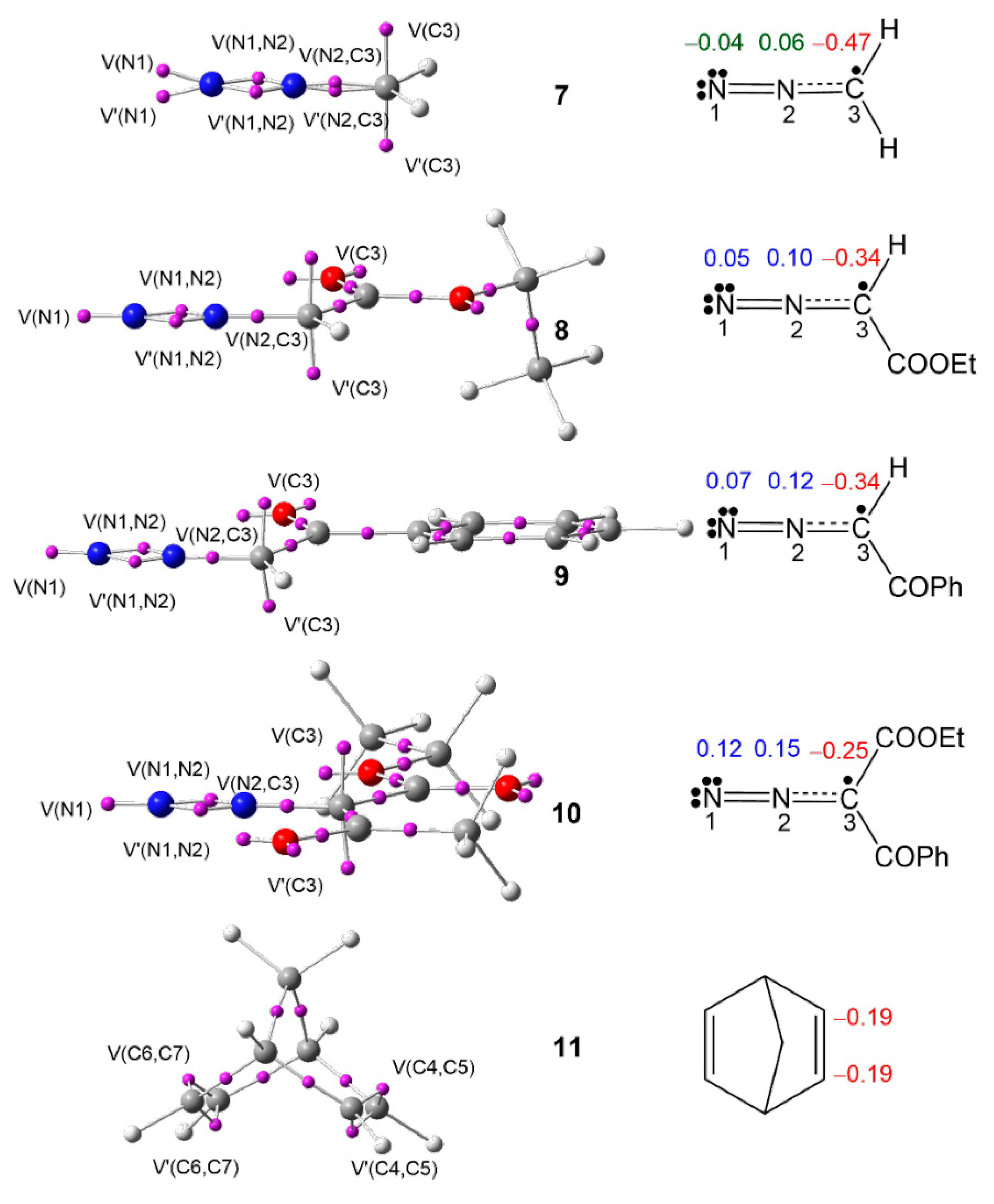
3.1. Analysis of the ELF Topology at the Ground State Electronic Structures of DAAs 7–10 and NBD 11
ELF [
41,
42] topology allows quantitative characterization of chemical regions of a molecule, which is used to classify the TACs, and then to correlate their electronic structure with their molecular reactivity within the MEDT framework. Proposed by Domingo in 2019 [
36], this standard classification of TACs into
pseudodiradical,
pseudo(mono)radical, carbenoid, or zwitterionic type has characterized 32CA reactions into
pmr-type,
pdr-type,
cb-type, and
zw-type, respectively, based on the respective TAC participation [
36]. Herein, the ELF topology of DAAs
7–
10 and NBD
11 is studied to assess their electronic structures. The ELF valence basin populations are given in
Table 1, while the ELF basin attractor positions are shown in
Figure 1.
The ELF topological analysis of DAAs
7–
10 shows the presence of two monosynaptic basins, V(N1) and V′(N1), in
7 and one V(N1) monosynaptic basin in
8–
10, integrating a total population of 3.78 e (
7), 3.61 e (
8), 3.58 e (
9), and 3.47 e (
10), associated with the non-bonding electron density on N1 nitrogen; two disynaptic basins, V(N1,N2) and V′(N1, N2), integrating a total population of 3.74 e (
7), 3.88 e (
8), 3.90 e (
9), and 3.97 e (
10), associated with the N1–N2 double bond; and one V(C3,N2) disynaptic basin, integrating 3.01 e (
7), 2.90 e (
9), 2.88 e (
9), and 2.82 e (
10), associated with the C3–N2 overpopulated single bond, which shows the influence of substitution on the electronic distribution of the N1–N2–C3 moiety of these DAAs. Finally, the presence of two monosynaptic basins, V(C3) and V′(C3), integrating a total of 0.92 e (
7), 0.92 e (
8), 0.88 e (
9), and 0.98 e (
10), associated with a
pseudoradical center at C3, classifies the DAAs
7–
10 as
pseudo(mono)radical TACs, thus suggesting their participation in
pmr-type 32CA reactions [
36].
ELF topology of NBD 11 shows the presence of two pairs of disynaptic basins, V(C4,C5) and V′(C4,C5), and V(C6,C7) and V′(C6,C7), integrating a total population of 3.48 e each pair, associated with the C4–C5 and C6–C7 double bonds of NBD 11.
The proposed Lewis-like structures, together with the natural atomic charges of compounds
7–
11, are represented in
Figure 1. The
pseudoradical C3 carbon is negatively charged by −0.47 e (
7), −0.34 e (
8), −0.34 e (
9), and −0.25 e (
10), suggesting the influence of the EW substitution. The N1 and N2 nitrogen nuclei show negligible charges in simplest DAA
7, while the N2 nitrogen shows a charge slightly greater than 0.10 e at DAAs
8,
9, and
10. The charge distribution pattern in the DAAs
7–
10 does not follow the commonly accepted 1,2-zwitterionic concept for DAAs.
3.2. Analysis of the CDFT Indices of DAAs 7–10 and NBD 11
The analysis of the reactivity indices based on CDFT [
43,
44] has become a useful tool for studying reactivity in polar reactions [
44]. Therefore, an analysis of CDFT reactivity indices was performed to predict the reactivity of the reagents in these 32CA reactions. The CDFT indices were calculated at the B3LYP/6-31G (d) computational level since it was used to define the electrophilicity and nucleophilicity scales [
43,
68,
69]. The electronic chemical potential
μ, chemical hardness
η, electrophilicity
ω, and nucleophilicity
N indices of compounds
7–
11 are gathered in
Table 2. The MPWB1K/6-311G (d,p) reactivity indices are gathered in
Table S3 in Supplementary Material.
The electronic chemical potentials
μ of DAAs
7−
10, μ = −3.63 (
7), −4.27 (
8), −4.20 (
9), and −4.58 (
10) eV, are lower than that of NBD
11, μ = −2.94 eV, suggesting that along a polar process the electron density will flux from NBD
11 to DAAs
7–
10 via reverse electron density flux (REDF) reactions [
70].
The chemical hardness η of DAAs 7–10, ranging from 4.70 to 4.87 eV, is also lower than that of NBD 11, 5.88 eV, which means that the DAAs are more prone to electron density deformation than NBD 11, i.e., they are softer. Interestingly, DAA 10, having more EW substituents, is chemically harder than the simplest DAA 7.
Within the electrophilicity scale [
68], simplest DAA
7, ω = 1.40 eV, is classified as a moderate electrophile, while DAAs
8–
10, ω > 1.85 eV, are classified as strong electrophiles. However, it is worth noting that among the strongly electrophilic DAAs, only DAA
10 would be electrophilically activated enough to work well experimentally, which is against the experimental findings. On the other hand, within the nucleophilicity scale [
69], the simplest DAA
7,
N = 3.13 eV, is classified as a strong nucleophile, while DAAs
8–
10,
N < 2.53 eV, are classified as moderate nucleophiles. As expected, the inclusion of EW groups at the C3 carbon of simplest DAA
7 increases the electrophilicity ω index and decreases the nucleophilicity
N index of the DAA derivatives.
NBD 11 is classified as a marginal electrophile, ω = 0.74 eV, and a strong nucleophile, N = 3.23 eV. Both the strain caused by the bicyclic system and the presence of two C–C double bonds considerably increase the nucleophilicity N index of 11 with respect to that of ethylene 26, N = 1.87 eV.
The analysis of the CDFT indices predicts that along a polar process, DAAs
7–
10 will behave as electrophilic species while NBD
11 as the nucleophilic one. However, the experimental outcome [
22] revealed that the EW substitution in the DAA decreases its reactivity towards NBD
11. Along this series, the fastest 32CA reaction was that involving simplest DAA
7, while strongly electrophilic DAA
10 having two EW –COOMe and –COPh groups was completely inactive. In addition, except for the reaction involving DAA
10, CDFT predicts the other reactions to have a low-polar character as the DAAs are not sufficiently electrophilically activated. Thus, this CDFT analysis does not agree with the experimental results of this series of reactions.
A comparison of the B3LYP/6-31G (d) and MPWB1K/6-311G (d,p) reactivity indices shows that although they have different values, both computational levels give similar trends of reactivity.
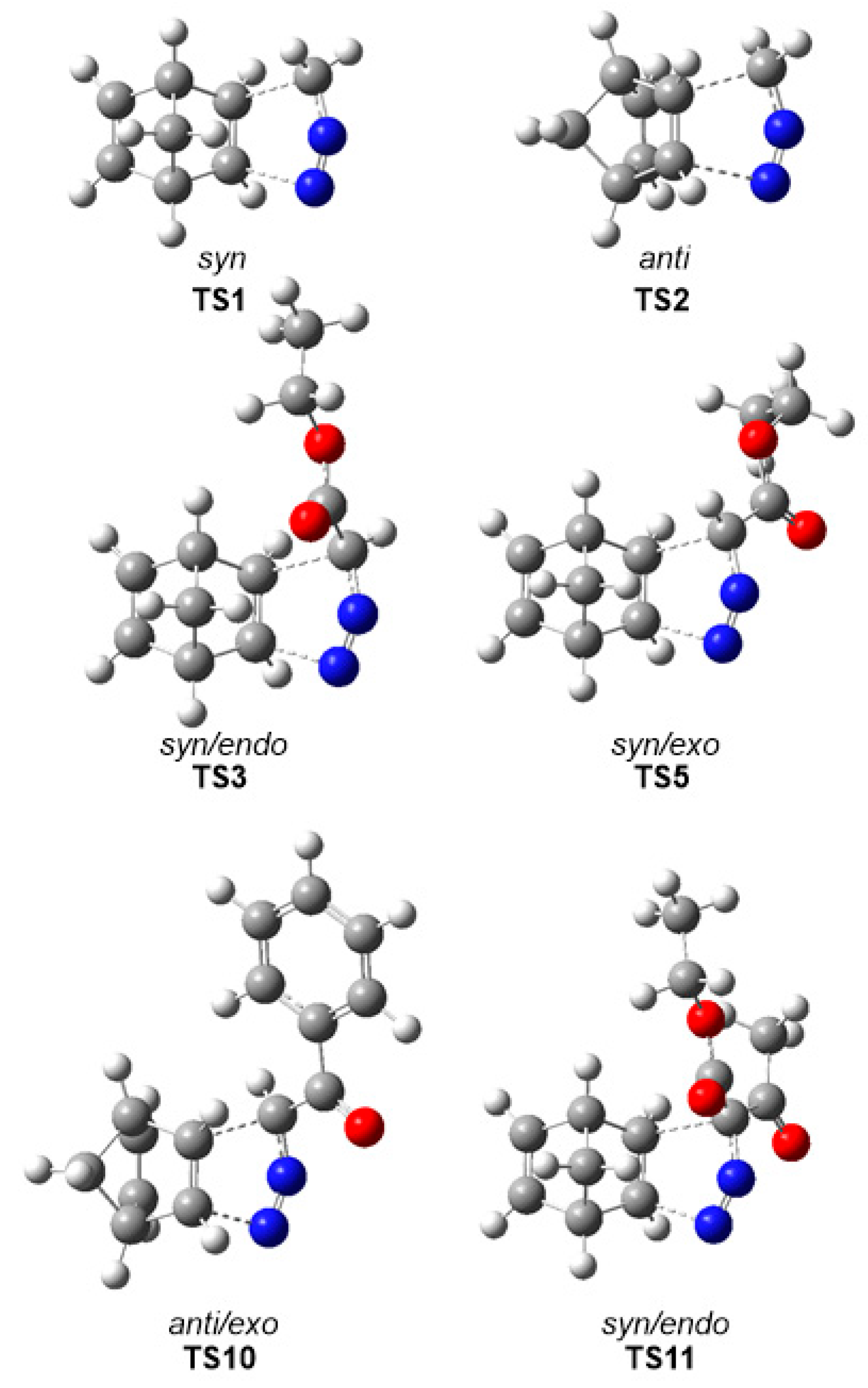
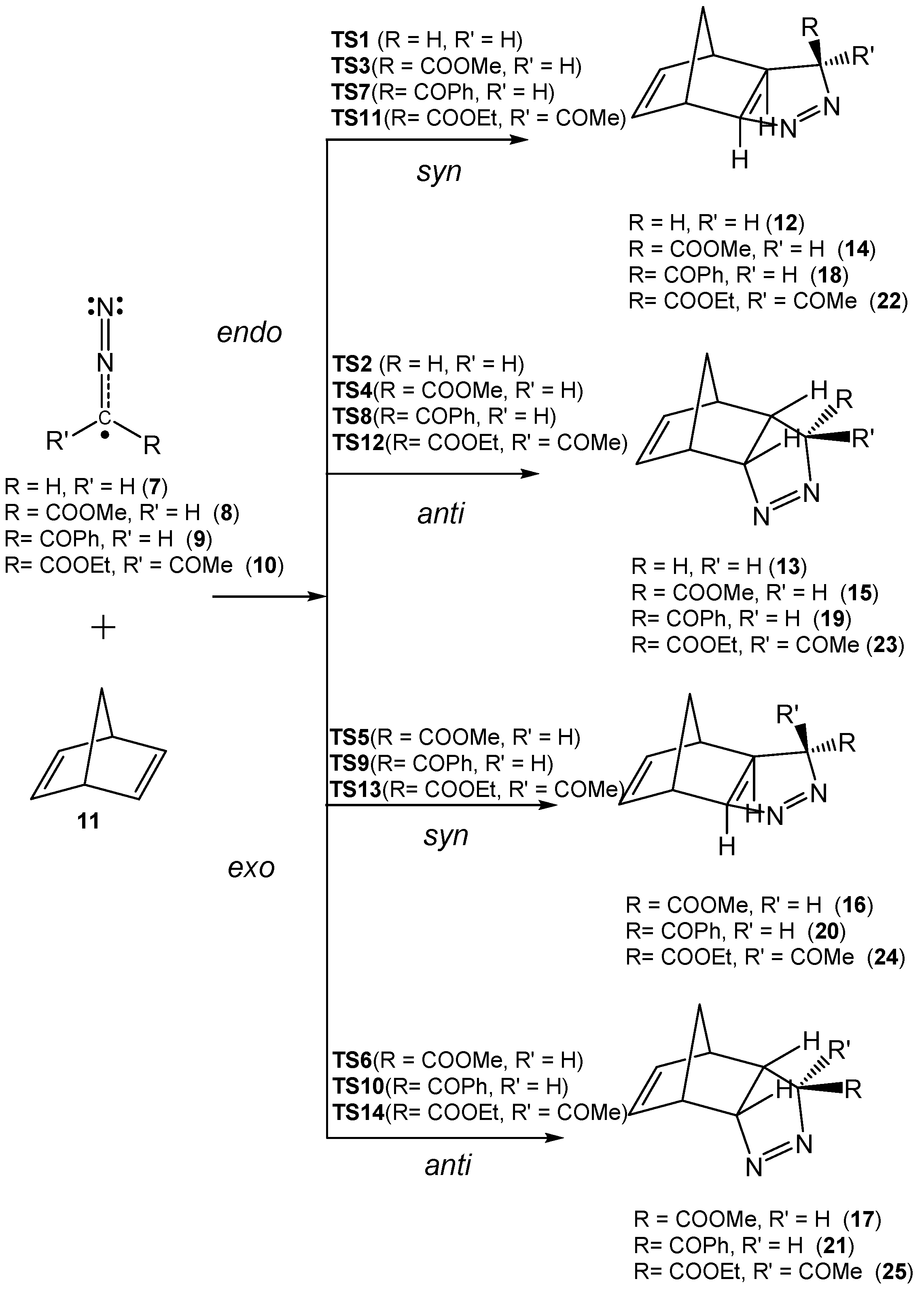
3.3. Analysis of the Reaction Paths Associated with the 32CA Reactions between DAAs 7–10 and NBD 11
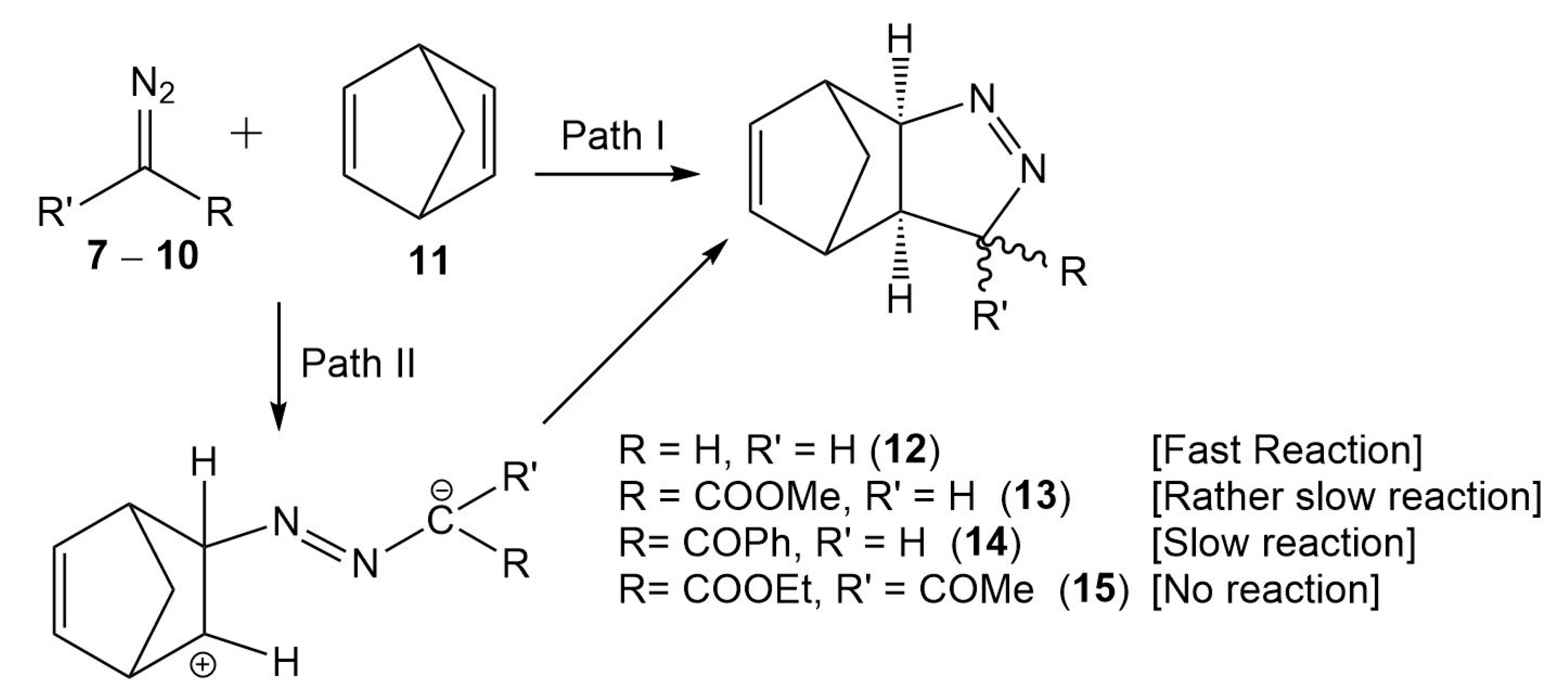
Due to the molecular symmetry of the simplest DAA
7 and NBD
11, two competitive stereoisomeric reaction paths are feasible for this 32CA reaction. They are related to the two approach modes of DAA
7 with respect to the two stereoisomeric faces of the C–C double bonds of NBD
11. These reaction paths are called
syn and
anti. For DAAs
8–
10, which present a different substitution at the C3 carbon, two additional stereoisomeric reaction paths are feasible, the
endo and the
exo ones. Consequently, for the 32CA reactions involving DAAs
8–
10, four competitive reaction paths were considered (see
Scheme 4). For the 32CA reaction of simplest DAA
7, the
syn and
anti reaction paths lead to two diastereoisomeric pyrazolines
12 and
13, respectively, via
TS1 and
TS2. For the 32CA reactions involving substituted DAAs
8–
10, four diastereoisomeric pyrazolines
14–
17,
18–
21, and
22–
25 can be formed via
TS3–
TS6,
TS7–
TS10, and
TS11–
TS14, respectively (
Scheme 4). The four 32CA reactions follow a one-step mechanism. Relative electronic energies, enthalpies, entropies, and Gibbs free energies of the TSs and adducts are given in
Table 3.
From the energy results given in
Table 3, a series of appealing conclusions can be obtained: (i) The gas phase activation energies associated with the energetically most favorable reaction paths of these 32CA reactions are found between 15.9 (
TS1) and 23.3 (
TS11) kcal·mol
−1; (ii) the increase of the activation energies in the order
TS1 (
7) <
TS3 (
8) <
TS10 (
9) <
TS11 (
10) is in complete agreement with the experimental outcomes [
22]; (iii) these reactions are strongly exothermic by more than 30 kcal·mol
−1; (iv) inclusion of solvent effects increases the activation energies by between 0.3 (
TS11) and 1.9 (
TS7) kcal·mol
−1, and increases the exothermic character of the reactions by between 2.0 (
12) and 4.1 (
22) kcal·mol
−1. Formation of
18 is disfavored by 1.2 kcal·mol
−1; (v) inclusion of the thermal corrections increases the activation enthalpies in acetonitrile by between 0.8 (
TS3) and 1.1 (
TS11) kcal·mol
−1 and decreases the reaction enthalpies by between 3.6 (
22) and 4.8 (
12) kcal·mol
−1; (vi) inclusion of entropies to enthalpies increases the activation Gibbs free energies in acetonitrile between 29.9 (
TS1) and 43.0 (
TS14) kcal·mol
−1, and decreases the reaction Gibbs free energies between −16.2 (
22) and −37.1 (
12) kcal·mol
−1; (vii) the strong exergonic character of these 32CA reactions makes them irreversible, and consequently, the products are obtained by kinetic control; (viii) the
syn/
anti facial stereoselectivity ranges from 0.7 (
7) to 3.1 (
8) kcal·mol
−1. While DAAs
7,
8, and
10 prefer the
syn attack, DAA
9 prefers the
anti one; and (ix) these 32CA reactions present a low
endo/
exo stereoselectivity; lower than 0.8 kcal·mol
−1. While the 32CA reactions of DAAs
7,
8, and
10 are
endo selective, that of DAA
9 is
exo selective.
The gas phase optimized geometries of the more favorable TSs are given in
Figure 2, while the distances between the C–C and C–N interacting nuclei are given in
Table 4. Some appealing conclusions can be drawn for these geometrical parameters: (i) Considering that C–C bond formation begins slightly earlier than the C–N one, [
36] these distances at
TS1–
TS10 indicate that, from a geometrical point of view, they correspond to hardly asynchronous TSs; (ii) a comparison of these distances at
TS11–
TS14 with respect to those at the other TSs shows that the TSs involving DAA
10 are clearly more asynchronous. At
TS11–
TS14, the C–N distances are shorter, while the C–C ones are longer, suggesting more delayed C–C bond formation but more advanced C–N bond formation; i.e., more asynchronous processes; (iii) as all these distances are greater than 2.0 Å in the gas phase and acetonitrile, and considering that the C–C and C–N single bond formation take place at the short range of 2.0–1.9 and 1.9–1.8 Å, respectively, [
36] they indicate that formation of the new C–C and C–N single bonds has not started yet at any TS (see
Section 3.6); (iv) inclusion of solvent effects in the optimizations does not considerably modify the gas phase geometries. Note that for the 32CA reactions of DAAs
7–
9, these differences are lower than 0.02 Å.
Finally, the GEDT at the TSs was calculated to assess the polarity of these 32CA reactions, and is given in
Table 3. Reactions with GEDT values lower than 0.05 e correspond to non-polar processes, while values higher than 0.20 e correspond to polar processes. The 32CA reaction between simplest DAA
7 and NBD
11, with GEDT of 0.10 e (
TS1) and 0.12 (
TS2), in the gas phase and in acetonitrile, is predicted as a 32CA reaction with some polar character, with the electron density fluxing from DAA
7 to NBD
11. This GEDT analysis suggests that this 32CA reaction is classified as a forward electron density flux (FEDF) reaction [
70]. Interestingly, this finding is against the analysis of the electronic chemical potential μ, electrophilicity ω, and nucleophilicity
N indices of DAA
7 and NBD
11, which predict that the reaction would not work experimentally but, if so, it should be a reaction of reverse electron density flux (REDF) [
70] (see
Table 2 and
Section 3.2).
For the 32CA reactions of DAAs
8–
10 with NBD
11, the GEDT values are found between 0.02–0.05 e, predicting non-polar 32CA reactions, thus, being classified as reactions of null electron density flux (NEDF) [
71]. This decrease in polarity is consistent with the increase in the energy barrier of the corresponding 32CA reactions. For the 32CA reaction of DAA
10 with NBD
11 in acetonitrile, which does not take place experimentally, the GEDT at the TSs is 0.08 e, showing a very low polar character. At this TS, the low electron density that fluxes from the NBD framework to the DAA ones, might permit to classify this 32CA reaction as a reaction of REDF [
70]. However, note that this 32CA does not take place experimentally.
Interestingly, the 32CA reaction of simplest DAA
7, the least electrophilic DAA of the series, with NBD
11 presents the highest GEDT. In addition, the electron density fluxes from DAA
7 to NBD
11, against the analysis of the CDFT indices (see
Section 3.2). Similar behavior was found along the
pdr-type 32CA reaction between the simplest azomethine ylide and ethylene
26, GEDT = 0.10 e [
37], which was expected to be a reaction of NEDF. Note that neither NBD
11 nor ethylene
26 have any tendency to act as electrophiles. It seems that the presence of a non-bonding electron density at the ends of the TACs, which accounts for their high nucleophilic character, is responsible for the local electron density transfer taking place from these terminal carbon and nitrogen atoms to the double bonds of NBD
11 and ethylene
26, a local phenomenon that cannot be anticipated by the analysis of the electronic chemical potential μ of the reagents.
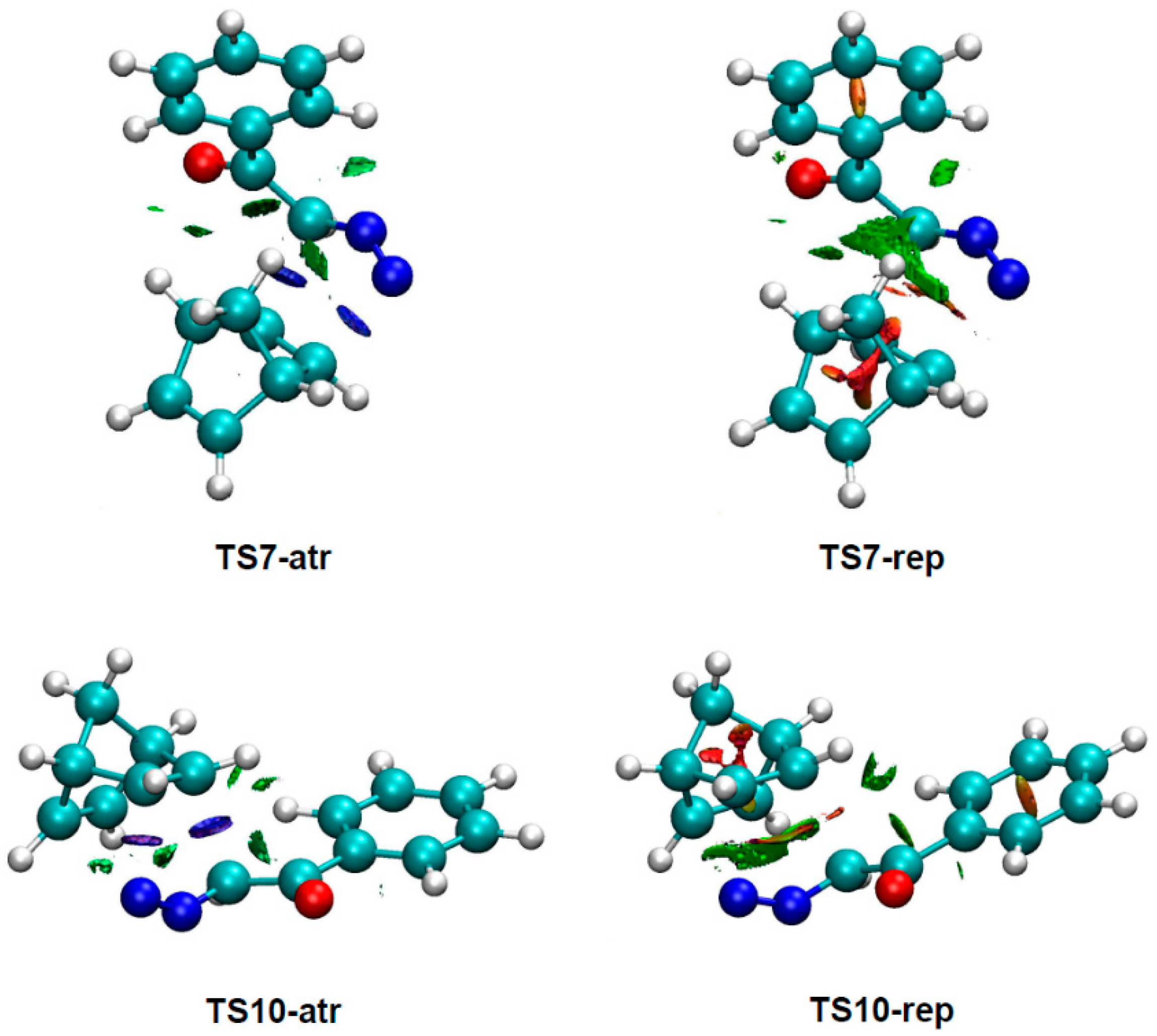
3.4. NCI Analysis of the Preferred Anti/Exo Stereoselectivity in the 32CA Reaction of DAA 9 with NBD 11
To understand the origin of the
anti/exo stereoselectivity in the 32CA reaction of DAA
9 with NBD
11, a comparative topological analysis of the attractive and repulsive NCIs taking place at
syn/endo TS7 and
anti/exo TS10 was performed by means of the NCI approach. Note that the
syn/endo stereoselectivity is preferred in every 32CA reaction except that involving DAA
9. The corresponding isosurfaces are represented in
Figure 3, while the plots of the reduced density gradient
s vs. sign(λ
2) ρ(r) are given in
Figure 4. A continuous color-coding scheme based on the second eigenvalue of the Hessian is used, where strong, attractive interactions are represented in blue, weak interactions in green, and strong repulsive interactions in red.
As can be observed in
Figure 3, the surfaces associated with repulsive density overlap are greater than those associated with attractive interactions. This suggests that repulsive interactions have a greater role than the attractive ones in the preference for the
anti/exo approach. A closer look reveals that considering only the surfaces associated with intermolecular interactions and skipping those associated with bond formation, the repulsive overlap spreads wider at
TS7 than at
TS10.
The plots of the reduced density gradient
s vs. sign(λ
2) ρ show that there are no qualitative differences between the density characteristics of the interactions (see
Figure 4). The only noticeable change is that the peaks associated with the weak NCIs, both the attractive and the repulsive ones, reach higher values of
s at
TS10 than at
TS7. This feature only indicates that these interactions are not associated with critical points in the electron density, [
72] thus being only different in strength. Consequently, this analysis suggests that the weaker unfavorable interactions present at
TS10 are responsible for the
anti/exo stereoselectivity in the 32CA reaction between DAA
9 and NBD
11.
3.5. BET Study for the 32CA Reactions of DAAs 7 and 10 with NBD 11
The BET has established the molecular mechanism of a great number of organic reactions by means of the analysis of the bonding changes along the reaction path, which is possible due to the straightforward connection between the electron density distribution and the chemical structure [
42]. The detailed BET studies of the 32CA reactions of DAAs
7 and
10 with NBD
11 are given in the
Supplementary Material in
Section 1 and
Section 2. Herein, only the most relevant conclusions are analyzed and compared. The ELF basin attractor positions of the structures involved in the bond formation are shown in
Figure 5.
From the BET study of the 32CA reaction of simplest DAA
7 with NBD
11, we conclude that: (i) This 32CA reaction can be topologically divided into nine phases starting from structure
S0-I and ending in pyrazoline
12; (ii)
Phases I and
II are characterized by the depopulation of the N1–N2, N2–C3 and C4–C5 bonding regions. These changes in electron density demand an energy cost (EC) of 15.4 kcal mol
−1, which accounts for 97% of the activation energy needed to reach
TS1, found in
Phase III; (iii) the depopulation of these bonding regions creates the N2 nitrogen non-bonding electron density at structure
S1-I, and the C4 and C5
pseudoradical centers at
S3-I and
S4-I, respectively (see
S4-I in
Figure 5); (v) formation of the first C3–C4 single bond takes place at
S5-I, by the C-to-C coupling of the two C3 and C4
pseudoradical centers present in
S4-I, at a C–C distance of 2.05 Å and with an initial population of 1.36 e (see
S5-I in
Figure 5). Note that the C3
pseudoradical center was already present at DAA
7; (vi) formation of the second N1–C5 single bond takes place at
S8-I, by sharing part of the non-bonding electron density of the N1 nitrogen and that of the C5
pseudoradical center present in
S7-I, at a C–N distance of 1.84 Å and with an initial population of 1.51 e (see
S8-I in
Figure 5); (vii) the formation of the second N1–C5 single bond begins when the first C3–C4 single bond formation has been completed by up to 93%, suggesting a
two-stage one-step mechanism [
73].
The BET study of the 32CA reaction of DAA 10 with NBD 11 draws some significant mechanistic conclusions: (i) This 32CA reaction can be divided into seven topological phases starting from S0-II and ending in pyrazoline 22; (ii) Phases I–III are characterized by the depopulation of the N1–N2, N2–C3 and C4–C5 bonding regions. These changes in electron density demand an EC of 20.9 kcal mol−1. This accounts for 90% of the activation energy needed to reach TS11, which is found in Phase IV; (iv) the depopulation of N1–N2 and N2–C3 bonding regions along Phase I creates the non-bonding electron density at N2 nitrogen at S1-II, while the depopulation of the C4–C5 bonding region along Phases I–III creates the C4 and C5 pseudoradical centers; (v) formation of the first C3–C4 single bond takes place at S5-II, by the C-to-C coupling of the two C3 and C4 pseudoradical centers present in S4-II, at a C–C distance of 2.11 Å, and with an initial population of 1.25 e. Note that the C3 pseudoradical center was already present at DAA 10; (vii) formation of the second N1–C5 single bond takes place at S6-II, by sharing part of the non-bonding electron density of the N1 nitrogen and that of the C5 pseudoradical center present in S5-I, at a C–N distance of 1.82 Å, and with an initial population of 1.38 e; (vii) the formation of second N1–C5 single bond begins when the first C3–C4 single bond formation has been completed by up to 70%, suggesting an asynchronous one-step mechanism.
From this comparative analysis of the two BET studies, we conclude: (i) These 32CA reactions follow an asynchronous one-step mechanism, the highest asynchronicity in the bond formation being observed for the most favorable 32CA reaction of simplest DAA
7 with NBD
11, which takes place via a
two-stage one-step mechanism [
73]. This finding confirms that any analysis based on the geometries of TSs, such as bond orders, is not suitable to assess the asynchronicity of a reaction; (ii) the initial phases of both 32CA reactions are related to the rupture of the N1–N2, N2–C3 and C4–C5 double bonds, demanded the formation of the N1, C3 and C5 non-bonding electron densities; (iii) while at the 32CA reaction involving simplest DAA
7 these bonding changes demand an EC of 15.4 kcal mol
−1 with a GEDT of 0.10 e, at the 32CA involving DAA
10 these changes demand an EC of 20.9 kcal mol
−1 with a GEDT of 0.01 e; and finally, (iv) formation of the C3–C4 single bond takes place before the N1–C5 bond formation, by coupling of two C3 and C4
pseudoradical centers. While the former is already present at the reagents, the latter is created along the reaction path. This behavior confirms the
pmr-type mechanism [
36].
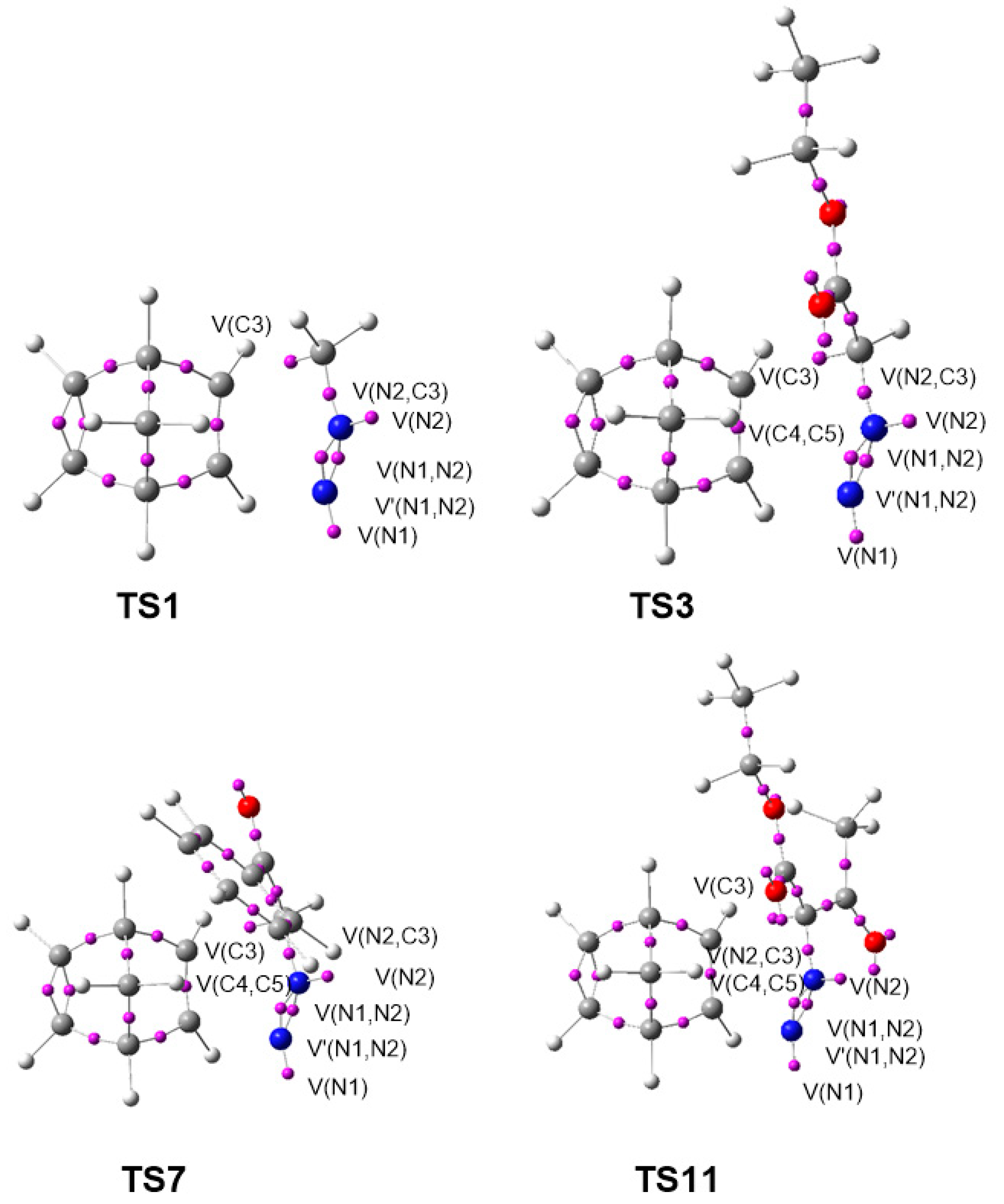
3.6. ELF Study at the TSs Associated with the 32CA Reactions of DAAs 7–10 with NBD 11
A comparative topological analysis of the ELF of the TSs involved in the 32CA reactions of DAAs
7–
10 with NBD
11 was performed. The ELF valence basin populations at
TS1–
TS14 associated with the 32CA reactions of DAAs
7–
10 with NBD
11 are given in
Table 5, while the ELF basin attractor positions of the
syn/endo TSs,
TS1,
TS3,
TS7, and
TS11, are represented in
Figure 6.
TS1–TS14 show the presence of a V(N2) monosynaptic basin integrating between 1.74 and 2.01 e, which is absent at the ground state electronic structures of the DAAs 7–10. The V(N1,N2) and V′(N1,N2) disynaptic basins are depopulated from 3.74 e at DAA 7 to 3.15 e at TS1 and 3.16 e at TS2, from 3.88 e at DAA 8 to 3.08–3.12 e at TS3–TS6, from 3.90 e at DAA 9 to 3.08–3.10 e at TS7–TS10, and from 3.97 e at DAA 10 to 3.01–3.07 e at TS11–TS14. On the other hand, the V(C3,N2) disynaptic basin integrating 3.01 e at DAA 7 is depopulated to 2.04 e at TS1 and 2.03 e at TS2, from 2.90 e at 8 to 2.00–2.04 e at TS3–TS6, from 2.88 e at 9 to 1.99–2.05 e at TS7–TS10, and from 2.82 e at 10 to 1.99–2.01 e at TS11–TS14. Thus, the N1–N2 and C3–N2 bonding regions are depopulated at the TSs to create the V(N2) monosynaptic basin associated with the N2 non-bonding electron density.
The V(C4,C5) and V′(C4,C5) disynaptic basins, integrating 3.48 e at NBD 11, are merged into one V(C4,C5) disynaptic basin, integrating 3.16–3.32 e at the TSs, due to the depopulation of that region.
As shown in
Figure 6, the four TSs,
TS1,
TS3,
TS7, and
TS11, present an identical electronic structure. Only small differences in the basin populations are observed as a consequence of the different substitutions at the DAA framework (see
Table 5).
The activation energies of the TSs is, therefore, associated with the EC for the formation of the N2 non-bonding electron density and the rupture of C4–C5 double bond of NBD
11. The TSs do not involve the formation of the new C3–C4 and N1–C5 single bonds, indicated by the absence of any V(C3,C4) and V(N1,C5) disynaptic basin. This is consistent with the distances between the C–C and N–C interacting centers, which are greater than 2 Å (see
Figure 2) and the positive Laplacian of electron density at the bond critical points (BCPs) of the interatomic bonding regions at the TSs, discussed in
Section 3.7.
3.8. Analysis of the Origin of the Deceleration of These 32CA Reactions with the Increase of the EW Character of the Substituents Present in These DAA
Many MEDT studies of cycloaddition reactions have shown that the activation energies of non-polar and polar reactions are mainly associated with the EC demanded the depopulation, i.e., rupture, of the X–Y double bond regions involved in the reaction [
36]. In polar reactions, this EC is decreased by the GEDT taking place along the reaction path [
74]. The very low GEDT found at the TSs associated with the 32CA reactions of DAA
8–
10, lower than 0.05 e (see
Table 3), indicates that they have a non-polar character. Consequently, the corresponding activation energies can mainly be associated with the changes in electron density in the N1–N2–C3 and C4–C5 bonding regions. Analysis of the total population of the V(N1), V′(N1), V(N1,N2), V′(N1,N2), and V(C3,N2) valence basins of DAAs
7–10, 3.01 e
7, 2.90 e
8, 2.88 e
9, and 2.82 e
10, shows a continuous depopulation of the N1–N2–C3 bonding region of these TACs with the increase of the EW character of the substituents present at the C3 carbon (see
Table 1).
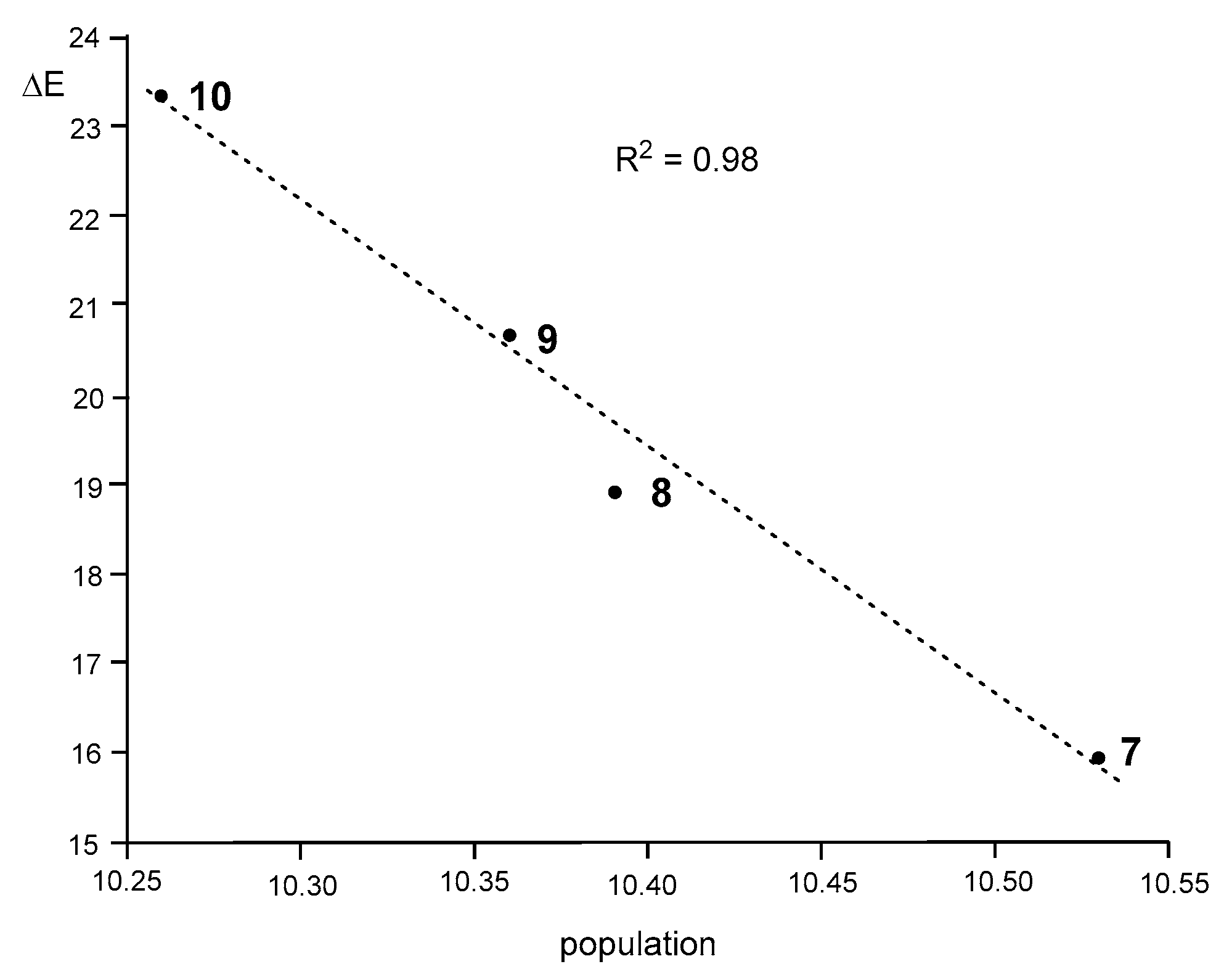
Interestingly, as
Figure 7 shows, a very good correlation between the total populations of the V(N1), V′(N1), V(N1,N2), V′(N1,N2), and V(C3,N2) valence basins and the activation energies of these 32CA reactions can be established (R
2 = 0.98).
As can be seen, the activation energies of this series of 32CA reactions linearly increase with the depopulation of the N1–N2–C3 bonding region, which could be understood as a drawback to using the electron density needed for the formation of the new bonds. Consequently, the lowest population found at the disubstituted DAA
10 provokes that the corresponding 32CA reaction presents the highest EC for the bonding changes, thus presenting the highest activation energy (see
Section 3.3). Note that the chemical hardness of
10 is the highest (see
Table 1). This finding justifies the observation that the 32CA reaction involving the most electrophilic species of this series does not take place experimentally.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}