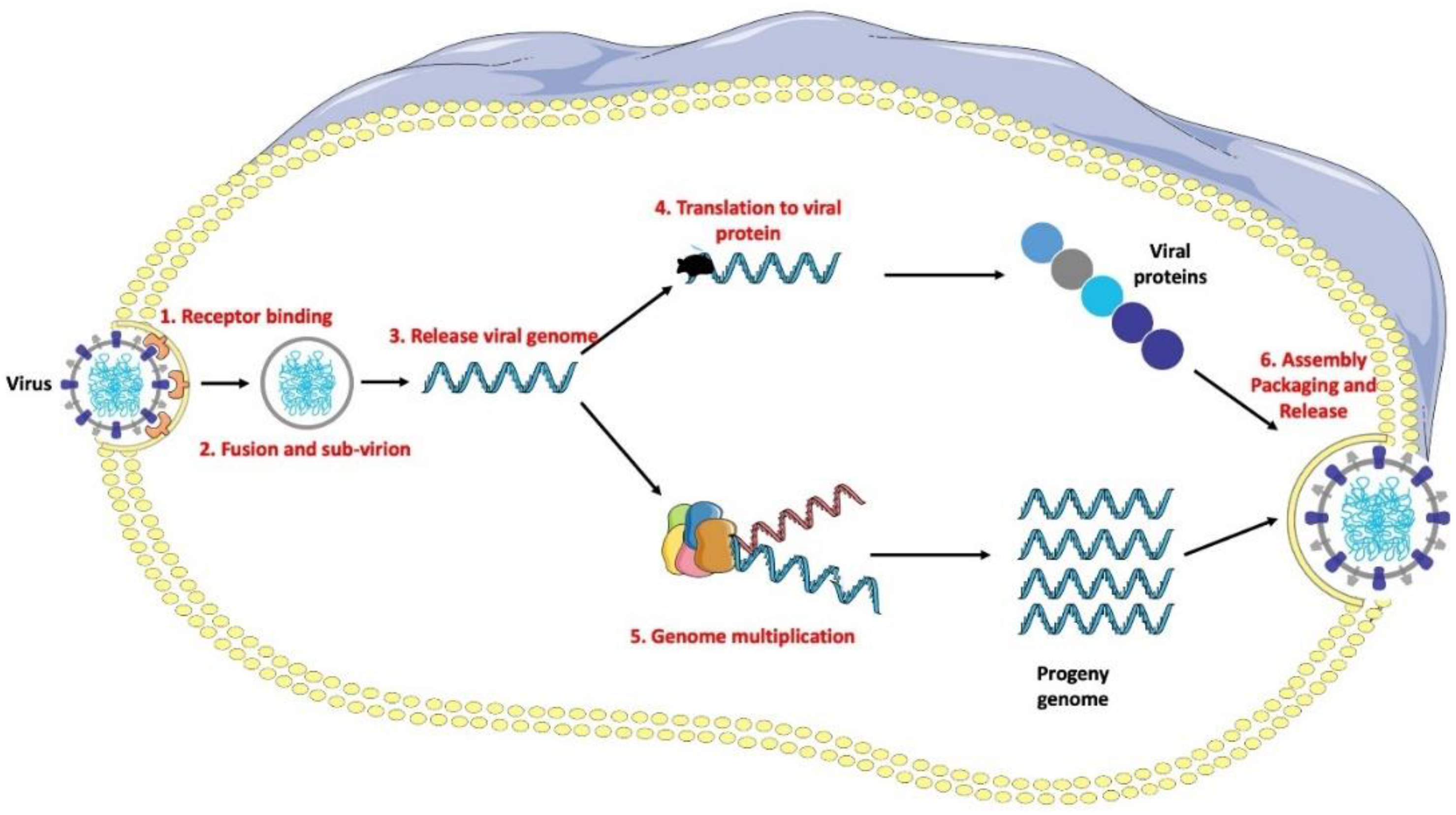
3. RNA Viruses: Structure of IV, HIV, and SARS-CoV-2
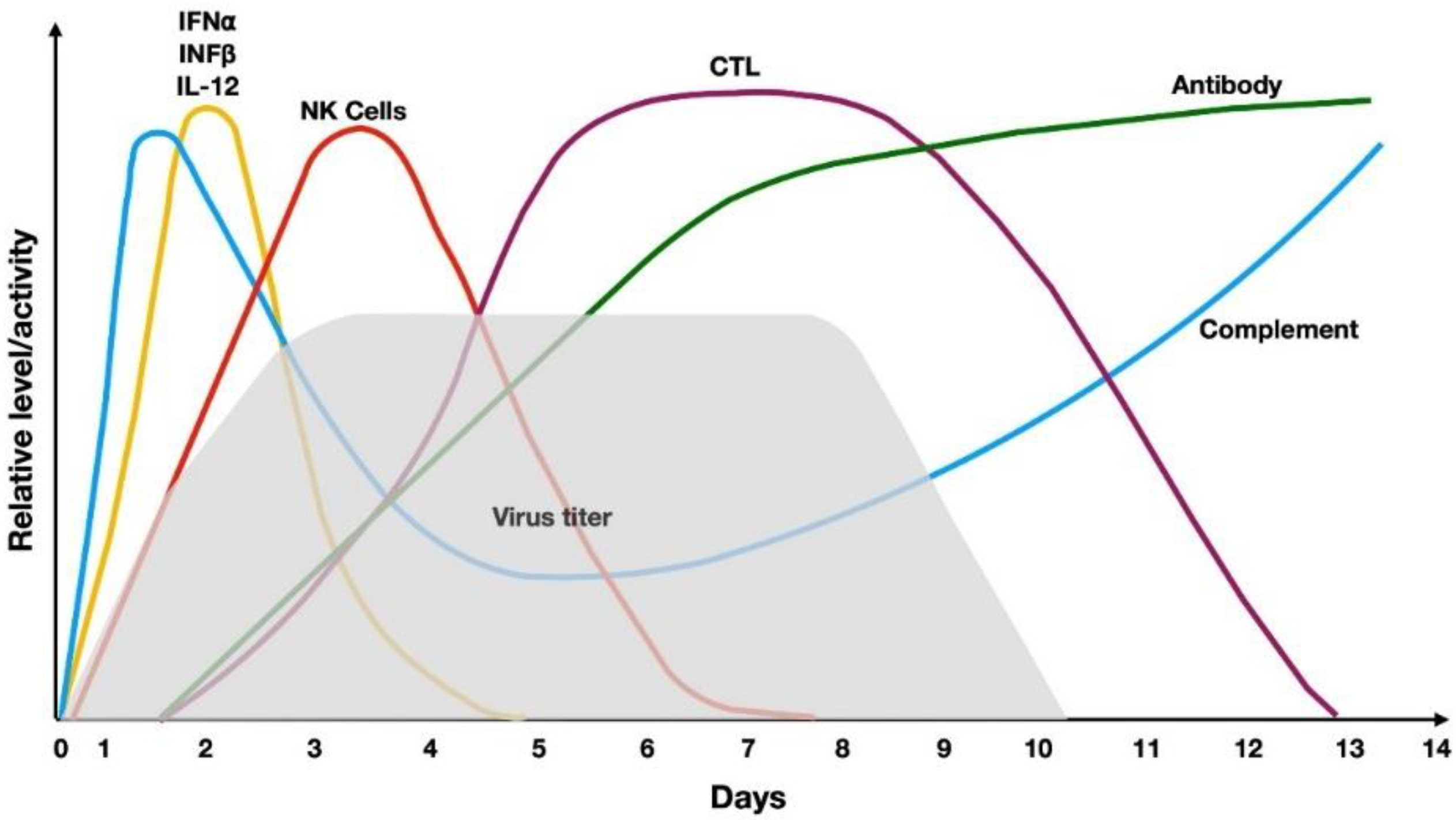
4. Host Immune Response against Viruses
4.1. The ‘One Fits All’ Immunology
4.2. Individual Response to Pathogens
5. Prevention of Viral Infections by Vaccination
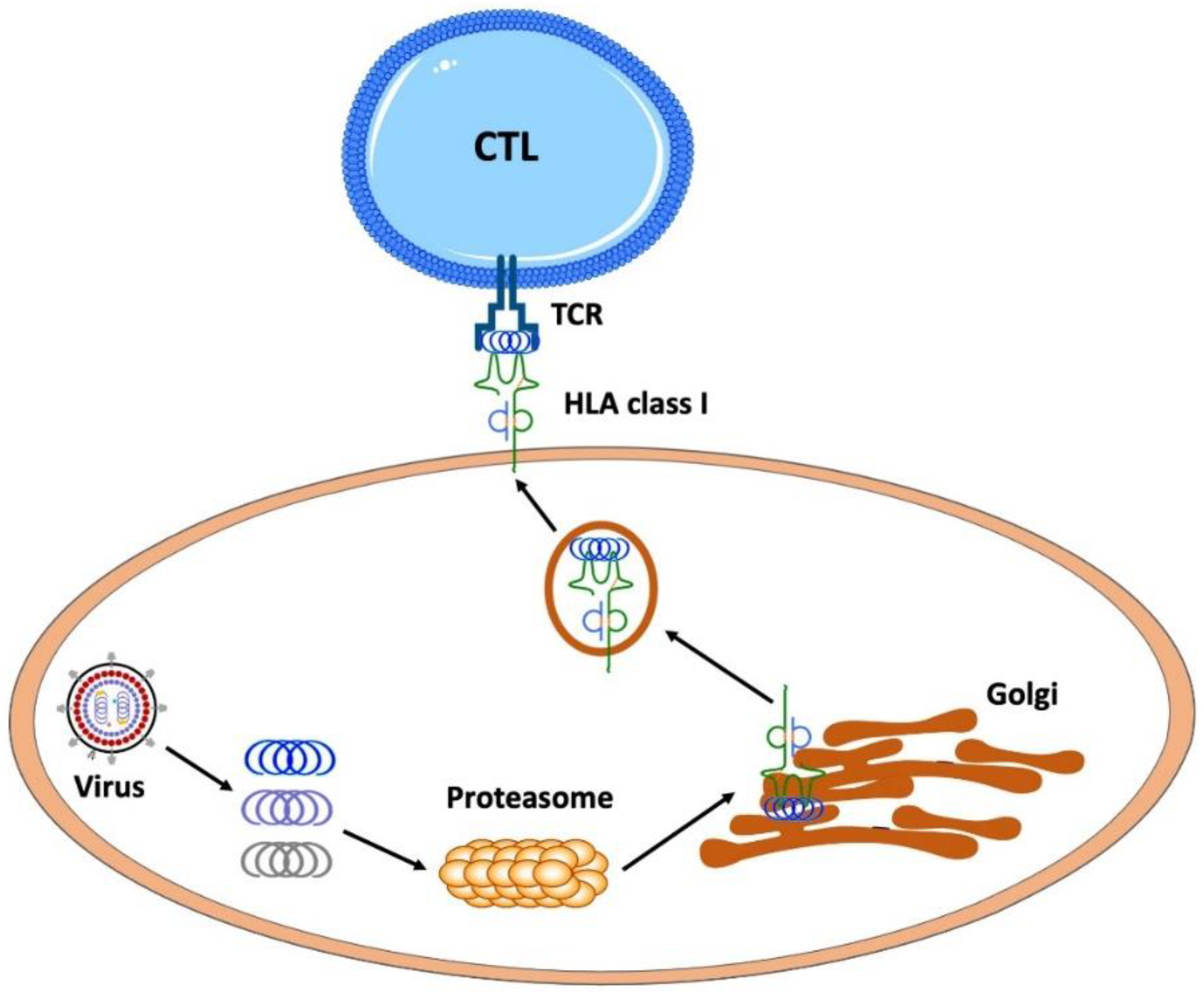
5.1. Essential Immunology for Vaccine Production
- A good vaccine ensures that the immune system ‘remembers’ the pathogen. The immune system, however, can only form an immune memory against proteins, therefore a viral protein has to be selected to be used as a vaccine;
- To trigger an immune response and consequent immune memory, the pathogenic protein has to be presented to the immune cells as short peptides with the aid of HLA class I as well as HLA class II proteins where the presented pathogenic peptides have to fit into the clefts of the presenting HLA molecules.
- ∘
- The shapes of HLA clefts are therefore highly important;
- ∘
- ∘
- Receptors of the immune cells (T and B cell receptors) that need to recognize the presented peptides are also shaped by both genetic background and the environment, T, and B cell development and selection, as well as exposure to pathogens [26].
5.2. Challenges in Vaccine Production
- Active vaccination (pathogen proteins to trigger an immune response)
- ∘
- traditional vaccine production (attenuated and inactivated pathogens) is still amongst the actively used methods (e.g., influenza vaccines) to prevent infection or at least to reduce the seriousness of symptoms caused by infection of the wild type pathogen;
- ∘
- biotechnology revolutionized vaccine production decades ago, but the revolution was only recognized when SARS-CoV-2 started its deadly spread across the world in 2019.
- Passive vaccination (antibodies against pathogenic proteins)
- ∘
- passive vaccination aims to provide anti-pathogen antibodies for immune-compromised individuals traditionally; antibodies are purified from sera of people who have recovered from the disease;
- ∘
- Antigen-drift is the result of regularly occurring point mutations. Although antigen-drift causes relatively small-scale changes, these changes may be sufficient to ensure that the virus avoids being detected by cytotoxic memory T cells and antibodies produced by memory B cells formed during a previous infection or vaccination.
- Antigen-shift is the result of recombination events between the RNA genomes of different types of viruses infecting the same cell at the same time. Consequently, our immune system becomes vulnerable to the new pathogen, and as the results of these random recombination events are unpredictable, they further complicate vaccine development.
- (i)
- Targeting the host’s immune response
- (ii)
- Pharmacotherapy of viral infection
- Binding to the host’s cellular receptors;
- ‘Unpacking’ of the viral genome;
- Replication of the RNA/DNA of the virus;
- Synthesis of viral proteins;
- Release of virions.

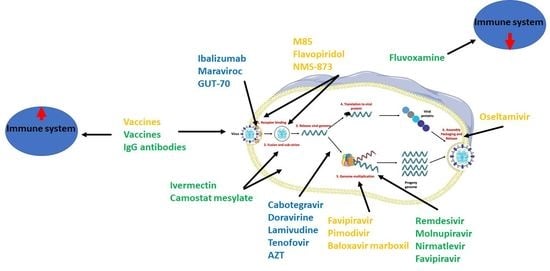
6. Anti-HIV, Anti-IV, and Anti-SARS-CoV-2 Agents
- Virus- and host-targeting antiviral agents
- (i)
- Direct-acting or virus-based antiviral agents that act directly on viruses to inhibit their replication.
- (ii)
- Host (indirect)-acting or host-based antiviral agents that act on host-based factors that are needed for viral replication.
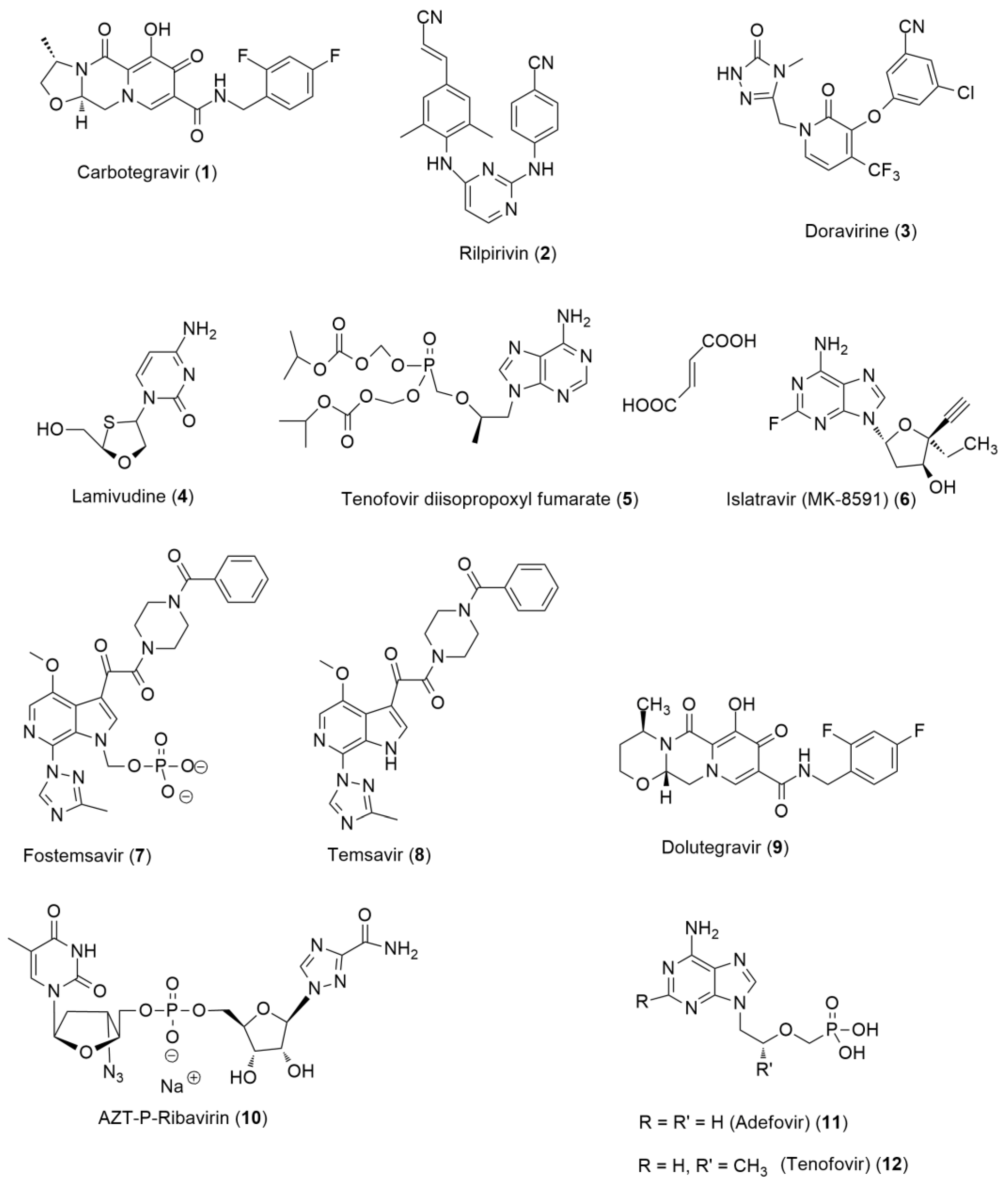
6.1. Anti-HIV Agents
- Non-nucleoside reverse transcriptase inhibitors (NNRTIs);
- Nucleoside reverse transcriptase inhibitors (NRTIs);
- Protease inhibitors (PIs);
- Fusion inhibitors;
- CCR5 antagonists;
- Integrase strand transfer inhibitors (INSTIs);
- Post-attachment inhibitors.
6.1.1. Direct-Acting Anti-HIV Agents
6.1.2. Host-Based Anti-HIV Agents
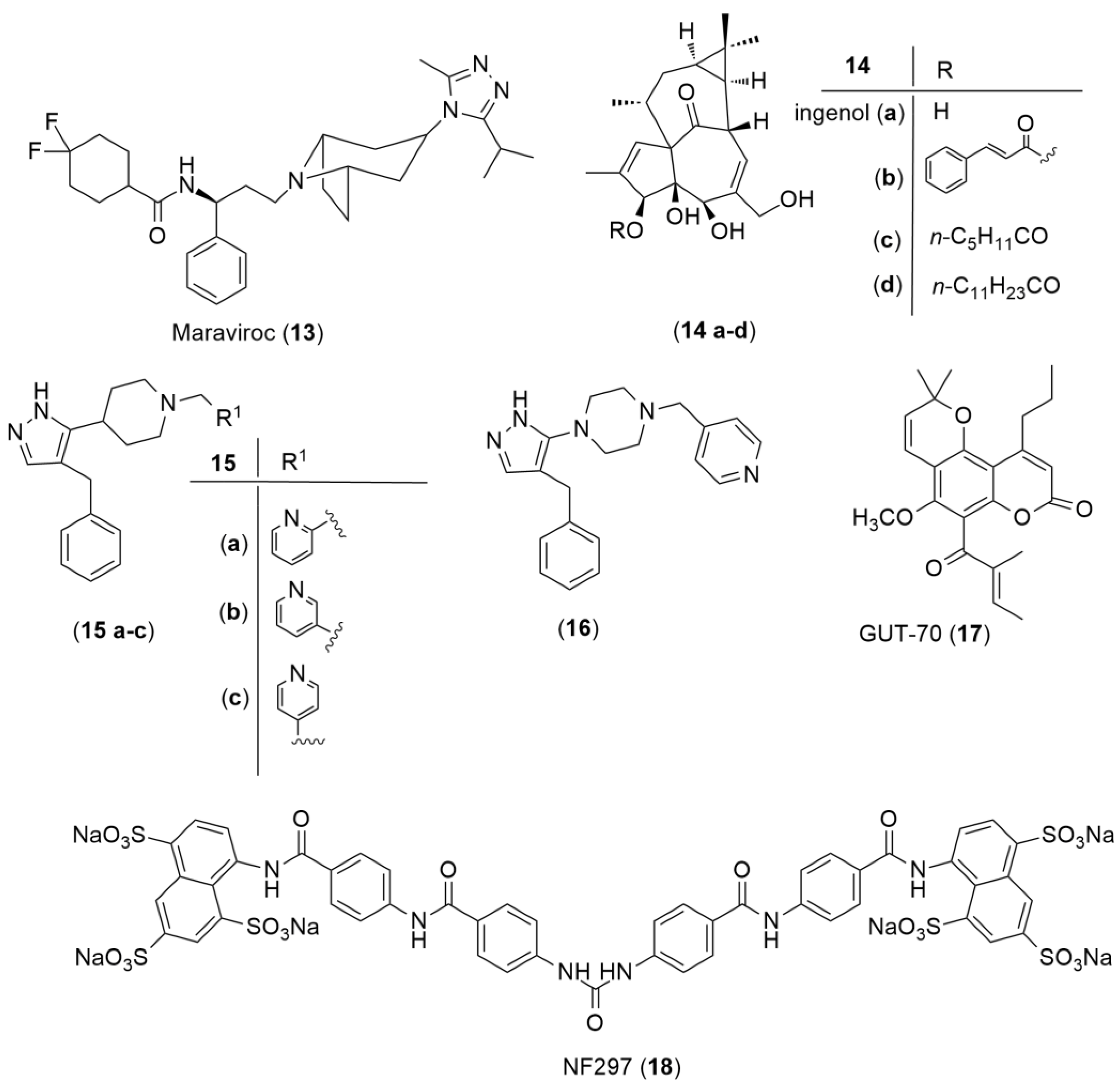
Dual CCR5 and CXCR4 Antagonists
Purine-Receptor Antagonists
Post-Attachment Inhibitors
New Host-Based Targets
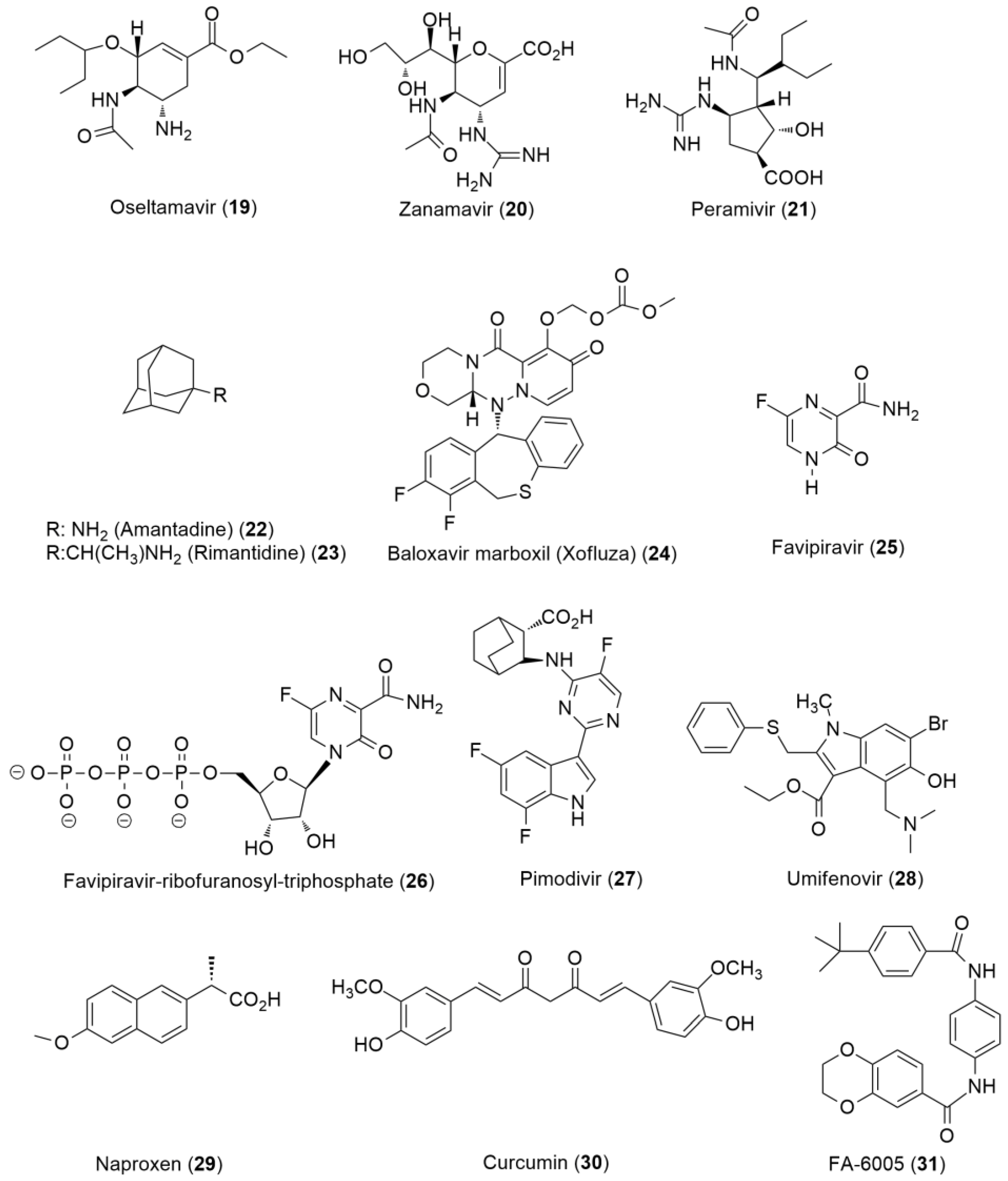
6.2. Anti-Influenza Agents
6.2.1. Direct-Acting Anti-Influenza Agents
6.2.2. Host-Based Anti-Influenza Agents
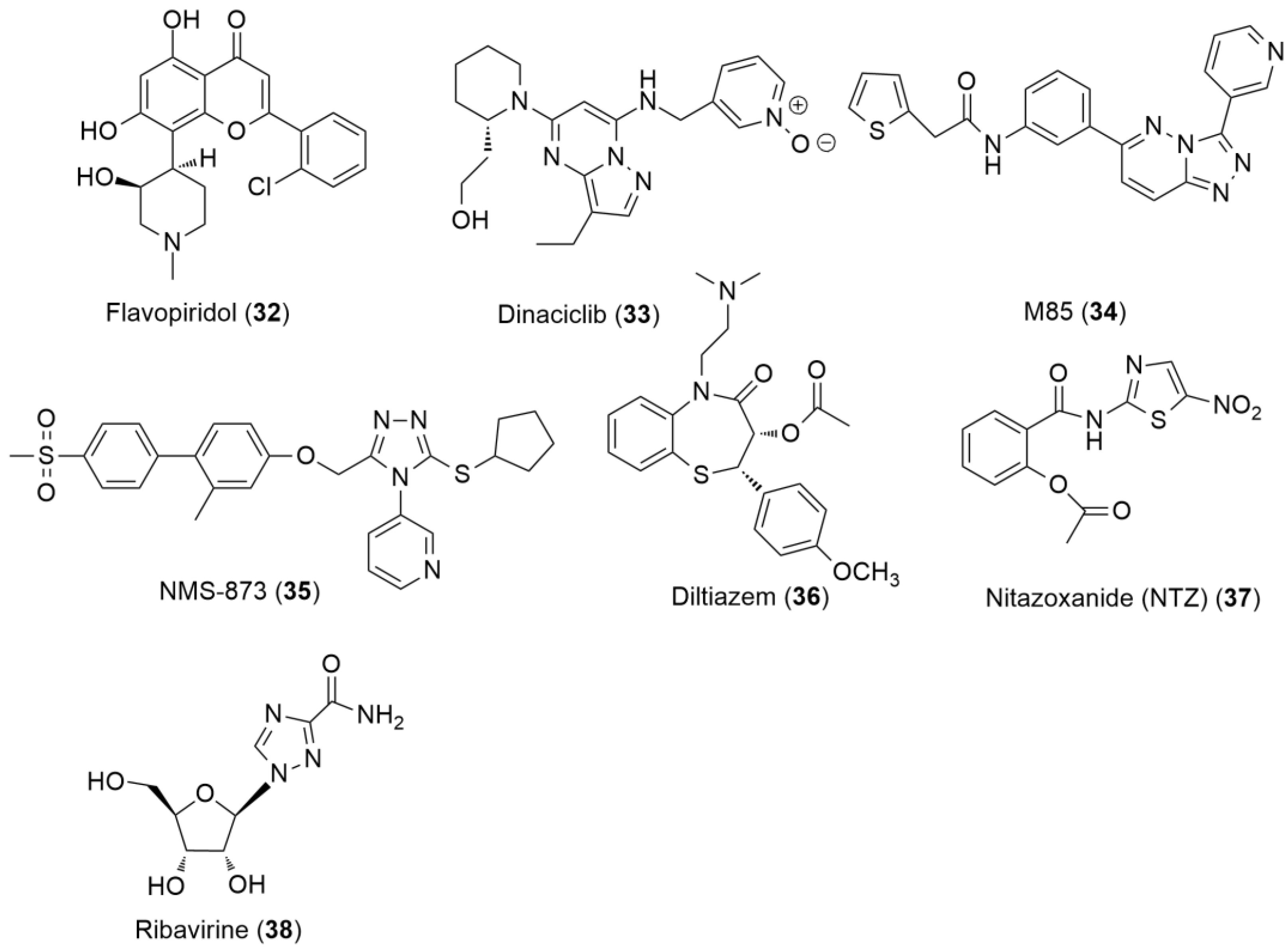
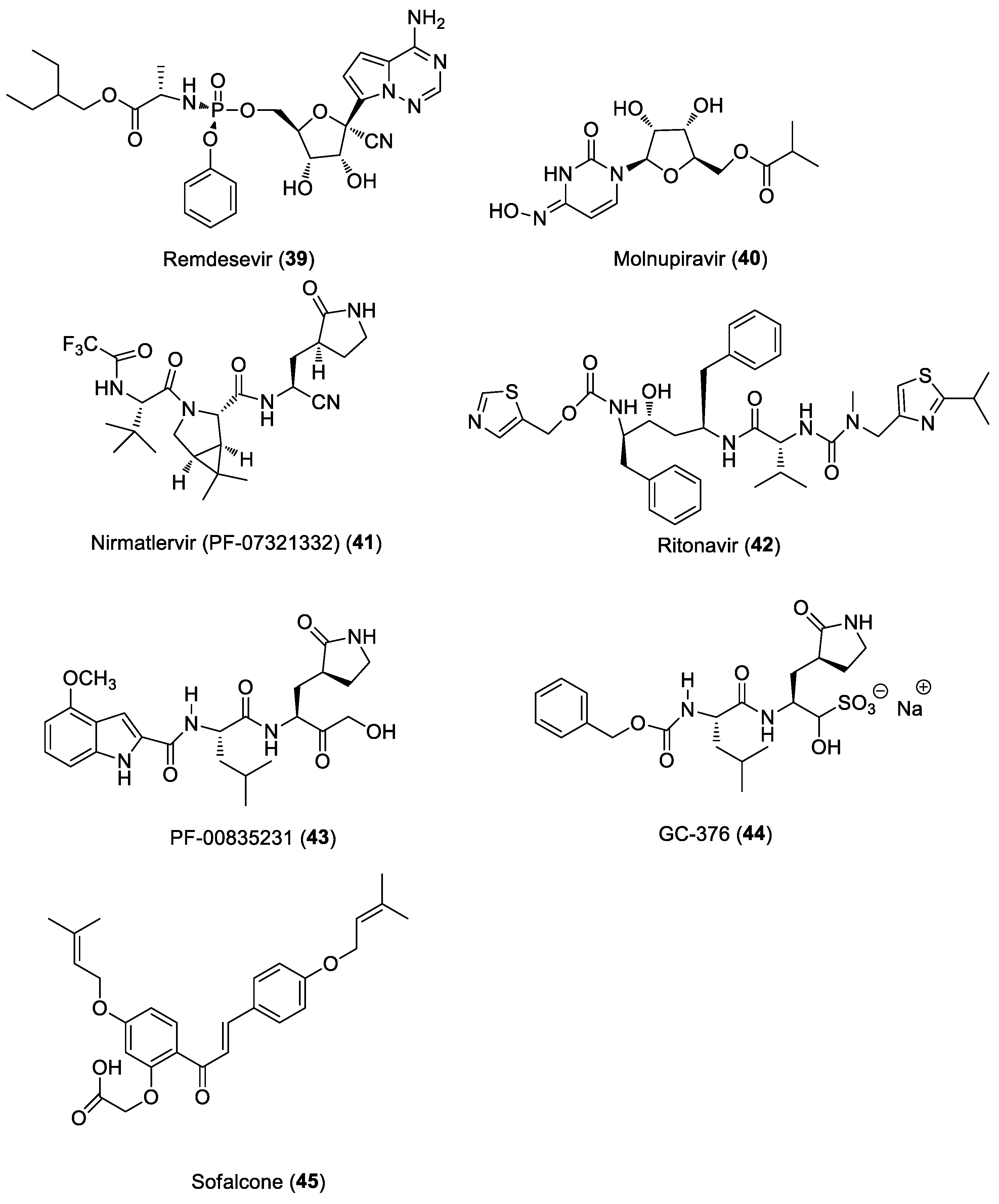
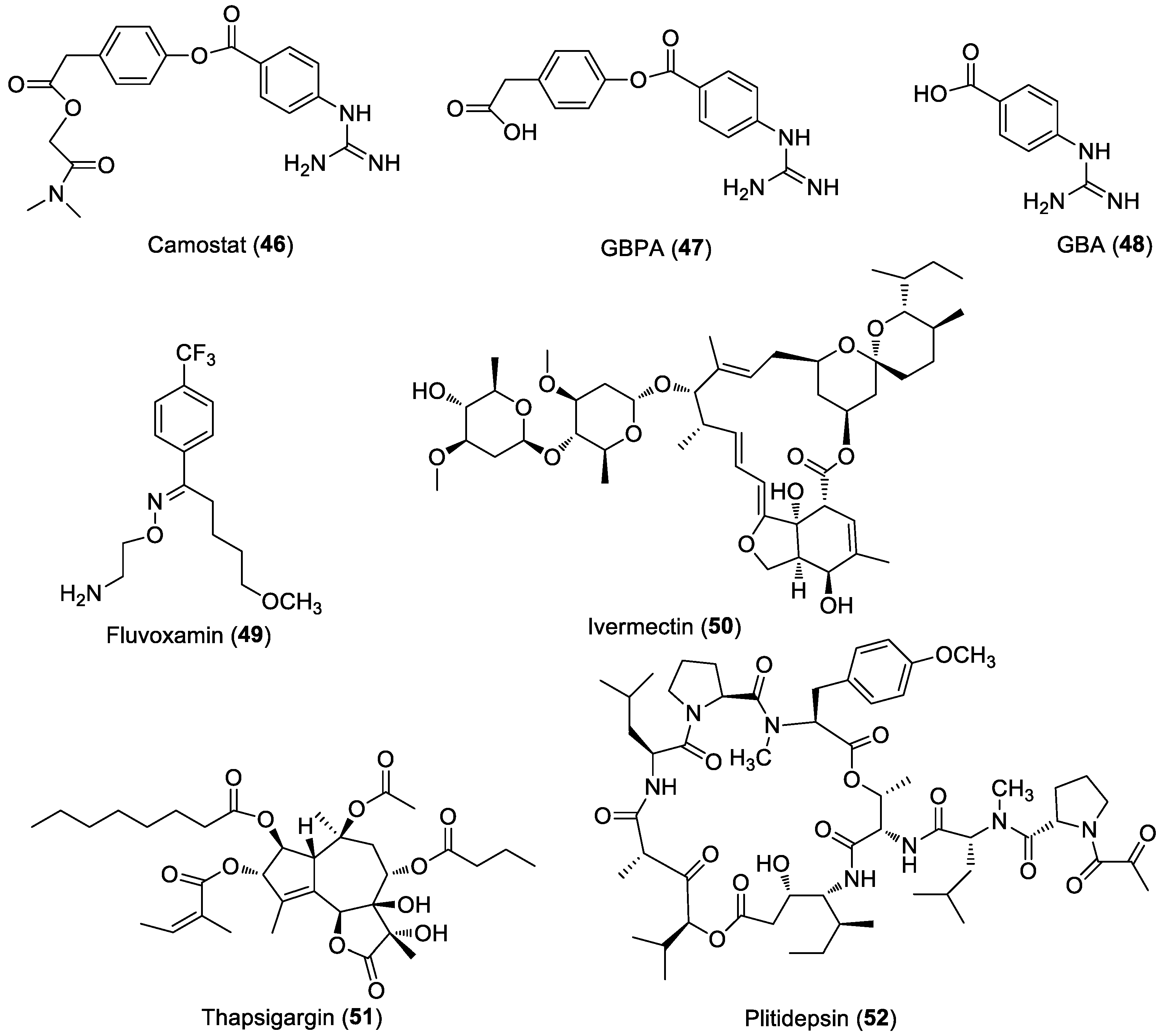
6.3. Anti-SARS-CoV-2 Agents
6.3.1. Direct-Acting Antivirals for COVID-19
6.3.2. Host-Based Antivirals for COVID-19
6.4. Multi-Targeting Approach
7. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woolhouse, M.E.J.; Brierley, L. Epidemiological Characteristics of Human-Infective RNA Viruses. Sci. Data 2018, 5, 180017. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Petkova, E.; Shaman, J. The 1918 Influenza Pandemic in New York City: Age-Specific Timing, Mortality, and Transmission Dynamics. Influenza Other Respir. Viruses 2014, 8, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Spreeuwenberg, P.; Kroneman, M.; Paget, J. Reassessing the Global Mortality Burden of the 1918 Influenza Pandemic. Am. J. Epidemiol. 2018, 187, 2561–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickol, M.E.; Kindrachuk, J. A Year of Terror and a Century of Reflection: Perspectives on the Great Influenza Pandemic of 1918–1919. BMC Infect. Dis. 2019, 19, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilbourne, E.D. Influenza Pandemics of the 20th Century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.-Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated Global Mortality Associated with the First 12 Months of 2009 Pandemic Influenza A H1N1 Virus Circulation: A Modelling Study. Lancet Infect. Dis. 2012, 12, 687–695. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Home Page. Available online: https://www.who.int/ (accessed on 11 December 2021).
- OECDiLibrary. Demography—Elderly Population. Available online: https://www.oecd-ilibrary.org/social-issues-migration-health/elderly-population/indicator/english_8d805ea1-en (accessed on 11 February 2022).
- Zheng, W.; Tao, Y.J. Structure and Assembly of the Influenza A Virus Ribonucleoprotein Complex. FEBS Lett. 2013, 587, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Mackenzie, J.S.; Fimmel, P.J. The Effect of ABO Blood Groups on the Incidence of Epidemic Influenza and on the Response to Live Attenuated and Detergent Split Influenza Virus Vaccines. Epidemiol. Infect. 1978, 80, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Falfán-Valencia, R.; Narayanankutty, A.; Reséndiz-Hernández, J.M.; Pérez-Rubio, G.; Ramírez-Venegas, A.; Nava-Quiroz, K.J.; Bautista-Félix, N.E.; Vargas-Alarcón, G.; Castillejos-López, M.D.J.; Hernández, A. An Increased Frequency in HLA Class I Alleles and Haplotypes Suggests Genetic Susceptibility to Influenza A (H1N1) 2009 Pandemic: A Case-Control Study. J. Immunol. Res. 2018, 2018, 3174868. [Google Scholar] [CrossRef]
- Ochoa, E.E.; Huda, R.; Scheibel, S.F.; Nichols, J.E.; Mock, D.J.; El-Daher, N.; Domurat, F.M.; Roberts, N.J. HLA-Associated Protection of Lymphocytes during Influenza Virus Infection. Virol. J. 2020, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00510–e00520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augusto, D.G.; Yusufali, T.; Peyser, N.D.; Butcher, X.; Marcus, G.M.; Olgin, J.E.; Pletcher, M.J.; Maiers, M.; Hollenbach, J.A. HLA-B*15:01 Is Associated with Asymptomatic SARS-CoV-2 Infection. medRxiv 2021. preprint. [Google Scholar] [CrossRef]
- McLaren, P.J.; Fellay, J. HIV-1 and Human Genetic Variation. Nat. Rev. Genet. 2021, 22, 645–657. [Google Scholar] [CrossRef]
- McLaren, P.J.; Coulonges, C.; Ripke, S.; van den Berg, L.; Buchbinder, S.; Carrington, M.; Cossarizza, A.; Dalmau, J.; Deeks, S.G.; Delaneau, O.; et al. Association Study of Common Genetic Variants and HIV-1 Acquisition in 6300 Infected Cases and 7200 Controls. PLoS Pathog. 2013, 9, e1003515. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.; McLaren, P.J.; Dorrell, L.; Shianna, K.V.; Stemke, A.; Pelak, K.; Moore, S.; Oldenburg, J.; Alvarez-Roman, M.T.; Angelillo-Scherrer, A.; et al. A Genome-Wide Association Study of Resistance to HIV Infection in Highly Exposed Uninfected Individuals with Hemophilia A. Hum. Mol. Genet. 2013, 22, 1903–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackelprang, R.D.; Bamshad, M.J.; Chong, J.X.; Hou, X.; Buckingham, K.J.; Shively, K.; deBruyn, G.; Mugo, N.R.; Mullins, J.I.; McElrath, M.J.; et al. Whole Genome Sequencing of Extreme Phenotypes Identifies Variants in CD101 and UBE2V1 Associated with Increased Risk of Sexually Acquired HIV-1. PLoS Pathog. 2017, 13, e1006703. [Google Scholar] [CrossRef] [Green Version]
- Bouloc, A.; Bagot, M.; Delaire, S.; Bensussan, A.; Boumsell, L. Triggering CD101 Molecule on Human Cutaneous Dendritic Cells Inhibits T Cell Proliferation via IL-10 Production. Eur. J. Immunol. 2000, 30, 3132–3139. [Google Scholar] [CrossRef]
- Xia, Z.-P.; Sun, L.; Chen, X.; Pineda, G.; Jiang, X.; Adhikari, A.; Zeng, W.; Chen, Z.J. Direct Activation of Protein Kinases by Unanchored Polyubiquitin Chains. Nature 2009, 461, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 Is an Innate Immune Sensor for the Retrovirus Capsid Lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Sturniolo, T.; Bono, E.; Ding, J.; Raddrizzani, L.; Tuereci, O.; Sahin, U.; Braxenthaler, M.; Gallazzi, F.; Protti, M.P.; Sinigaglia, F.; et al. Generation of Tissue-Specific and Promiscuous HLA Ligand Databases Using DNA Microarrays and Virtual HLA Class II Matrices. Nat. Biotechnol. 1999, 17, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Gras, S.; Mobbs, J.I.; Vivian, J.P.; Cortes, A.; Barber, T.; Kuttikkatte, S.B.; Jensen, L.T.; Attfield, K.E.; Dendrou, C.A.; et al. Structural and Regulatory Diversity Shape HLA-C Protein Expression Levels. Nat. Commun. 2017, 8, 15924. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Vargas, E.; Barker, A.P.; Zhou, Z.; He, X.; Jensen, P.E. HLA-DM Catalytically Enhances Peptide Dissociation by Sensing Peptide–MHC Class II Interactions throughout the Peptide-Binding Cleft. J. Biol. Chem. 2020, 295, 2959–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Nourishirazi, E.; Guinet, E.; Nouri-Shirazi, M. The Genetic Background Influences the Cellular and Humoral Immune Responses to Vaccines. Clin. Exp. Immunol. 2016, 186, 190–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Goronzy, J.J. Aging of the Immune System. Mechanisms and Therapeutic Targets. Ann. ATS 2016, 13, S422–S428. [Google Scholar] [CrossRef]
- Warren, L.A.; Rossi, D.J. Stem Cells and Aging in the Hematopoietic System. Mech. Ageing Dev. 2009, 130, 46–53. [Google Scholar] [CrossRef]
- Thomas, R.; Su, D.-M. Age-Related Thymic Atrophy: Mechanisms and Outcomes. In Thymus; Rezaei, N., Ed.; IntechOpen: London, UK, 2020; ISBN 978-1-78985-133-5. [Google Scholar]
- Kvell, K.; Pongracz, J.E. Central Immune Senescence, Reversal Potentials. In Senescence; Nagata, T., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0144-4. [Google Scholar]
- Farheen, S.; Agrawal, S.; Zubair, S.; Agrawal, A.; Jamal, F.; Altaf, I.; Kashif Anwar, A.; Umair, S.M.; Owais, M. Patho-Physiology of Aging and Immune-Senescence: Possible Correlates with Comorbidity and Mortality in Middle-Aged and Old COVID-19 Patients. Front. Aging 2021, 2, 748591. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage Display-Derived Human Antibodies in Clinical Development and Therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [Green Version]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef]
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef]
- Johnson, A.R. In Vitro and In Vivo Assays. In Drug Discovery; Li, J.J., Corey, E.J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 67–98. ISBN 978-1-118-35448-3. [Google Scholar]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring MRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Flemming, A. MRNA Vaccine Shows Promise in Autoimmunity. Nat. Rev. Immunol. 2021, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A Guide to Immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhao, G.; Liu, C.; Fan, W.; Zhou, X.; Zeng, L.; Guo, Y.; Kou, Z.; Yu, H.; Li, J.; et al. Treatment With Anti-C5a Antibody Improves the Outcome of H7N9 Virus Infection in African Green Monkeys. Clin. Infect. Dis. 2015, 60, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Kendall, L.; Boyd, T.; Sillau, S.; Bosco-Lauth, A.; Markham, N.; Fong, D.; Clarke, P.; Tyler, K.; Potter, H. GM-CSF Promotes Immune Response and Survival in a Mouse Model of COVID-19. Res. Sq. 2022. preprint. [Google Scholar] [CrossRef]
- Ataya, A.; Knight, V.; Carey, B.C.; Lee, E.; Tarling, E.J.; Wang, T. The Role of GM-CSF Autoantibodies in Infection and Autoimmune Pulmonary Alveolar Proteinosis: A Concise Review. Front. Immunol. 2021, 12, 752856. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Chapter 1. Overview of Antiviral Drug Discovery and Development: Viral Versus Host Targets. In Drug Discovery; Muñoz-Fontela, C., Delgado, R., Eds.; Royal Society of Chemistry: Cambridge, UK, 2021; pp. 1–27. ISBN 978-1-78801-564-6. [Google Scholar]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-Directed Antiviral Therapy. Clin. Microbiol. Rev. 2020, 33, e00168-19. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Chahroudi, A.; Bosinger, S.E.; Vanderford, T.H.; Paiardini, M.; Silvestri, G. Natural SIV Hosts: Showing AIDS the Door. Science 2012, 335, 1188–1193. [Google Scholar] [CrossRef] [Green Version]
- Kolata, G. FDA Approves AZT. Science 1987, 235, 1570. [Google Scholar] [CrossRef]
- Meanwell, N.A.; D’Andrea, S.V.; Cianci, C.W.; Dicker, I.B.; Yeung, K.-S.; Belema, M.; Krystal, M. Antiviral Drug Discovery. In Drug Discovery; Li, J.J., Corey, E.J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 439–515. ISBN 978-1-118-35448-3. [Google Scholar]
- Tseng, A.; Seet, J.; Phillips, E.J. The Evolution of Three Decades of Antiretroviral Therapy: Challenges, Triumphs and the Promise of the Future: Three Decades of Antiretroviral Therapy. Br. J. Clin. Pharmacol. 2015, 79, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J. History of Drug Discovery. In Drug Discovery; Li, J.J., Corey, E.J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 1–42. ISBN 978-1-118-35448-3. [Google Scholar]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for Viral Persistence in the Presence of Antiviral Immune Responses and Antiretroviral Therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, H.; Wang, Q.; Hua, C.; Zhang, H.; Li, W.; Jiang, S.; Lu, L. Advancements in Developing Strategies for Sterilizing and Functional HIV Cures. Biomed Res. Int. 2017, 2017, 6096134. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Moreno, J.; Trapero-Bertran, M. Economic Impact of HIV in the Highly Active Antiretroviral Therapy Era—Reflections Looking Forward. AIDS Rev. 2019, 20, 428. [Google Scholar] [CrossRef]
- U.S Food & Drug Administration FDA Approves First Injectable Treatment for HIV Pre-Exposure Prevention. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-injectable-treatment-hiv-pre-exposure-prevention (accessed on 17 February 2022).
- Kozal, M.; Aberg, J.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.-M.; Grinsztejn, B.; Diaz, R.; Castagna, A.; Kumar, P.; et al. Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2020, 382, 1232–1243. [Google Scholar] [CrossRef]
- Lataillade, M.; Lalezari, J.P.; Kozal, M.; Aberg, J.A.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.-M.; Moreno, S.; Grinsztejn, B.; et al. Safety and Efficacy of the HIV-1 Attachment Inhibitor Prodrug Fostemsavir in Heavily Treatment-Experienced Individuals: Week 96 Results of the Phase 3 BRIGHTE Study. Lancet HIV 2020, 7, e740–e751. [Google Scholar] [CrossRef]
- Ackerman, P.; Thompson, M.; Molina, J.-M.; Aberg, J.; Cassetti, I.; Kozal, M.; Castagna, A.; Martins, M.; Ramgopal, M.; Sprinz, E.; et al. Long-Term Efficacy and Safety of Fostemsavir among Subgroups of Heavily Treatment-Experienced Adults with HIV-1. AIDS 2021, 35, 1061–1072. [Google Scholar] [CrossRef]
- Turkova, A.; White, E.; Mujuru, H.A.; Kekitiinwa, A.R.; Kityo, C.M.; Violari, A.; Lugemwa, A.; Cressey, T.R.; Musoke, P.; Variava, E.; et al. Dolutegravir as First- or Second-Line Treatment for HIV-1 Infection in Children. N. Engl. J. Med. 2021, 385, 2531–2543. [Google Scholar] [CrossRef]
- Roy, U.; Drozd, V.; Durygin, A.; Rodriguez, J.; Barber, P.; Atluri, V.; Liu, X.; Voss, T.G.; Saxena, S.; Nair, M. Characterization of Nanodiamond-Based Anti-HIV Drug Delivery to the Brain. Sci. Rep. 2018, 8, 1603. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Kraft, J.C.; Yu, D.; Ho, R.J.Y. Recent Developments of Nanotherapeutics for Targeted and Long-Acting, Combination HIV Chemotherapy. Eur. J. Pharm. Biopharm. 2019, 138, 75–91. [Google Scholar] [CrossRef]
- Cunha, R.F.; Simões, S.; Carvalheiro, M.; Pereira, J.M.A.; Costa, Q.; Ascenso, A. Novel Antiretroviral Therapeutic Strategies for HIV. Molecules 2021, 26, 5305. [Google Scholar] [CrossRef] [PubMed]
- Muheem, A.; Baboota, S.; Ali, J. An In-Depth Analysis of Novel Combinatorial Drug Therapy via Nanocarriers against HIV/AIDS Infection and Their Clinical Perspectives: A Systematic Review. Expert Opin. Drug Deliv. 2021, 18, 1025–1046. [Google Scholar] [CrossRef] [PubMed]
- Mátyus, P. Több Támadáspontú Gyógyszerek: Múlt, Jelen És Jövő. Orv. Hetil. 2020, 161, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- De Castro, S.; Camarasa, M.-J. Polypharmacology in HIV Inhibition: Can a Drug with Simultaneous Action against Two Relevant Targets Be an Alternative to Combination Therapy? Eur. J. Med. Chem. 2018, 150, 206–227. [Google Scholar] [CrossRef]
- Stelitano, G.; Sammartino, J.C.; Chiarelli, L.R. Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules 2020, 25, 1239. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, S.; Alvarez, R.; San-Felix, A.; Jimeno, M.L.; De Clercq, E.; Balzarini, J.; Camarasa, M.J. Synthesis and Anti-HIV Activity of [AZT]-[TSAO-T] and [AZT]-[HEPT] Dimers as Potential Multifunctional Inhibitors of HIV-1 Reverse Transcriptase. J. Med. Chem. 1995, 38, 1641–1649. [Google Scholar] [CrossRef]
- Singal, A.K.; Anand, B.S. Management of Hepatitis C Virus Infection in HIV/HCV Co-Infected Patients: Clinical Review. World J. Gastroenterol. 2009, 15, 3713. [Google Scholar] [CrossRef]
- Meleddu, R.; Corona, A.; Distinto, S.; Cottiglia, F.; Deplano, S.; Sequeira, L.; Secci, D.; Onali, A.; Sanna, E.; Esposito, F.; et al. Exploring New Scaffolds for the Dual Inhibition of HIV-1 RT Polymerase and Ribonuclease Associated Functions. Molecules 2021, 26, 3821. [Google Scholar] [CrossRef]
- Nickoloff-Bybel, E.A.; Festa, L.; Meucci, O.; Gaskill, P.J. Co-Receptor Signaling in the Pathogenesis of NeuroHIV. Retrovirology 2021, 18, 24. [Google Scholar] [CrossRef]
- Abreu, C.M.; Price, S.L.; Shirk, E.N.; Cunha, R.D.; Pianowski, L.F.; Clements, J.E.; Tanuri, A.; Gama, L. Dual Role of Novel Ingenol Derivatives from Euphorbia Tirucalli in HIV Replication: Inhibition of De Novo Infection and Activation of Viral LTR. PLoS ONE 2014, 9, e97257. [Google Scholar] [CrossRef] [Green Version]
- Grande, F.; Occhiuzzi, M.; Rizzuti, B.; Ioele, G.; De Luca, M.; Tucci, P.; Svicher, V.; Aquaro, S.; Garofalo, A. CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy. Molecules 2019, 24, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.U.; Saadabadi, A.; Vanmeert, M.; Salo-Ahen, O.M.H.; Abdullah, I.; Claes, S.; De Jonghe, S.; Schols, D.; Ahmad, S.; Froeyen, M. Discovery of HIV Entry Inhibitors via a Hybrid CXCR4 and CCR5 Receptor Pharmacophore-based Virtual Screening Approach. Eur. J. Pharm. Sci. 2020, 155, 105537. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.; Du, Y.; Giroud, C.; Kim, J.H.; Qui, M.; Fu, H.; Melikyan, G.B. High-Throughput HIV–Cell Fusion Assay for Discovery of Virus Entry Inhibitors. Assay Drug Dev. Technol. 2015, 13, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Hattori, S.; Kariya, R.; Komizu, Y.; Kudo, E.; Goto, H.; Taura, M.; Ueoka, R.; Kimura, S.; Okada, S. Inhibition of HIV-1 Entry by the Tricyclic Coumarin GUT-70 through the Modification of Membrane Fluidity. Biochem. Biophys. Res. Commun. 2015, 457, 288–294. [Google Scholar] [CrossRef]
- Overeem, N.J.; Vries, E.; Huskens, J. A Dynamic, Supramolecular View on the Multivalent Interaction between Influenza Virus and Host Cell. Small 2021, 17, 2007214. [Google Scholar] [CrossRef]
- Drugs.com. Leronlimab FDA Approval Status. Available online: https://www.drugs.com/history/leronlimab.html (accessed on 17 January 2022).
- Ivanov, S.; Lagunin, A.; Filimonov, D.; Tarasova, O. Network-Based Analysis of OMICs Data to Understand the HIV–Host Interaction. Front. Microbiol. 2020, 11, 1314. [Google Scholar] [CrossRef]
- Durães-Carvalho, R.; Salemi, M. In-Depth Phylodynamics, Evolutionary Analysis and in Silico Predictions of Universal Epitopes of Influenza A Subtypes and Influenza B Viruses. Mol. Phylogenet. Evol. 2018, 121, 174–182. [Google Scholar] [CrossRef]
- Hurt, A.C. Antiviral Therapy for the Next Influenza Pandemic. Trop. Med. Int. Health 2019, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Parra-Rojas, C.; Nguyen, V.; Hernandez-Mejia, G.; Hernandez-Vargas, E. Neuraminidase Inhibitors in Influenza Treatment and Prevention–Is It Time to Call It a Day? Viruses 2018, 10, 454. [Google Scholar] [CrossRef] [Green Version]
- Groaz, E.; De Clercq, E.; Herdewijn, P. Anno 2021: Which Antivirals for the Coming Decade? In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2021; Volume 57, pp. 49–107. ISBN 978-0-323-91511-3. [Google Scholar]
- Yin, H.; Jiang, N.; Shi, W.; Chi, X.; Liu, S.; Chen, J.-L.; Wang, S. Development and Effects of Influenza Antiviral Drugs. Molecules 2021, 26, 810. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Jones, J.C.; Wong, S.-S.; Zanin, M. Antivirals Targeting the Surface Glycoproteins of Influenza Virus: Mechanisms of Action and Resistance. Viruses 2021, 13, 624. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Chibale, K.; Goss, R.J.M.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral Drug Discovery: Preparing for the next Pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Brooke, C.B. What’s New with Flu? An Overview. Viruses 2019, 11, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotey; Lukosaityte; Quaye; Ampofo; Awandare; Iqbal Current and Novel Approaches in Influenza Management. Vaccines 2019, 7, 53. [CrossRef] [PubMed] [Green Version]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In Vitro Characterization of Baloxavir Acid, a First-in-Class Cap-Dependent Endonuclease Inhibitor of the Influenza Virus Polymerase PA Subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Ng, K.E. Xofluza (Baloxavir Marboxil) for the Treatment Of Acute Uncomplicated Influenza. Pharm. Ther. 2019, 44, 9–11. [Google Scholar]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Dufrasne, F. Baloxavir Marboxil: An Original New Drug against Influenza. Pharmaceuticals 2021, 15, 28. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Lai, C.-C.; Wang, Y.-H.; Chen, C.-H.; Wang, C.-Y. Clinical Efficacy and Safety of Baloxavir Marboxil in the Treatment of Influenza: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Microbiol. Immunol. Infect. 2021, 54, 865–875. [Google Scholar] [CrossRef]
- Liu, J.-W.; Lin, S.-H.; Wang, L.-C.; Chiu, H.-Y.; Lee, J.-A. Comparison of Antiviral Agents for Seasonal Influenza Outcomes in Healthy Adults and Children: A Systematic Review and Network Meta-Analysis. JAMA Netw. Open 2021, 4, e2119151. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Wang, D. Advanced Researches on the Inhibition of Influenza Virus by Favipiravir and Baloxavir. Biosaf. Health 2020, 2, 64–70. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a Broad Spectrum Inhibitor of Viral RNA Polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, O.P.; Chernyshova, A.I. Favipiravir: The Hidden Threat of Mutagenic Action. J. Microbiol. Epidemiol. Immunobiol. 2021, 98, 213–220. [Google Scholar] [CrossRef]
- Naesens, L.; Guddat, L.W.; Keough, D.T.; van Kuilenburg, A.B.P.; Meijer, J.; Vande Voorde, J.; Balzarini, J. Role of Human Hypoxanthine Guanine Phosphoribosyltransferase in Activation of the Antiviral Agent T-705 (Favipiravir). Mol. Pharmacol. 2013, 84, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newdrugapprovals. Tag Archives: PIMODIVIR. Available online: https://newdrugapprovals.org/tag/pimodivir/ (accessed on 11 February 2022).
- Pizzorno, A.; Padey, B.; Terrier, O.; Rosa-Calatrava, M. Drug Repurposing Approaches for the Treatment of Influenza Viral Infection: Reviving Old Drugs to Fight Against a Long-Lived Enemy. Front. Immunol. 2019, 10, 531. [Google Scholar] [CrossRef]
- Jennings, M.R.; Parks, R.J. Curcumin as an Antiviral Agent. Viruses 2020, 12, 1242. [Google Scholar] [CrossRef]
- Pizzorno, A.; Terrier, O.; Nicolas de Lamballerie, C.; Julien, T.; Padey, B.; Traversier, A.; Roche, M.; Hamelin, M.-E.; Rhéaume, C.; Croze, S.; et al. Repurposing of Drugs as Novel Influenza Inhibitors From Clinical Gene Expression Infection Signatures. Front. Immunol. 2019, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Terrier, O.; Dilly, S.; Pizzorno, A.; Chalupska, D.; Humpolickova, J.; Bouřa, E.; Berenbaum, F.; Quideau, S.; Lina, B.; Fève, B.; et al. Antiviral Properties of the NSAID Drug Naproxen Targeting the Nucleoprotein of SARS-CoV-2 Coronavirus. Molecules 2021, 26, 2593. [Google Scholar] [CrossRef]
- Yang, F.; Pang, B.; Lai, K.K.; Cheung, N.N.; Dai, J.; Zhang, W.; Zhang, J.; Chan, K.-H.; Chen, H.; Sze, K.-H.; et al. Discovery of a Novel Specific Inhibitor Targeting Influenza A Virus Nucleoprotein with Pleiotropic Inhibitory Effects on Various Steps of the Viral Life Cycle. J. Virol. 2021, 95, e01432-20. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, Y.; Hau, R.; Musharrafieh, R.; Ma, C.; Zhou, X.; Chen, Y.; Wang, J. Identification of NMS-873, an Allosteric and Specific P97 Inhibitor, as a Broad Antiviral against Both Influenza A and B Viruses. Eur. J. Pharm. Sci. 2019, 133, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Perwitasari, O.; Yan, X.; O’Donnell, J.; Johnson, S.; Tripp, R.A. Repurposing Kinase Inhibitors as Antiviral Agents to Control Influenza A Virus Replication. ASSAY Drug Dev. Technol. 2015, 13, 638–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schor, S.; Einav, S. Repurposing of Kinase Inhibitors as Broad-Spectrum Antiviral Drugs. DNA Cell Biol. 2018, 37, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Meineke, R.; Rimmelzwaan, G.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- O’Hanlon, R.; Leyva-Grado, V.H.; Sourisseau, M.; Evans, M.J.; Shaw, M.L. An Influenza Virus Entry Inhibitor Targets Class II PI3 Kinase and Synergizes with Oseltamivir. ACS Infect. Dis. 2019, 5, 1779–1793. [Google Scholar] [CrossRef]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.-K.; Wu, Z.Y.; et al. The P97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer. Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Trial to Evaluate the Efficacy and Safety of Nitazoxanide in the Treatment of Acute Uncomplicated Influenza. Available online: https://clinicaltrials.gov/ct2/show/NCT02612922?term=NCT02612922&draw=2&rank=1 (accessed on 24 December 2021).
- Pires de Mello, C.P.; Drusano, G.L.; Adams, J.R.; Shudt, M.; Kulawy, R.; Brown, A.N. Oseltamivir-Zanamivir Combination Therapy Suppresses Drug-Resistant H1N1 Influenza A Viruses in the Hollow Fiber Infection Model (HFIM) System. Eur. J. Pharm. Sci. 2018, 111, 443–449. [Google Scholar] [CrossRef]
- Beigel, J.H.; Bao, Y.; Beeler, J.; Manosuthi, W.; Slandzicki, A.; Dar, S.M.; Panuto, J.; Beasley, R.L.; Perez-Patrigeon, S.; Suwanpimolkul, G.; et al. Oseltamivir, Amantadine, and Ribavirin Combination Antiviral Therapy versus Oseltamivir Monotherapy for the Treatment of Influenza: A Multicentre, Double-Blind, Randomised Phase 2 Trial. Lancet Infect. Dis. 2017, 17, 1255–1265. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, W.; Jia, H.; Zheng, L.; Xing, J.; Liu, A.; Du, G. Discovery of Multitarget-Directed Ligands Against Influenza A Virus From Compound Yizhihao Through a Predictive System for Compound-Protein Interactions. Front. Cell. Infect. Microbiol. 2020, 10, 16. [Google Scholar] [CrossRef]
- Wieczorek, K.; Szutkowska, B.; Kierzek, E. Anti-Influenza Strategies Based on Nanoparticle Applications. Pathogens 2020, 9, 1020. [Google Scholar] [CrossRef]
- Chan, Y.; Ng, S.W.; Mehta, M.; Anand, K.; Kumar Singh, S.; Gupta, G.; Chellappan, D.K.; Dua, K. Advanced Drug Delivery Systems Can Assist in Managing Influenza Virus Infection: A Hypothesis. Med. Hypotheses 2020, 144, 110298. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Shinn, P.; Itkin, Z.; Eastman, R.T.; Bostwick, R.; Rasmussen, L.; Huang, R.; Shen, M.; Hu, X.; Wilson, K.M.; et al. Drug Repurposing Screen for Compounds Inhibiting the Cytopathic Effect of SARS-CoV-2. Front. Pharmacol. 2021, 11, 592737. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Agoramoorthy, G.; Lee, S.-S. The Drug Repurposing for COVID-19 Clinical Trials Provide Very Effective Therapeutic Combinations: Lessons Learned From Major Clinical Studies. Front. Pharmacol. 2021, 12, 704205. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The Coronavirus Is Mutating—Does It Matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Tramontano, E.; Summa, V. A Focus on Severe Acute Respiratory Syndrome (SARS) Coronavirus (SARS-CoVs) 1 and 2. In New Drug Development for Known and Emerging Viruses; Rübsamen-Schaeff, H., Buschmann, H., Eds.; Wiley: Weinheim, Germany, 2022; ISBN 978-3-527-34337-9. [Google Scholar]
- Jena, N.R. Drug Targets, Mechanisms of Drug Action, and Therapeutics against SARS-CoV-2. Chem. Phys. 2021, 2, 100011. [Google Scholar] [CrossRef]
- National Institutes of Health The COVID-19 Treatment Guidelines Panel’s Statement on Therapies for High-Risk, Nonhospitalized Patients with Mild to Moderate COVID-19. Available online: https://www.https://www.covid19treatmentguidelines.nih.gov/therapies/statement-on-therapies-for-high-risk-nonhospitalized-patients/-patients (accessed on 11 February 2022).
- Noor, R. A Review on the Effectivity of the Current COVID-19 Drugs and Vaccines: Are They Really Working Against the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants? Curr. Clin. Microbiol. Rep. 2021, 8, 186–193. [Google Scholar] [CrossRef]
- Okoli, G.N.; Rabbani, R.; Al-Juboori, A.; Copstein, L.; Askin, N.; Abou-Setta, A.M. Antiviral Drugs for Coronavirus Disease 2019 (COVID-19): A Systematic Review with Network Meta-Analysis. Expert Rev. Anti Infect. Ther. 2022, 20, 267–278. [Google Scholar] [CrossRef]
- Azerang, P.; Yazdani, M.; RayatSanati, K.; Tahghighi, A. Newly Identified COVID-19 Drug Candidates Based on Computational Strategies. J. Comput. Biophys. Chem. 2022, 21, 123–137. [Google Scholar] [CrossRef]
- Bobrowski, T.; Chen, L.; Eastman, R.T.; Itkin, Z.; Shinn, P.; Chen, C.Z.; Guo, H.; Zheng, W.; Michael, S.; Simeonov, A.; et al. Synergistic and Antagonistic Drug Combinations against SARS-CoV-2. Mol. Ther. 2021, 29, 873–885. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64, e00819–e00820. [Google Scholar] [CrossRef]
- Yadav, D.K.; Singh, D.D.; Han, I.; Kumar, Y.; Choi, E.-H. Current Potential Therapeutic Approaches against SARS-CoV-2: A Review. Biomedicines 2021, 9, 1620. [Google Scholar] [CrossRef] [PubMed]
- Bolarin, J.A.; Oluwatoyosi, M.A.; Orege, J.I.; Ayeni, E.A.; Ibrahim, Y.A.; Adeyemi, S.B.; Tiamiyu, B.B.; Gbadegesin, L.A.; Akinyemi, T.O.; Odoh, C.K.; et al. Therapeutic Drugs for SARS-CoV-2 Treatment: Current State and Perspective. Int. Immunopharmacol. 2021, 90, 107228. [Google Scholar] [CrossRef] [PubMed]
- Malani, M.; Salunke, P.; Kulkarni, S.; Jain, G.K.; Sheikh, A.; Kesharwani, P.; Nirmal, J. Repurposing Pharmaceutical Excipients as an Antiviral Agent against SARS-CoV-2. J. Biomater. Sci. Polym. Ed. 2022, 33, 110–136. [Google Scholar] [CrossRef] [PubMed]
- Mule, S.; Singh, A.; Greish, K.; Sahebkar, A.; Kesharwani, P.; Shukla, R. Drug Repurposing Strategies and Key Challenges for COVID-19 Management. J. Drug Target. 2022, 30, 413–429. [Google Scholar] [CrossRef]
- Ledford, H. Hundreds of COVID Trials Could Provide a Deluge of New Drugs. Nature 2022, 603, 25–27. [Google Scholar] [CrossRef]
- Alencar, W.L.M.; da Silva Arouche, T.; Neto, A.F.G.; de Castro Ramalho, T.; de Carvalho Júnior, R.N.; de Jesus Chaves Neto, A.M. Interactions of Co, Cu, and Non-Metal Phthalocyanines with External Structures of SARS-CoV-2 Using Docking and Molecular Dynamics. Sci. Rep. 2022, 12, 3316. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Pourhanifeh, M.H.; Hamblin, M.R.; Shahrzad, M.K.; Mirzaei, H. RdRp Inhibitors and COVID-19: Is Molnupiravir a Good Option? Biomed. Pharmacother. 2022, 146, 112517. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic Efficacy of the Small Molecule GS-5734 against Ebola Virus in Rhesus Monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir Is a Direct-Acting Antiviral That Inhibits RNA-Dependent RNA Polymerase from Severe Acute Respiratory Syndrome Coronavirus 2 with High Potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [Green Version]
- Naydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-Dependent RNA Polymerase in the Presence of Favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef]
- Padhi, A.K.; Dandapat, J.; Saudagar, P.; Uversky, V.N.; Tripathi, T. Interface-based Design of the Favipiravir-binding Site in SARS-CoV-2 RNA-dependent RNA Polymerase Reveals Mutations Conferring Resistance to Chain Termination. FEBS Lett. 2021, 595, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- WHO Solidarity Trial Consortium. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The ‘Very, Very Bad Look’ of Remdesivir, the First FDA-Approved COVID-19 Drug. Science 2020. [Google Scholar] [CrossRef]
- Cully, M. A Tale of Two Antiviral Targets—And the COVID-19 Drugs That Bind Them. Nat. Rev. Drug. Discov. 2022, 21, 3–5. [Google Scholar] [CrossRef]
- Pourkarim, F.; Pourtaghi-Anvarian, S.; Rezaee, H. Molnupiravir: A New Candidate for COVID-19 Treatment. Pharmacol. Res. Perspect. 2022, 10, e00909. [Google Scholar] [CrossRef]
- Halford, B. How Pfizer Scientists Transformed an Old Drug Lead into a COVID-19 Antiviral. Available online: https://cen.acs.org/pharmaceuticals/drug-discovery/How-Pfizer-scientists-transformed-an-old-drug-lead-into-a-COVID-19-antiviral/100/i3 (accessed on 18 February 2022).
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 M pro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Li, P.; Wang, Y.; Lavrijsen, M.; Lamers, M.M.; de Vries, A.C.; Rottier, R.J.; Bruno, M.J.; Peppelenbosch, M.P.; Haagmans, B.L.; Pan, Q. SARS-CoV-2 Omicron Variant Is Highly Sensitive to Molnupiravir, Nirmatrelvir, and the Combination. Cell Res. 2022, 32, 322–324. [Google Scholar] [CrossRef]
- Takashita, E.; Kinoshita, N.; Yamayoshi, S.; Sakai-Tagawa, Y.; Fujisaki, S.; Ito, M.; Iwatsuki-Horimoto, K.; Chiba, S.; Halfmann, P.; Nagai, H.; et al. Efficacy of Antibodies and Antiviral Drugs against COVID-19 Omicron Variant. N. Engl. J. Med. 2022, 386, 995–998. [Google Scholar] [CrossRef]
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like Protease Inhibitors Block Coronavirus Replication in Vitro and Improve Survival in MERS-CoV–Infected Mice. Sci. Transl. Med. 2020, 12, eabc5332. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA Proofreading Machine Regulates Replication Fidelity and Diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Rona, G.; Zeke, A.; Miwatani-Minter, B.; de Vries, M.; Kaur, R.; Schinlever, A.; Garcia, S.F.; Goldberg, H.V.; Wang, H.; Hinds, T.R.; et al. The NSP14/NSP10 RNA Repair Complex as a Pan-Coronavirus Therapeutic Target. Cell. Death Differ. 2022, 29, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Tampere, M.; Pettke, A.; Salata, C.; Wallner, O.; Koolmeister, T.; Cazares-Körner, A.; Visnes, T.; Hesselman, M.C.; Kunold, E.; Wiita, E.; et al. Novel Broad-Spectrum Antiviral Inhibitors Targeting Host Factors Essential for Replication of Pathogenic RNA Viruses. Viruses 2020, 12, 1423. [Google Scholar] [CrossRef] [PubMed]
- Chitalia, V.C.; Munawar, A.H. A Painful Lesson from the COVID-19 Pandemic: The Need for Broad-Spectrum, Host-Directed Antivirals. J. Transl. Med. 2020, 18, 390. [Google Scholar] [CrossRef] [PubMed]
- Mei, M.; Tan, X. Current Strategies of Antiviral Drug Discovery for COVID-19. Front. Mol. Biosci. 2021, 8, 671263. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat Mesylate Inhibits SARS-CoV-2 Activation by TMPRSS2-Related Proteases and Its Metabolite GBPA Exerts Antiviral Activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef]
- Han-soo, L. Daewoong Fails to Reach Primary Endpoint in Covid-19 Treatment Trials. Available online: http://www.koreabiomed.com/news/articleView.html?idxno=11753 (accessed on 18 February 2022).
- Breining, P.; Frølund, A.L.; Højen, J.F.; Gunst, J.D.; Staerke, N.B.; Saedder, E.; Cases-Thomas, M.; Little, P.; Nielsen, L.P.; Søgaard, O.S.; et al. Camostat Mesylate against SARS-CoV-2 and COVID-19—Rationale, Dosing and Safety. Basic Clin. Pharmacol. Toxicol. 2021, 128, 204–212. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Sukhatme, V.P.; Reiersen, A.M.; Vayttaden, S.J.; Sukhatme, V.V. Fluvoxamine: A Review of Its Mechanism of Action and Its Role in COVID-19. Front. Pharmacol. 2021, 12, 652688. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Suzuki, T.; Hashimoto, K. Mechanisms of Action of Fluvoxamine for COVID-19: A Historical Review. Mol. Psychiatry 2022, 7, 1–10. [Google Scholar] [CrossRef]
- Crump, A.; Omura, S. Ivermectin, “Wonder Drug” from Japan: The Human Use Perspective. Proc. Jpn. Acad. Ser. B 2011, 87, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Yagisawa, M.; Foster, P.J.; Hanaki, H.; Ōmura, S. Global Trends in Clinical Studies of Ivermectin in COVID-19. Jpn. J. Antibiot. 2021, 74, 44–95. [Google Scholar]
- Chaccour, C.; Hammann, F.; Ramón-García, S.; Rabinovich, N.R. Ivermectin and COVID-19: Keeping Rigor in Times of Urgency. Am. J. Trop. Med. 2020, 102, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-Approved Drug Ivermectin Inhibits the Replication of SARS-CoV-2 in Vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.K.; Dehgani-Mobaraki, P. The Mechanisms of Action of Ivermectin against SARS-CoV-2—An Extensive Review. J. Antibiot. 2022, 75, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Heidary, F.; Gharebaghi, R. Ivermectin: A Systematic Review from Antiviral Effects to COVID-19 Complementary Regimen. J. Antibiot. 2020, 73, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Mega, E.R. Latin America’s Embrace of an Unproven COVID Treatment Is Hindering Drug Trials. Nature 2020, 586, 481–482. [Google Scholar] [CrossRef]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin Inhibits the Sarcoplasmic or Endoplasmic Reticulum Ca-ATPase Family of Calcium Pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [CrossRef]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin—From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef]
- Al-Beltagi, S.; Goulding, L.V.; Chang, D.K.E.; Mellits, K.H.; Hayes, C.J.; Gershkovich, P.; Coleman, C.M.; Chang, K.-C. Emergent SARS-CoV-2 Variants: Comparative Replication Dynamics and High Sensitivity to Thapsigargin. Virulence 2021, 12, 2946–2956. [Google Scholar] [CrossRef]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-Level Inhibition of Coronavirus Replication by Chemical ER Stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin Has Potent Preclinical Efficacy against SARS-CoV-2 by Targeting the Host Protein EEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Eloy, P.; Le Grand, R.; Malvy, D.; Guedj, J. Combined Treatment of Molnupiravir and Favipiravir against SARS-CoV-2 Infection: One + Zero Equals Two? EBioMedicine 2021, 74, 103663. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.L.; Mátyus, P. One Size Does Not Fit All: Challenging Some Dogmas and Taboos in Drug Discovery. Future Med. Chem. 2016, 8, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Rawle, D.J.; Le, T.T.; Suhrbier, A. Simple Rapid in Vitro Screening Method for SARS-CoV-2 Anti-Virals That Identifies Potential Cytomorbidity-Associated False Positives. Virol. J. 2021, 18, 123. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Jill Lai, C.-Y.; Hastie, A.; Mondal, A.K.; Dehkordi, S.R.; van der Made, C.I.; Fedrigo, O.; Al-Ajli, F.; Jalnapurkar, S.; Byrska-Bishop, M.; et al. Optical Genome Mapping Identifies Rare Structural Variations as Predisposition Factors Associated with Severe COVID-19. iScience 2022, 25, 103760. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-Y.; Chien, C.-W.; Tung, T.-H. Healthcare Practice Strategies for Integrating Personalized Medicine: Management of COVID-19. World J. Clin. Cases 2021, 9, 8647–8657. [Google Scholar] [CrossRef]
- DeMerle, K.; Angus, D.C.; Seymour, C.W. Precision Medicine for COVID-19: Phenotype Anarchy or Promise Realized? JAMA 2021, 325, 2041. [Google Scholar] [CrossRef]
- Shrestha, G.S.; Paneru, H.R.; Vincent, J.-L. Precision Medicine for COVID-19: A Call for Better Clinical Trials. Crit. Care 2020, 24, 282. [Google Scholar] [CrossRef]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In Silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef]
- Ko, Y. Computational Drug Repositioning: Current Progress and Challenges. Appl. Sci. 2020, 10, 5076. [Google Scholar] [CrossRef]
- D’Souza, S.; Prema, K.V.; Balaji, S. Machine Learning Models for Drug–Target Interactions: Current Knowledge and Future Directions. Drug Discov. Today 2020, 25, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. The Race for Antiviral Drugs to Beat COVID—And the next Pandemic. Nature 2021, 592, 340–343. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Cell Surface Entry Point | Viral Genome | Revers Transcriptase | Primary Target Cell Type |
|---|---|---|---|---|
| Influenza | terminal α-sialic acid | one single-stranded, negative-sense segmented RNA | No | Epithelial cells (primarily bronchial) |
| SARS-CoV-2 | Angiotensin-Converting Enzyme 2 (ACE2), transmembrane Serin protease 2 (TMPRSS2) | one single-stranded positive-sense RNA | No | Epithelial cell (primarily bronchial, alveolar and intestinal) |
| HIV | CD4, CCR5 | two single-stranded, non- segmented RNA | Yes | T helper cell |
| Virus | Blood Group Susceptibility Highest to Infection | Blood Group Susceptibility Lowest to Infection |
|---|---|---|
| Influenza | A | 0 |
| SARS-CoV-2 | A | 0 |
| HIV | 0 | B |
| Production Method | Influenza | SARS-CoV-2 | HIV | ||
|---|---|---|---|---|---|
| Traditional for active vaccination | intact, inactivated virus | Yes | Yes | Yes, but any of the current vaccine trials produced no effective immune response | |
| Biotechnology based for active vaccination | 1. | mRNA | Research is ongoing, clinical trials recruiting soon | Yes | Research is ongoing, clinical trial starts recruiting |
| 2. | recombinant viral subunit protein | Yes | |||
| 3. | recombinant adenoviral delivery vector containing viral protein sequence | Yes | |||
| Traditional for passive vaccination | polyclonal antibody, purified from human sera recovered from the disease | Yes | |||
| Biotechnology based for passive vaccination | monoclonal antibodies, effective sequences selected by phage display | Yes |
| Drug Class | Generic Name (Other Names and Acronyms) | Brand Name | FDA Approval Date |
|---|---|---|---|
| NNRTI | doravirine | Pifeltro | 30 August 2018 |
| INSTI | cabotegravir | Vocabria | 22 January 2021 |
| AI | fostemsavir | Rukobia | 2 July 2020 |
| PAI | ibalizumab-uiyk | Trogarzo | 6 March 2018 |
| Generic Name | Brand Name | FDA Approval Date |
|---|---|---|
| bictegravir, emtricitabine, and tenofovir alafenamide (bictegravir sodium/emtricitabine/tenofovir alafenamide fumarate, BIC/FTC/TAF) | Biktarvy | 7 February 2018 |
| cabotegravir and rilpivirine (CAB and RPV, CAB plus RPV, Cabenuva kit, cabotegravir extended-release injectable suspension, and rilpivirine extended-release injectable suspension) | Cabenuva | 22 January 2021 |
| darunavir and cobicistat (darunavir ethanolate/cobicistat, DRV/COBI) | Prezcobix | 29 January 2015 |
| darunavir, cobicistat, emtricitabine, and tenofovir alafenamide (darunavir ethanolate/cobicistat/emtricitabine/tenofovir AF, darunavir ethanolate/cobicistat/emtricitabine/tenofovir alafenamide, darunavir/cobicistat/emtricitabine/tenofovir AF, darunavir/cobicistat/emtricitabine/tenofovir alafenamide fumarate, DRV/COBI/FTC/TAF) | Symtuza | 17 July 2018 |
| dolutegravir and lamivudine (dolutegravir sodium/lamivudine, DTG/3TC) | Dovato | 8 April 2019 |
| dolutegravir and rilpivirine (dolutegravir sodium/rilpivirine hydrochloride, DTG/RPV) | Juluca | 21 November 2017 |
| doravirine, lamivudine, and tenofovir disoproxil fumarate (doravirine/lamivudine/TDF, doravirine/lamivudine /tenofovir DF, DOR/3TC/TDF) | Delstrigo | 30 August 2018 |
| efavirenz, lamivudine, and tenofovir disoproxil fumarate (EFV/3TC/TDF) | Symfi Lo | 5 February 2018 |
| lamivudine and tenofovir disoproxil fumarate (Temixys, 3TC/TDF) | Cimduo | 28 February 2018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kálai, T.; Pongrácz, J.E.; Mátyus, P. Recent Advances in Influenza, HIV and SARS-CoV-2 Infection Prevention and Drug Treatment—The Need for Precision Medicine. Chemistry 2022, 4, 216-258. https://doi.org/10.3390/chemistry4020019
Kálai T, Pongrácz JE, Mátyus P. Recent Advances in Influenza, HIV and SARS-CoV-2 Infection Prevention and Drug Treatment—The Need for Precision Medicine. Chemistry. 2022; 4(2):216-258. https://doi.org/10.3390/chemistry4020019
Chicago/Turabian StyleKálai, Tamás, Judit Erzsébet Pongrácz, and Péter Mátyus. 2022. "Recent Advances in Influenza, HIV and SARS-CoV-2 Infection Prevention and Drug Treatment—The Need for Precision Medicine" Chemistry 4, no. 2: 216-258. https://doi.org/10.3390/chemistry4020019