1. Introduction
The concept of strain has long held interest for organic chemists, going back all the way to Baeyer [
1,
2,
3,
4,
5]. Strain refers to the amount by which the energy of a molecule exceeds that which one would expect if all bond lengths, bond angles, and dihedral angles could simultaneously hold their ideal values, and if no repulsive nonbonded interactions (steric repulsions) were present. As such, strain is generally assumed to be absent in molecules such as straight-chain alkanes in which the bond lengths, angles, and dihedral angles are not geometrically constrained, and in which the extended conformation avoids repulsive nonbonded interactions. Small rings, on the other hand, force bond angles to be smaller than ideal, and lead to other nonidealities (such as torsional strain) as well. In highly strained molecules, bond angle and steric strain are almost always the main contributors to the overall strain energy [
5]. Syntheses of a wide variety of highly strained compounds have been carried out in ingenious ways, allowing the experimental study of these elusive species. It is frequently of interest to quantify the strain, generally as an energy of some sort, and many approaches exist for doing so [
2,
3,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]. Most of these approaches rely, either explicitly or implicitly, on comparison of the molecular energy to that of a “strain-free” reference system. It is only in describing such a procedure that the somewhat fuzzy concept of strain becomes precisely, if also somewhat arbitrarily, defined.
One straightforward approach is to use isodesmic [
16], homodesmotic [
14], or group equivalent reactions [
17], in which the reactants and products of a hypothetical reaction are paired so to isolate the source of strain from other contributing factors. Thus, for instance, one can design a reaction in which the reactant and product sides have equal numbers of bonds of a given type, and all compounds but the single compound of interest can reasonably be assumed to be free of strain. The energy of the reaction, obtained either by experimental or computational means, can then be associated with the strain. Of course, different levels of exactitude are possible regarding what is meant by “bond type”. Wheeler et al. have provided careful and elegant definitions of different orders of homodesmotic reactions that provide progressively more complete definitions of “bond types” and “atom types,” and thus, in principle, more precisely defined strain energies [
18].
Another approach involves comparing the experimental heat of formation of a given compound to a hypothetical “strain free” value derived from a more general model. These models rely on the additivity of the energies of molecular fragments, a phenomenon that has long been recognized and often used, and that holds remarkably accurately for even quite generic fragments [
19,
20,
21,
22,
23,
24,
25,
26,
27]. For instance, both Franklin [
24] and Benson [
19,
20,
21] pioneered the notion of “group equivalents” that could be used to estimate the heat of formation for a novel structure, based on patterns in the known experimental data. A simple approach is to use the number of methyl, methylene, methane, and quaternary carbon groups, plus the number of alkene functional groups, to estimate an enthalpy of formation for a hydrocarbon. As the increments are based on data for unstrained compounds, one can define as the strain energy the difference between the actual, experimental enthalpy of formation and the estimate obtained by summing the unstrained increments. Along similar lines, Benson defined a far more extensive set of group equivalents, permitting a more precise prediction of strain-free enthalpy. This procedure is, for instance, presented in a leading advanced organic chemistry textbook [
28].
Computational methods for assessing strain follow the same patterns as experimental methods. A common and versatile approach is simply to compute energies for the components of an isodesmic or homodesmotic reaction using electronic structure theory. Another approach is to use computational methods to obtain an enthalpy of formation, which can then be compared to strain-free estimates generated by Franklin’s or Benson’s methods [
29]. A still more direct approach, however, is to use computational group equivalents: that is, to develop group increments that permit the estimation of a strain-free electronic energy, that can then be directly compared to the result of an actual electronic structure calculation for the compound of interest. The intermediate step of predicting an enthalpy of formation is thus avoided. Wiberg [
30,
31] first used such an approach in 1984, when HF/6-31G(d) represented a fairly high level of calculation, and Schleyer [
32] further elaborated the scheme.
This direct computational technique offers several advantages. First, once the group increments for a given calculational level are available, only one electronic structure calculation is required to obtain a strain energy for a new molecule of interest: a calculation of that molecule. That stands in contrast to the isodesmic/homodesmotic approach, in which all components of the reaction must be computed. Perhaps more importantly, the approach is conceptually more direct; it removes the unnecessary intermediate step of estimating an experimental heat of formation from an electronic structure calculation, as well as the additional labor and potential sources of error thereby introduced. Finally, one might argue that chemists are most interested, conceptually speaking, in the strain as defined in the pure essence of an electronic energy, without the complications of thermodynamic factors that affect enthalpies at 298K. In such a sense, the ability to define strain energies in terms of energy/enthalpy at absolute zero (with only the zero-point energy as a thermodynamic correction), and in the absence of medium effects, is perhaps a conceptual advantage.
The computational group equivalent approach first explored by Wiberg is thus a valuable one. However, the original version involves electronic structure methods that are suboptimal by today’s standards (HF/6-31G(d)), as well as a very simple and thus somewhat limited definition of the strain-free reference. Here, the approach is updated and expanded in two ways. First, a much wider variety of group equivalents is used, following the approach of Benson rather than of Franklin, permitting both a wider variety of hydrocarbons to be considered, and also providing a somewhat more precisely calibrated definition of the strain-free reference than is possible using more limited definitions. Second, the approach is modernized by using highly accurate compound procedures of the type available and routinely used today: W1BD [
33], G-4 [
34], CBS-APNO [
35], and CBS-QB3 [
36,
37], as well as a modern density functional method, M062X/6-31+G(2df,p) [
38], that was found in a previous study to offer results in generally good accord with the aforementioned multi-component procedures [
39].
3. Results
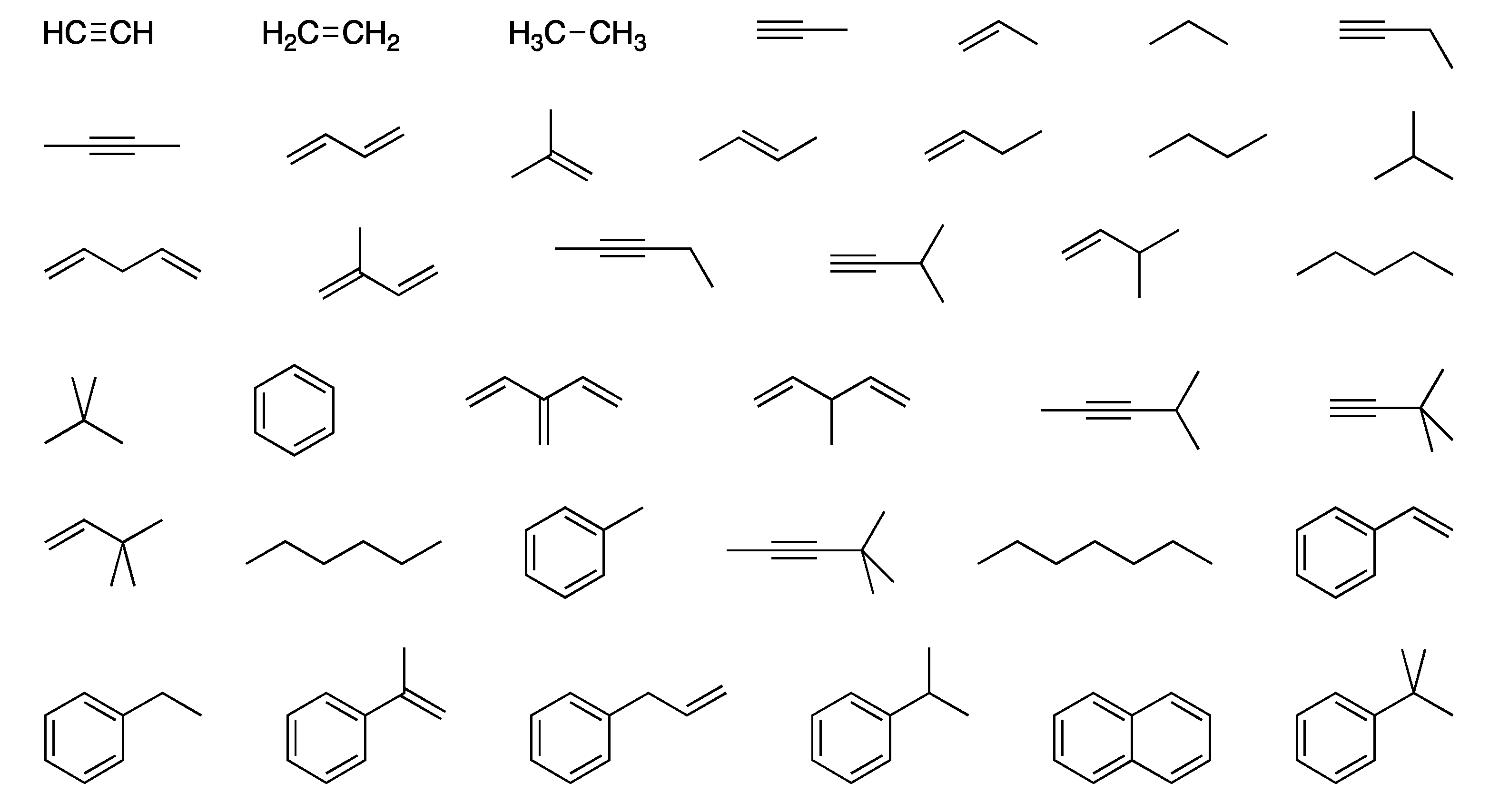
Table 1 lists increments in the calculated electronic energy for a methylene group on going progressively from ethane to octane. The increments are highly consistent: they vary by just a few tenths of a millihartree. However, there is a perceptible alternation in the numbers; e.g., the W1BD value is –39.29347 ± 0.00001 on going from an even to an odd chain, but –39.29359 ± 0.00000 on going from an odd to an even chain. By taking (heptane – propane)/4 to define methylene, we attempt to average out this alternation. More generally, however, the high degree of constancy of the increments lends credence to the approach of adding together largely context-independent group increment energies to obtain a strain-free reference energy for a molecule.
Table 2 lists definitions of the various group equivalents, which are generally based on the simplest example (or two, in some cases) providing the desired “type” of atom.
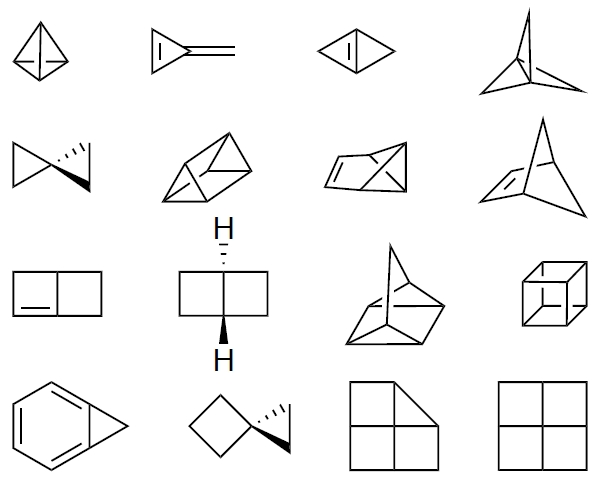
Figure 1 shows the full set of compounds used for this purpose. There is some indeterminacy that results from the fact that one can define more reasonable atom types than corresponding examples. Following Benson, we have chosen to consider all methyl groups equivalent, as a way to address this indeterminacy. We have also included some increments that are suitable for alkynes (Ct carbons), that Benson did not originally define.
Table 3 and
Table 4 list the values obtained for the group increments defined in
Table 2 using five electronic structure methods: W1BD, G-4, CBS-QB3, CBS-APNO, and M062X/6-31G(2df,p), as enthalpies either at 0 K (
Table 3) or at 298 K (
Table 4). To illustrate the approach, three worked examples are provided below, and are also illustrated in
Figure 2.
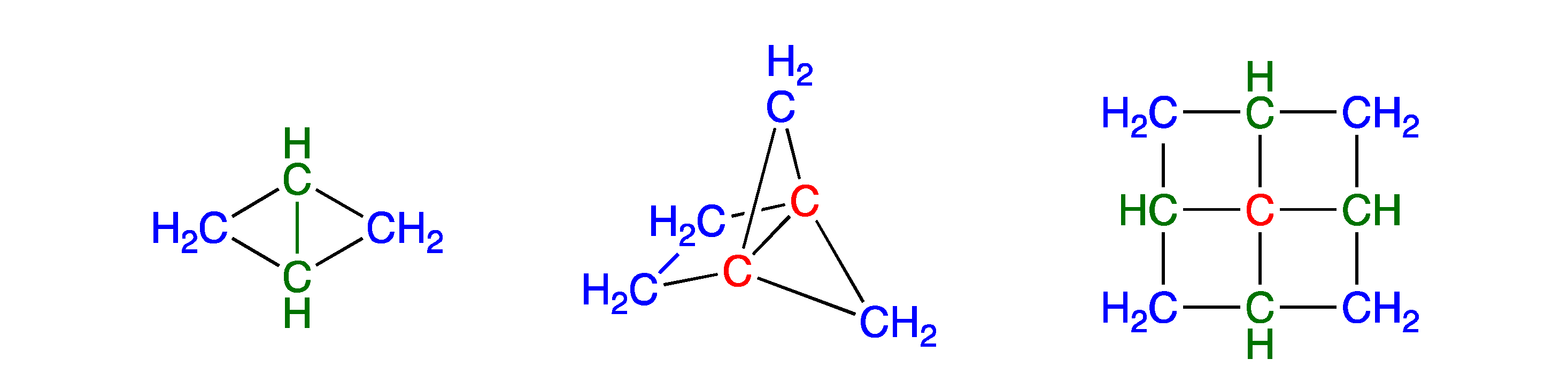
Example 1.
Bicyclobutane at 0 K using W1BD:| W1BD calculation: | −155.89922 |
| Increments: | |
| 2 × C–(H)2(C)2 | 2 × −39.29485 |
| 2 × C–(H)(C)3 | 2 × −38.70779 |
| Sum: | −156.00527 |
| Difference: | 0.10605 = 66.5 kcal/mol strain energy |
Example 2.[2.1.1]propellane at 298 K using G-4:| G-4 calculation: | −233.15571 |
| Increments: | |
| 4 × C–(H)2(C)2 | 4 × −39.27688 |
| 2 × C–(C)4 | 2 × −38.10499 |
| Sum: | −233.31748 |
| Difference: | 0.16178 = 101.5 kcal/mol strain energy |
Example 3.[4.4.4.4]fenestrane at 0 K using CBS-QB3:| CBS-QB3 calculation: | −349.250562 |
| Increments: | |
| 4 × C–(H)2(C)2 | 4 × −39.22572 |
| 4 × C–(H)(C)3 | 4 × −38.63907 |
| 1 × C–(C)4 | 1 × −38.05411 |
| Sum: | −349.51324 |
| Difference: | 0.26267 = 164.8 kcal/mol strain energy |
Table 5 and
Figure 3 show calculated strain energies for a variety of interesting hydrocarbons.
Table S1 in the Supporting Information lists the group equivalents used to define the strain-free reference for each molecule. Examples have been restricted to cases in which it is reasonable to assume a single conformation is dominant, obviating the need for conformational averaging or extensive conformational searching. Some molecules that are expected to be largely strain free, such as various cyclohexane and adamantane derivatives, have purposely been included.
4. Discussion
At least for the monocyclic cases, the strain energies in
Table 5 and
Figure 3 agree very well, typically within 1–2 kcal/mol, with previous estimates, both computational and experimental, such as those compiled by Liebman and Greenberg [
2,
3], Wiberg [
5], Anslyn and Dougherty [
29], Castaño and Notario [
42], Schleyer [
43], Ibrahim [
44], Davis [
10], Oth and Berson [
45], and Doering [
46], to name a few. Agreement is generally good (within 3 kcal/mol) for the more complex structures as well, although there are a few notable differences. For instance, the strain estimated here for quadricyclane (94.2 kcal/mol) agrees very closely with that reported by Doering and by Berson on an experimental basis (95–96 kcal/mol), although not at all well with that obtained by Davison on a purely computational basis (71 kcal/mol). The strain for norbornene (20.1) differs substantially from Schleyer’s (27.2), but agrees well with Wiberg’s (21.1). Similarly, the strain energies for spiropentane (62.9) and cubane (157.5) differ substantially from Schleyer’s (65.0 and 166.0), but are fairly close to Wiberg’s (63.2 and 154.7).
The spiroalkanes and cubane illustrate the principle of ring strain additivity and its limitations. The strain of spirohexane, spiro[3.3]octane, spiro[5.5]undecane, and cubane quite closely parallel the sums of the strain energies of the constituent rings: cyclopropane and cyclobutane (54.7 kcal/mol) for spirohexane (54.9 kcal/mol), twice cyclobutane (53.6 kcal/mol) for spiro[3.3]octane (51.0 kcal/mol), twice cyclohexane (4.2 kcal/mol) for spiro[5.5]undecane (3.6 kcal/mol), and six times cyclobutane (160.8 kcalmol) for cubane (157.5 kcal/mol. The strain energy of spiropentane (62.9 kcal.mol), however, significantly exceeds that of two cyclopropane rings (54.8 kcal/mol). Wiberg has noted that this happens because the central carbon is forced to adopt sp
3 hybridization, whereas in cyclopropane, the carbons are closer to sp
2 hybridization [
5]. A similar phenomenon is observed for bicyclobutane, for which the computed strain energy of 66.5 kcal/mol exceeds the sum for two cyclopropane rings by 10.7 kcal/mol. The strain of [2.1.0]bicyclopentane (55.6 kcal/mol), on the other hand, closely matches the sum for cyclopropane and cyclobutane (54.7 kcal/mol) (as well as Wiberg’s estimate of 54.7 kcal/mol) [
5].
Prismane is a somewhat intermediate case: its strain of 142.0 kcal/mol exceeds the sum of three cyclobutanes and two cyclopropanes (136.2 kcal/mol), but only by 5.8 kcal/mol. The small-ring fenestranes, on the other hand, exhibit strain far exceeding what would be expected on the basis of ring strain additivity. That is not surprising, given the tremendous distortion of the central carbon, which is forced to be close to planar. The strain energies of [3.4.4.4], [4.4.4.4], and [4.4.4.5]fenestrane exceed the corresponding sums of the strain energies of the constituent rings by 103.2, 57.5, and 17.7 kcal/mol, respectively.
The propellanes present another interesting comparison. The strain has previously been reported to increase from 98 to 104 to 105 kcal/mol on going from [1.1.1] to [2.1.1] to [2.2.1]propellane, before dropping to 89 kcal/mol for [2.2.2]propellane [
5]. This sequence seems surprising; one would expect that each replacement of a cyclopropane ring with a cyclobutane ring ought to decrease the strain by 1 kcal/mol or so, not increase it. The values computed here better match these expectations. The strain remains essentially constant, going from 99.2 to 99.6 to 99.7, along the sequence [1.1.1] to [2.1.1] to [2.2.1]propellane, before dropping to 93.9 for [2.2.2]propellane [
47].
The estimate here for norbornadiene, 28.6 kcal/mol, is substantially lower than several recent estimates that are in the range of 32–35 kcal/mol, [
42,
43,
48] although in good agreement with Doering’s original experimentally-based estimate of 29.0 kcal/mol [
46]. The difference results from somewhat alternative views of what the strain-free reference should be; for instance, are 1,4 interactions (such as a gauche butane interaction) to be considered part of the strain, or part of the reference against which strain is judged?
In the end, one cannot really view the differences in these strain estimates as “errors”. Of course, inaccuracies in either experimental measurements or calculated energies contribute to the differences, and that can particularly be true of older calculations performed at a time when large basis sets and proper accounting of electron correlation were not feasible. However, a significant amount of the difference also originates from differences in how the strain-free reference state is defined. Philosophically, the approach taken here follows very closely that described by Schleyer in 1970 [
43]. He recommended to use a wider set of parameters than just the number of CH
3, CH
2, CH, and C groups and alkene functionalities (as in the original Franklin scheme), and also to use what he termed “single conformation group increments”. Using experimental enthalpies of formation obtained at normal temperatures yields values that include some contributions from conformations higher in energy than the global minimum. He argued that including these contributions resulted in a somewhat inaccurate estimate of the true strain-free energy. The same view is taken here. While, when using experimental data, it is laborious to subtract out these contributions, using computational methods makes it simple and natural not to include them in the first place. From this perspective, the strain energies presented here, and the method used to compute them, should correspond especially closely to what organic chemists intuitively mean by the concept of strain.
One could of course imagine defining an even more extensive set of group equivalents designed to take into account non-next-nearest neighbor interactions. Arguably, doing so would provide an even more precise accounting for strain, at least if these non-next-nearest neighbor interactions are not regarded as part of the strain. However, taking such an approach would greatly increase the number of group equivalents required, and likely result in only very small changes to the computed strain energies. Furthermore, it is worth noting that the reference molecules listed in
Figure 1, and implicitly assumed to be strain free, were chosen so as to minimize any such non-next-nearest neighbor interactions. For instance, there are no alkane gauche interactions. The use of single, minimum-energy conformations means that the linear alkanes rigorously avoid such interactions, and the only branched alkanes (2-methylpropane and 2,2-dimethylpropane) lack a 4-carbon chain. Similarly, there are no cis alkenes. Unfortunately, there is likely some 1,3-allylic strain in 3,3-dimethyl-1-butene and in
t-butylbenzene, as well as perhaps in 2-methy-1,3-butadiene and alpha-methylstyrene. This could lead to a slight underestimate of strain energies when the parameters that rely on these four molecules are used, insofar as these particular parameters include a small amount of inherent strain.
It is interesting that the various cyclohexane derivatives, including trans-decalin and adamantane, are not calculated to be entirely strain free. Indeed, adamantane is calculated to have a rather substantial 6 kcal/mol of strain. Schleyer explored this issue in detail in 1970, and explained the strain in all these cases as resulting from a combination of angle strain, transannular C…C repulsion, and also an attractive interaction resulting from anti arrangements of CCCC fragments [
43]. The data from the present study fit these interpretations. Roughly speaking, each cyclohexane ring provides 2 kcal/mol of strain, but each methyl that is not axial (or gauche to another methyl) reduces the strain by 1 kcal/mol. Consistent with Schleyer’s explanation, each such methyl group indeed contributes two (in cyclohexane) or three (in adamantane) “anti-butane” configurations of the sort that he postulated to be stabilizing. In addition, the quaternary carbons that result from methyl substitution of the tertiary carbons in adamantane would be expected to have almost perfectly tetrahedral bond angles, thus reducing angle strain. A similar effect is likely at work in methyl-substituted cyclohexanes.







{kind=link}
{kind=link}
{kind=link}
{kind=link}