Synthesis of Naphthoxazinones in a One-Pot Two-Step Manner by the Application of Propylphosphonic Anhydride (T3P®)


Abstract
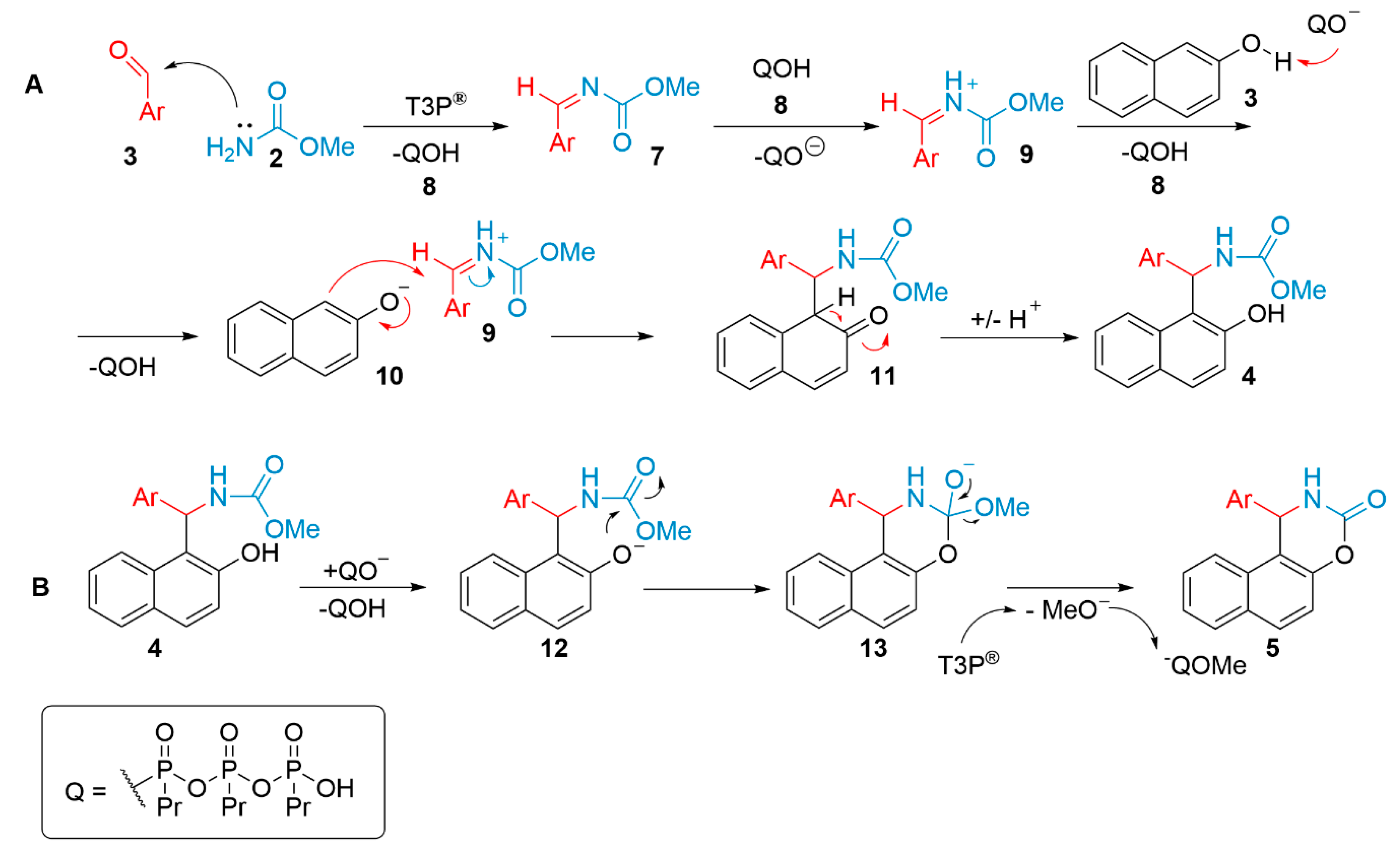
:
1. Introduction
2. Materials and Methods
2.1. General
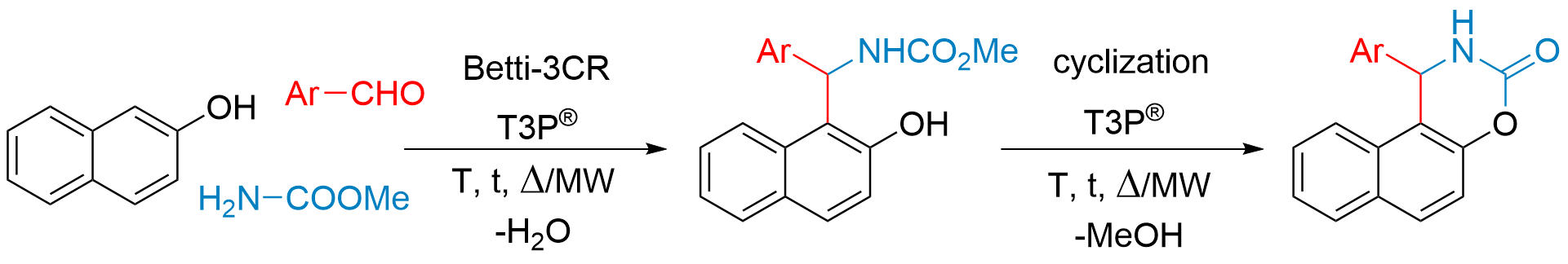
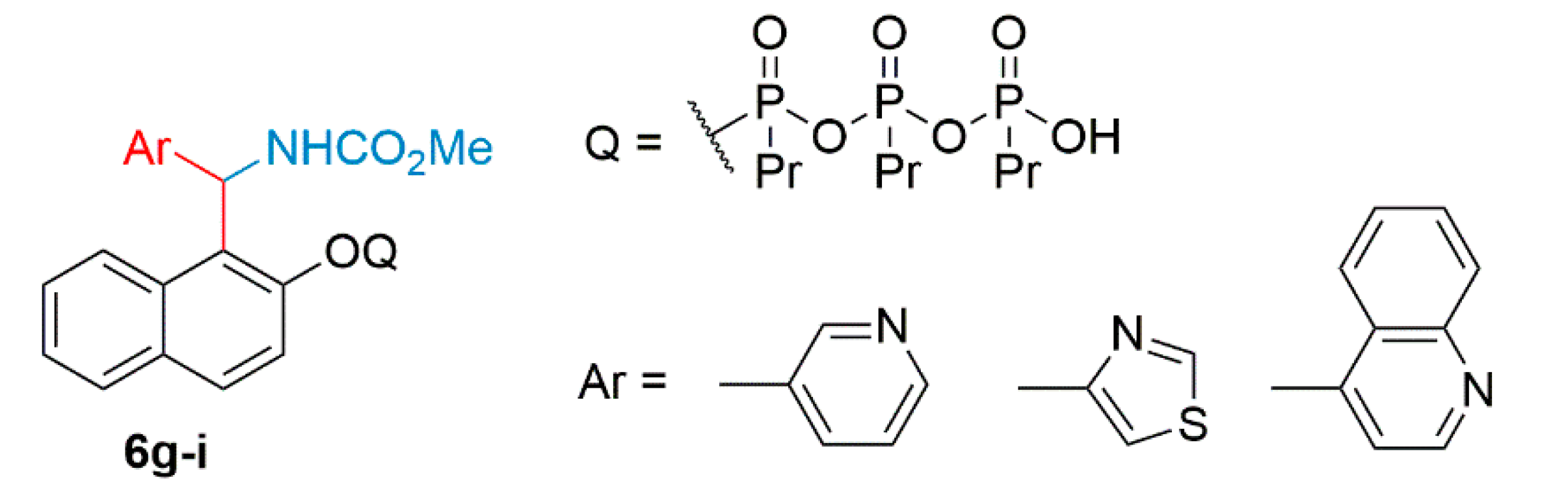
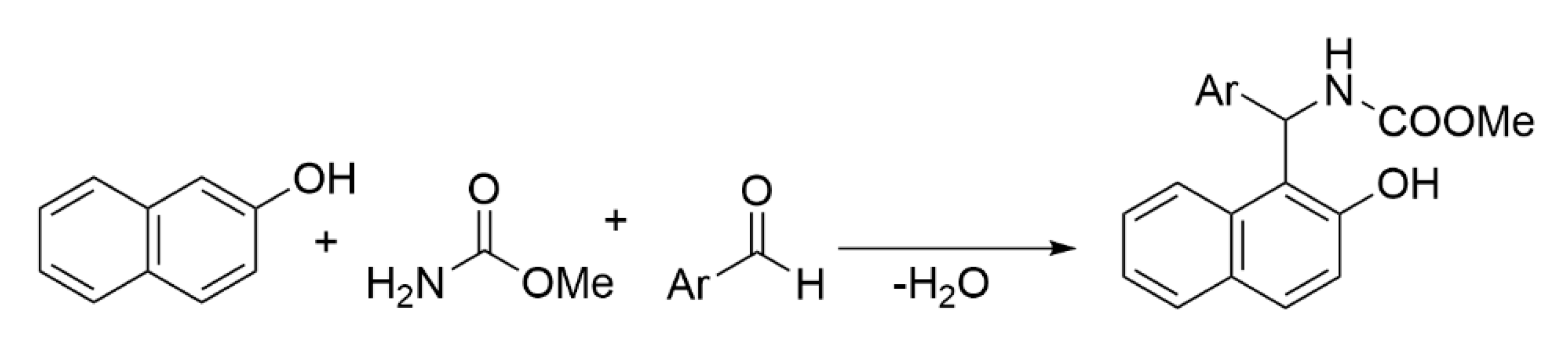
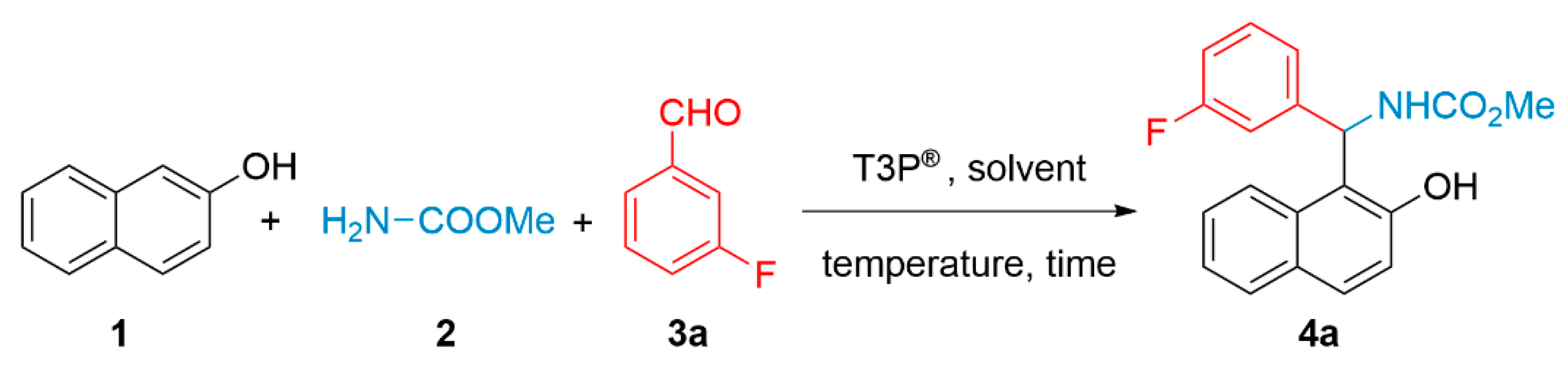
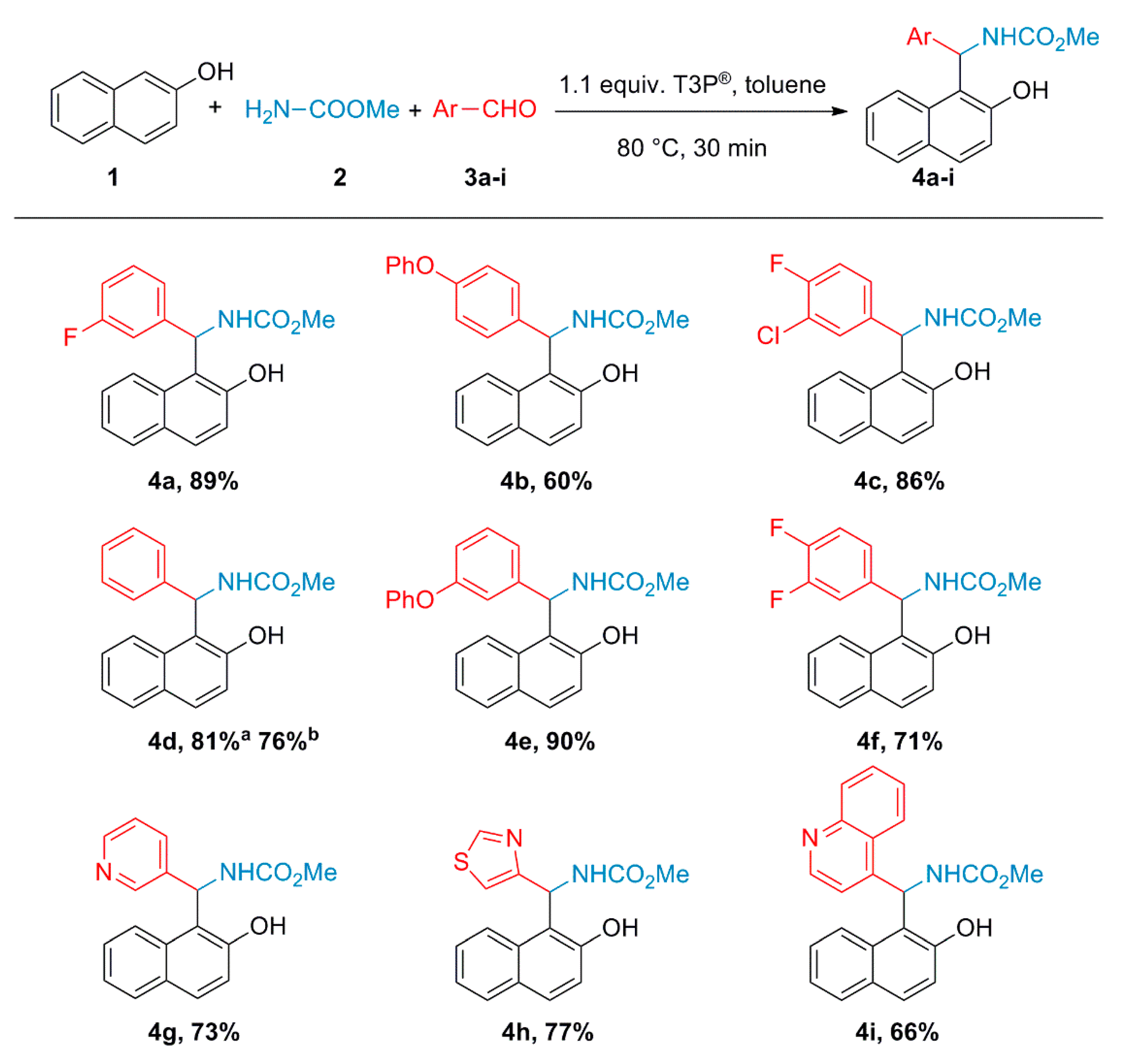
2.2. General Procedure for the Synthesis of 2-Hydroxynaphthalen-1-yl-carbamates (4a–i)
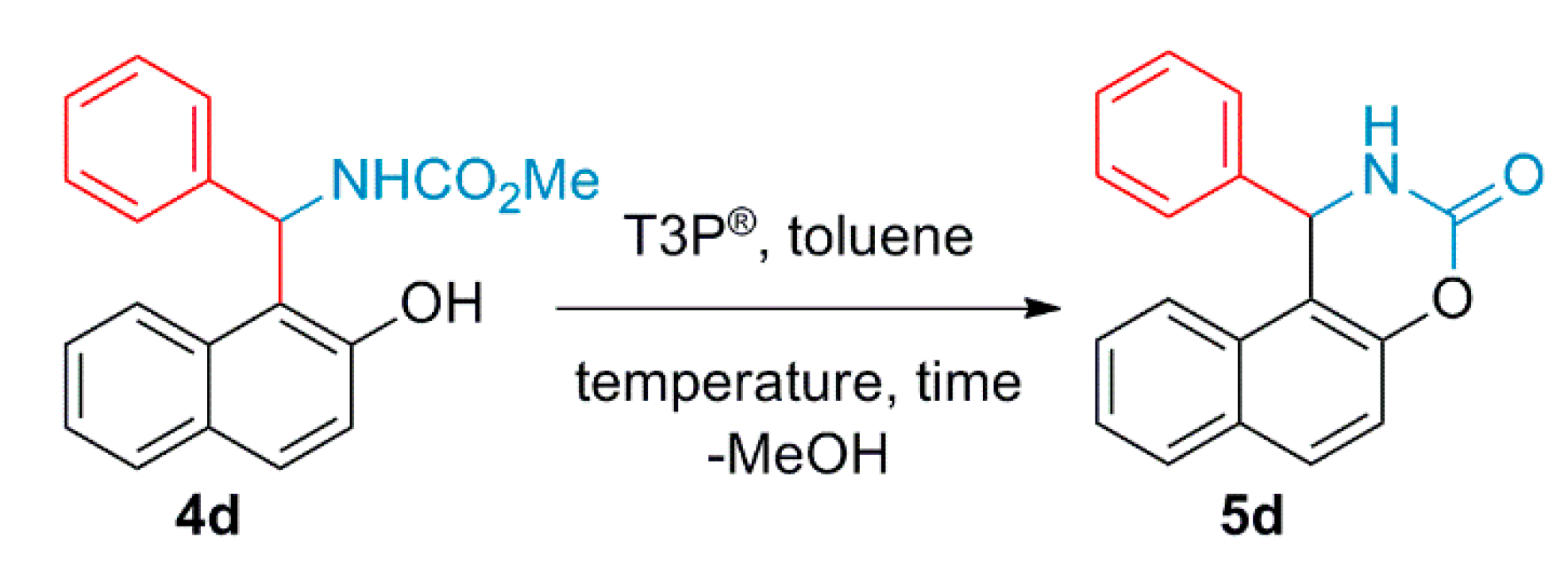
2.3. Ring-Closing Reaction from methyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)carbamate (4d) to naphta[1,2]oxazinone Derivatives (5d)
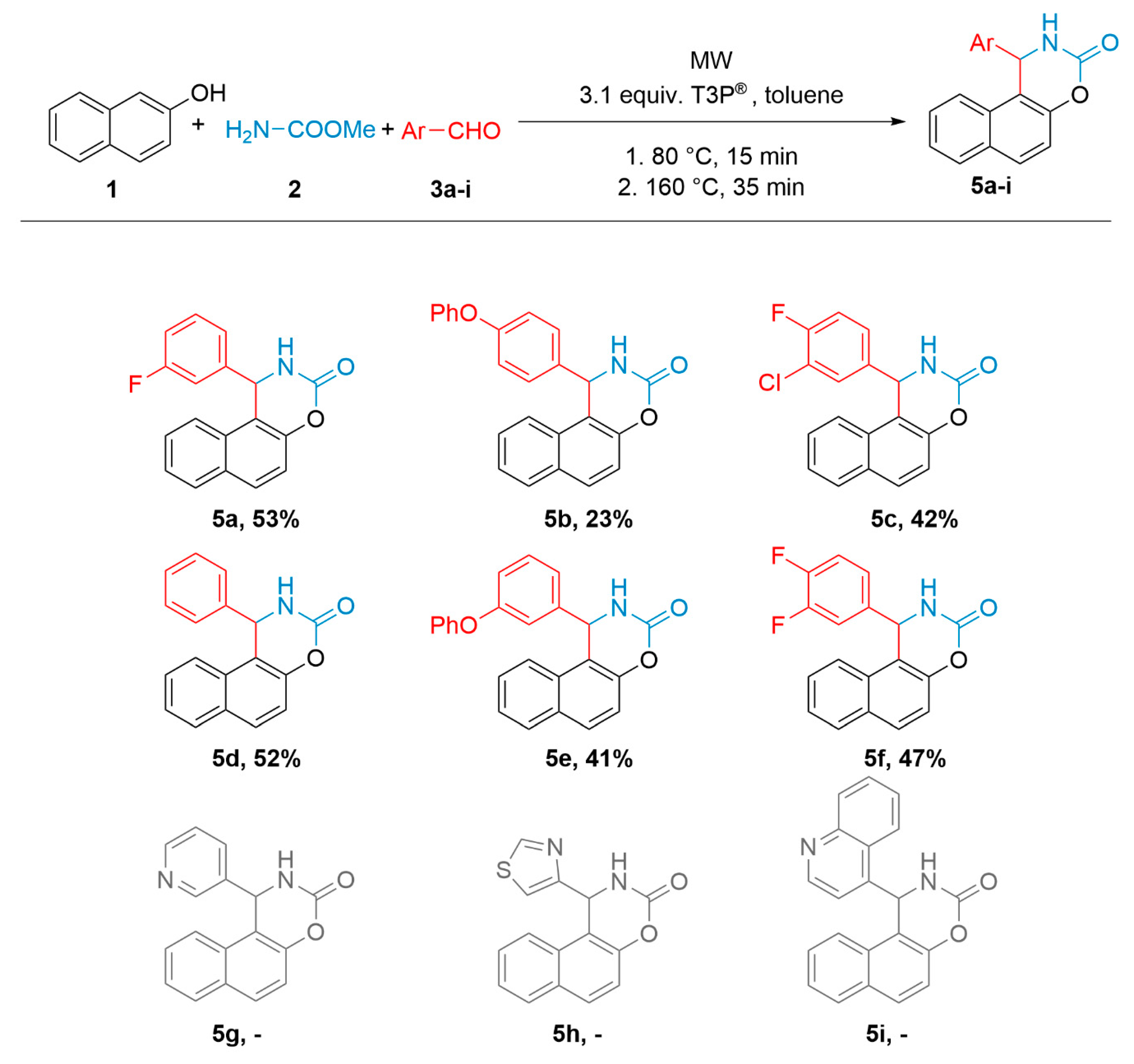
2.4. General Procedure for the Synthesis of naphta[1,2]oxazinones (5a–f)
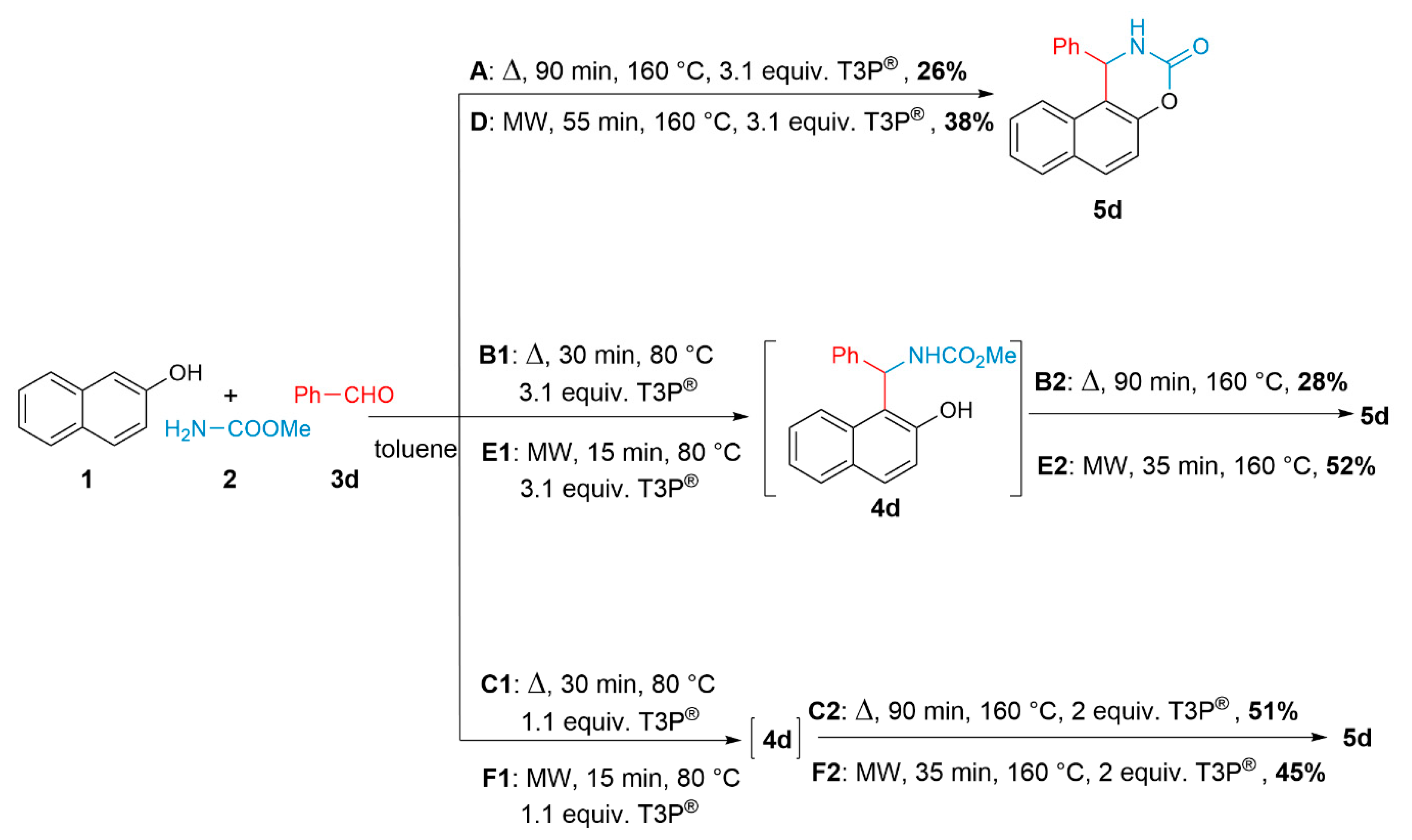
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ruijter, E.; Scheffelaar, R.; Orru, R.V.A. Multicomponent Reaction Design in the Quest for Molecular Complexity and Diversity. Angew. Chem. Int. Ed. 2011, 50, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Ruijter, E.; Orru, R.V.A. Recent applications of multicomponent reactions in medicinal chemistry. Med. Chem. Commun. 2012, 3, 1189–1218. [Google Scholar] [CrossRef]
- Afshari, R.; Shaabani, A. Materials Functionalization with Multicomponent Reactions: State of the Art. ACS Comb. Sci. 2018, 20, 499–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, Y.; Song, Z.; Zhao, S. A convenient three-component synthesis of carbamatoalkyl naphthols catalyzed by cerium ammonium nitrate. Bull. Chem. Soc. Ethiop. 2013, 27, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Zare, A.; Akbarzadeh, S.; Foroozani, E.; Kaveh, H.; Moosavi-Zare, A.R.; Hasaninejad, A.; Mokhlesi, M.H.; Zolfigol, M.A. Triethylamine-bonded sulfonic acid ([Et3N–SO3H]Cl): A highly efficient and homogeneous catalyst for the condensation of 2-naphthol with arylaldehydes and amides (alkyl carbamates or thioamides). J. Sulfur Chem. 2012, 33, 259–272. [Google Scholar] [CrossRef]
- Zare, A.; Merajoddin, M.; Moosavi-Zare, A.R.; Zarei, M.; Beyzavi, M.H.; Zolfigol, M.A. Design and characterization of nano-silica-bonded 3-n-propyl-1-sulfonic acid imidazolium chloride {nano-SB-[PSIM]Cl} as a novel, heterogeneous and reusable catalyst for the condensation of arylaldehydes with β–naphtol and alkyl carbamates. Res. Chem. Intermed. 2016, 42, 2365–2378. [Google Scholar] [CrossRef]
- Dadhania, H.N.; Raval, D.K.; Dadhania, A.N. Magnetically retrievable magnetite (Fe3O4) immobilized ionic liquid: An efficient catalyst for the preparation of 1-carbamatoalkyl-2-naphthols. Catal. Sci. Technol. 2015, 5, 4806–4812. [Google Scholar] [CrossRef]
- Dehghan, M.; Davoodnia, A.; Bozorgmehr, M.R.; Bamoharram, F.F. Evaluation of catalytic activity of two newly prepared functionalized sulfonic acids ionic liquids in the synthesis of carbamatoalkyl naphthols under mild conditions. Russ. J. Gen. Chem. 2017, 87, 311–318. [Google Scholar] [CrossRef]
- Tavakoli-Hoseini, N.; Heravi, M.M.; Bamoharram, F.F.; Davoodnia, A. Brønsted acidic ionic liquids as efficient catalysts for clean synthesis of carbamatoalkyl naphthols. Bull. Korean Chem. Soc. 2011, 32, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Dabiri, M.; Delbari, A.S.; Bazgir, A. A Simple and Environmentally Benign Method for the Synthesis of Naphthoxazin-3-one Derivatives. Heterocycles 2007, 71, 543–548. [Google Scholar] [CrossRef]
- Patel, N.; Katheriya, D.; Dadhania, H.; Dadhania, A. Graphene oxide supported dicationic ionic liquid: An efficient catalyst for the synthesis of 1-carbamatoalkyl-2-naphthols. Res. Chem. Intermed. 2019, 45, 5595–5607. [Google Scholar] [CrossRef]
- Kundu, D.; Majee, A.; Hajra, A. Zwitterionic-type molten salt: An efficient mild organocatalyst for synthesis of 2-amidoalkyl and 2-carbamatoalkyl naphthols. Catal. Commun. 2010, 11, 1157–1159. [Google Scholar] [CrossRef]
- Shafiee, M.R.M.; Molondi, R.; Ghashang, M. Preparation of Methyl (2-hydroxynaphthalen-1-yl)(aryl)methyl/benzylcarbamate derivatives using magnesium (II) 2,2,2-trifluoroacetate as an efficient catalyst. J. Chem. Res. 2011, 35, 622–625. [Google Scholar] [CrossRef]
- Yang, J.-M.; Jiang, C.-N.; Dong, H.; Fang, D. Synthesis of 1-carbamatoalkyl-2-naphthols in Tween® 20 Aqueous Micelles. J. Chem. Res. 2013, 37, 279–281. [Google Scholar] [CrossRef]
- Yarahmadi, H.; Shazerian, R. Sulfamic acid Functionalised Magnetic Nanoparticles: An Efficient Solid Acid for the Multicomponent Condensations. J. Chem. Res. 2012, 36, 52–55. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Q.L.; Zhao, S.; Wan, X. Solvent-free one-pot synthesis of 1-carbamatoalkyl-2-naphthols by a tin tetrachloride catalyzed multicomponent reaction. Monatsh. Chem. 2013, 144, 975–980. [Google Scholar] [CrossRef]
- Zare, A.; Kaveh, H.; Merajoddin, M.; Moosavi-Zare, A.R.; Hasaninejad, A.; Zolfigol, M.A. Saccharin Sulfonic Acid (SASA) as a Highly Efficient Catalyst for the Condensation of 2-Naphthol With Arylaldehydes and Amides (Thioamides or Alkyl Carbamates) Under Green, Mild, and Solvent-Free Conditions. J. Sulfur. Chem. 2013, 188, 573–584. [Google Scholar] [CrossRef]
- Shaterian, H.R.; Mohammadnia, M. Nanocrystalline TiO2–HClO4 catalyzed three-component preparation of derivatives of 1-amidoalkyl-2-naphtol, 1-carbamato-alkyl-2-naphthol, 1-(α-aminoalkyl)-2-naphthol, and 12-aryl-8,9,10,12-tetrahydrobenzo[α]-xanthen-11-one. Res. Chem. Intermed. 2013, 39, 4221–4237. [Google Scholar] [CrossRef]
- Ghashang, M. Preparation of methyl/benzyl(2-hydroxynaphthalen-1-yl)(aryl)methylcarbamate derivatives using magnesium hydrogen sulfate. Res. Chem. Intermed. 2014, 40, 1357–1364. [Google Scholar] [CrossRef]
- Shaterian, H.R.; Hosseinian, A.; Ghashang, M. Synthesis of New and Novel N-Protected 1-Aminoalkyl-2-naphtol Derivatives. Synth. Commun. 2009, 39, 2560–2574. [Google Scholar] [CrossRef]
- Khazaei, A.; Zolfigol, M.A.; Moosavi-Zare, A.R.; Abi, F.; Zare, A.; Kaveh, H.; Khakyzadeh, V.; Kazem-Rostami, M.; Parhami, A.; Torabi-Monfared, H. Discovery of an in situ carbocationic system using trityl chloride as a homogeneous organocatalyst for the solvent-free condensation of β-naphthol with aldehydes and amides/thioamides/alkyl carbamates in neutral media. Tetrahedron 2013, 69, 212–218. [Google Scholar] [CrossRef]
- Shaterian, H.R.; Hosseinian, A.; Ghashang, M. A three-component novel synthesis of 1-carbamato-alkyl-2-naphthol derivatives. Tetrahedron Lett. 2008, 49, 5804–5806. [Google Scholar] [CrossRef]
- Ghasemi, A.; Davoodnia, A.; Pordel, M.; Tavakoli-Hoseini, N. P2O5 supported on SiO2 as an efficient and reusable catalyst for rapid one-pot synthesis of carbamatoalkyl naphthols under solvent-free conditions. Cogent Chem. 2017, 3, 1317582. [Google Scholar] [CrossRef]
- Shen, A.Y.; Tsai, C.T.; Chen, C.L. Synthesis and cardiovascular evaluation of N-substituted 1-aminomethyl-2-naphthols. Eur. J. Med. Chem. 1999, 34, 877–882. [Google Scholar] [CrossRef]
- Latif, N.; Mishriky, N.; Assad, F.M. Carbonyl and thiocarbonyl compounds. XIX. Intramolecular cyclization of (2-nitroetheny1)aryl N-arylcarbamates: Synthesis of newer series of 3,4-dihydro-2H-1,3-oxazin-2-ones and their antimicrobial activities. Aust. J. Chem. 1982, 35, 1037–1043. [Google Scholar] [CrossRef]
- Wissmann, H.; Kleiner, H.-J. New Peptide Synthesis. Angew. Chem. Int. Ed. 1980, 19, 133–134. [Google Scholar] [CrossRef]
- Vishwanatha, B.T.M.; Panguluri, N.R.; Sureshbabu, V.V. Propanephosphonic Acid Anhydride (T3P®)—A Benign Reagent for Diverse Applications Inclusive of Large-Scale Synthesis. Synthesis 2013, 45, 1569–1601. [Google Scholar]
- Waghmare, A.A.; Hindupur, R.M.; Pati, H.N. Propylphosphonic anhydride (T3P®): An expedient reagent for organic synthesis. Rev. J. Chem. 2014, 4, 53–131. [Google Scholar] [CrossRef]
- Henyecz, R.; Milen, M.; Kánai, K.; Keglevich, G. Organophosphorus Chemistry: Novel Developments; De Gruyter: Berlin, Germany, 2018. [Google Scholar]
- Milen, M.; Ábrányi-Balogh, P.; Dancsó, A.; Frigyes, D.; Pongó, L.; Keglevich, G. T3P®-promoted Kabachnik–Fields reaction: An efficient synthesis of α-aminophosphonates. Tetrahedron Lett. 2013, 54, 5430–5433. [Google Scholar] [CrossRef]
- Milen, M.; Dancsó, A.; Földesi, T.; Slégel, P.; Volk, B. Propylphosphonic anhydride (T3P®) mediated one-pot three-component synthesis of racemic dialkyl (2-substituted-3-oxo-2,3-dihydro-1H-isoindol-1-yl)phosphonates. Tetrahedron 2016, 72, 5091–5099. [Google Scholar] [CrossRef]
- Milen, M.; Dancsó, A.; Földesi, T.; Volk, B. Study on the propylphosphonic anhydride (T3P®) mediated Ugi-type three-component reaction. Efficient synthesis of an α-amino amide library. Tetrahedron 2017, 73, 70–77. [Google Scholar] [CrossRef]
- Varga, V.; Milen, M.; Ábrányi-Balogh, P. Propylphosphonic anhydride (T3P®) mediated synthesis of 3-oxoisoindoline-1-carboxamides from 2-formylbenzoic acid, amines, and isocyanides. Preparation of isoindolinone alkaloids. Tetrahedron Lett. 2018, 59, 3683–3689. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Földesi, T.; Grün, A.; Volk, B.; Keglevich, G.; Milen, M. Synthetic study on the T3P®-promoted one-pot preparation of 1-substituted-3,4-dihydro-β-carbolines by the reaction of tryptamine with carboxylic acids. Tetrahedron Lett. 2016, 57, 1953–1957. [Google Scholar] [CrossRef]
- Rao, G.B.D.; Kaushik, M.P.; Halve, A.K. An efficient synthesis of naphtha[1,2-e]oxazinone and 14-substituted-14H-dibenzo[a,j]xanthene derivatives promoted by zinc oxide nanoparticle under thermal and solvent-free conditions. Tetrahedron Lett. 2012, 53, 2741–2744. [Google Scholar]
- Dabiri, M.; Delbari, A.S.; Bazgir, A. A Novel Three-Component, One-Pot Synthesis of 1,2-Dihydro-1-arylnaphtho[1,2-e][1,3]oxazine-3-one Derivatives under Microwave-Assisted and Thermal Solvent-Free Conditions. Synlett 2007, 5, 821–823. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 1/2/3a | T3P® (equiv.) | Time (h) | Solvent | Temperature (°C) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 1/1/1 | 1 | 26 | EtOAc | r.t. | 32 |
| 2 | 1/1/1 | 1 | 2 | EtOAc | 77 | 53 |
| 3 | 1/1/1 | 0 | 2 | EtOAc | 77 | 0 |
| 4 | 1/1.1/1.1 | 1.1 | 2 | EtOAc | 77 | 83 |
| 5 | 1/1.3/1.3 | 1.3 | 2 | EtOAc | 77 | 83 |
| 6 | 1/1.1/1.1 | 1.5 | 2 | EtOAc | 77 | 81 |
| 7 | 1/1.1/1.1 | 5 | 5 | EtOAc | 77 | 73 |
| 8 | 1/1.1/1.1 | 1.1 | 2 | THF | 66 | 90 |
| 9 | 1/1.1/1.1 | 1.1 | 2 | MeCN | 82 | 73 |
| 10 | 1/1.1/1.1 | 1.1 | 2 | CHCl3 | 62 | 89 |
| 11 | 1/1.1/1.1 | 1.1 | 2 | MTBE | 65 | 83 |
| 12 | 1/1.1/1.1 | 1.1 | 2 | MeTHF | 80 | 85 |
| 13 | 1/1.1/1.1 | 1.1 | 2 | PhMe | 80 | 91 |
| 14 | 1/1.1/1.1 | 1.1 | 0.5 | PhMe | 80 | 89 |
| Entry | T3P® (equiv.) | Temperature (°C) | Reaction Time | Yield (%) |
|---|---|---|---|---|
| 1 | - | 80 | 26 h | n.r. a |
| 2 | 1.1 | 80 | 30 min | 0 b |
| 3 | 1.1 | 135 | 4 h | 0 b |
| 4 | 2 | 135 | 70 min | 0 b |
| 5 | 2 | 160 | 60 min | 75 |
| 6 | 1.1 | 160 | 90 min | 47 |
| 7 | 3 | 160 | 45 min | 50 |
| 8 | 2 | 135 c | 90 min | 18 |
| 9 | 2 | 160 c | 30 min | 36 |
| Method | Number of Steps | Reaction Time (min) | Temperature (°C) | T3P® (equiv.) | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| MW/∆ | 1 or 2 | Step1 | Step2 | Step1 | Step2 | Step1 | Step2 | ||
| A | ∆ | 1 | 90 | - | 160 | - | 3.1 | - | 26 |
| B | ∆ | 2 | 30 | 90 | 80 | 160 | 3.1 | - | 28 |
| C | ∆ | 2 | 30 | 90 | 80 | 160 | 1.1 | 2.0 | 51 |
| D | MW | 1 | 55 | - | 160 | - | 3.1 | - | 38 |
| E | MW | 2 | 15 | 35 | 80 | 160 | 3.1 | - | 52 |
| F | MW | 2 | 15 | 35 | 80 | 160 | 1.1 | 2.0 | 45 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varga, V.; Ábrányi-Balogh, P.; Milen, M. Synthesis of Naphthoxazinones in a One-Pot Two-Step Manner by the Application of Propylphosphonic Anhydride (T3P®). Chemistry 2020, 2, 600-612. https://doi.org/10.3390/chemistry2020037
Varga V, Ábrányi-Balogh P, Milen M. Synthesis of Naphthoxazinones in a One-Pot Two-Step Manner by the Application of Propylphosphonic Anhydride (T3P®). Chemistry. 2020; 2(2):600-612. https://doi.org/10.3390/chemistry2020037
Chicago/Turabian StyleVarga, Valentina, Péter Ábrányi-Balogh, and Mátyás Milen. 2020. "Synthesis of Naphthoxazinones in a One-Pot Two-Step Manner by the Application of Propylphosphonic Anhydride (T3P®)" Chemistry 2, no. 2: 600-612. https://doi.org/10.3390/chemistry2020037