
Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Cause of the UPR
3. The UPR in Physiological and Pathological Conditions (Particularly, IBD)
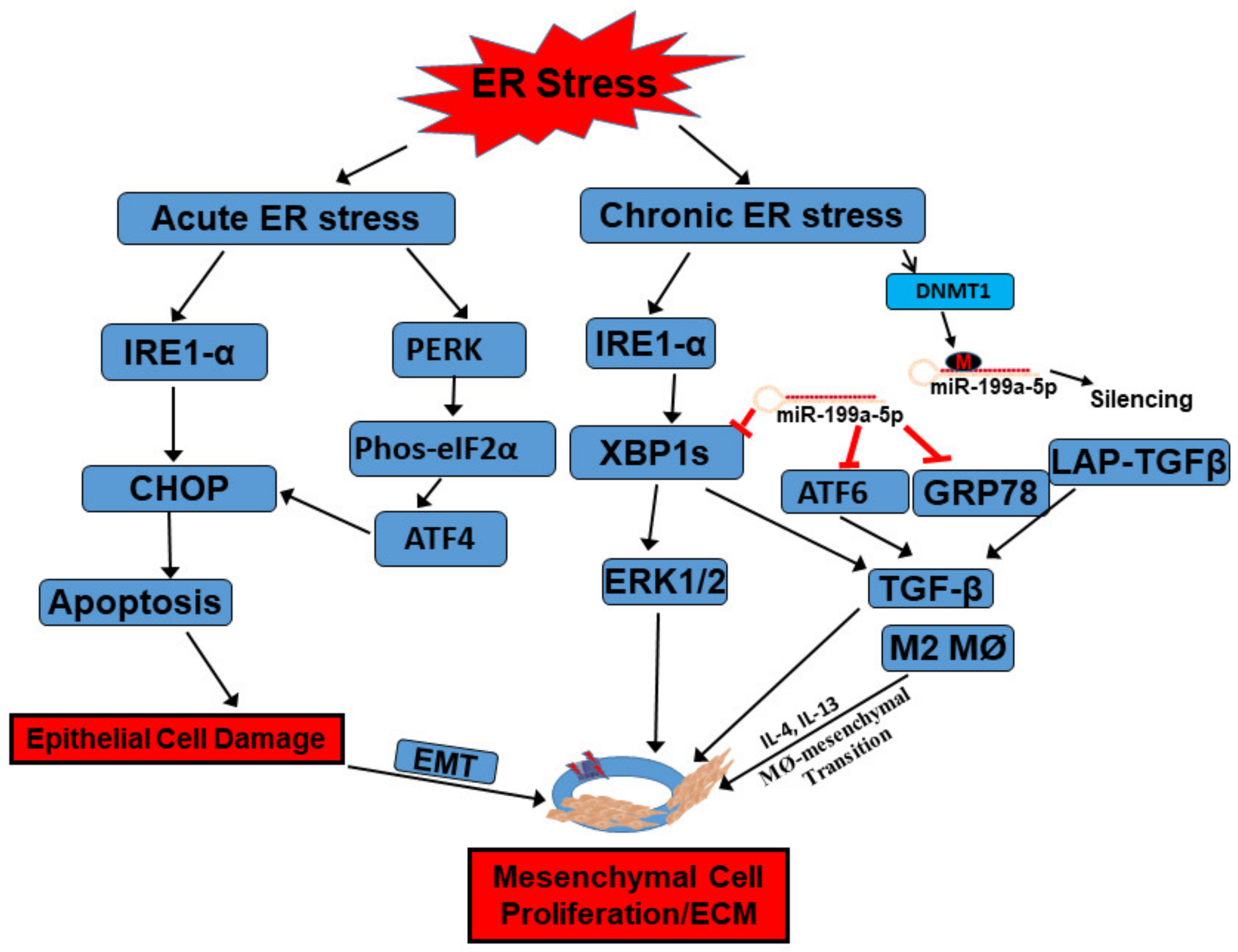
UPR and IBD
4. The UPR in Macrophage and Mesenchymal Cells during the Immune Response
5. Epigenetic Regulation of the UPR
6. Crosstalk between the UPR, Senescence, and Autophagy
6.1. Senescence and ER Stress
6.2. Autophagy and ER Stress
7. The UPR as a Therapeutic Target
8. Future Directions
- What is the direct cause of ER stress or unfolded protein response in human disease?
- How do the different binding partners and modifiers of the UPR components regulate their activity and contribute to cell type- and tissue-specific functions?
- How do cells decide when to initiate apoptosis, at what point, and are these mechanisms important in developmental regulation?
- What is the role of the UPR in adipose tissue and mesenchymal cells where ER stress is less well characterized?
- How do different cytokines affect ER stress response, such as IL-6 and IL-10, for exam-ple?
- Is there a cytokine or any other unknown stimulant that can directly activate ER stress?
- How does misfolded protein in the ER cause oxidative stress?
- How does the UPR establish the crosstalk with senescence and autophagy?
- How do we decide which animal model of ER stress can closely recapitulate the patho-genesis of the disease we study?
9. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-SMA | alpha-smooth muscle actin |
| ATF | activating transcription factor |
| BiP | immunoglobulin heavy chain-binding protein |
| CHOP | C⁄EBP homologous protein |
| DNMT1 | DNA-methyltransferase 1 |
| eIF2 a | a-subunit of eukaryotic translational initiation factor 2 |
| EGCG | epigallocatechin-3-gallate |
| ER | endoplasmic reticulum |
| ERAD | ER-associated degradation; |
| GRP78 | glucose-regulated protein 78 kDa |
| GWAS | Genome-wide association studies |
| IRE1 | inositol requirement 1 |
| JNK | Jun N-terminal kinase |
| IBD | Inflammatory Bowel Disease |
| LRRK2 | Leucine-rich repeat kinase 2; |
| NLR | NOD-like receptor |
| ORMDL3 | Orosmucoid-like 3 |
| PBA | 4-phenyl butyrate |
| PDI | protein disulfide isomerase |
| PERK | PRKR-like endoplasmic reticulum kinase |
| PKR | double stranded RNA-dependent protein kinase |
| SASP | senescence-associated secretory phenotype |
| SVIP | small p97/VCP-interacting protein |
| TNF | tumor necrosis factor |
| TUDCA | tauroursodeoxycholic acid |
| XBP1 | x-box binding protein 1 |
References
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug. Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dai, Z.; Sun, K.; Zhang, Y.; Chen, J.; Yang, Y.; Tso, P.; Wu, G.; Wu, Z. Intestinal Epithelial Cell Endoplasmic Reticulum Stress and Inflammatory Bowel Disease Pathogenesis: An Update Review. Front. Immunol. 2017, 8, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Li, C.; Grider, J.R.; Murthy, K.S.; Bohl, J.; Rivet, E.; Wieghard, N.; Kuemmerle, J.F. Endoplasmic Reticulum Stress in Subepithelial Myofibroblasts Increases the TGF-β1 Activity That Regulates Fibrosis in Crohn’s Disease. Inflamm. Bowel Dis. 2020, 26, 809–819. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, D.P.; Gardet, A.; Törkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.H.; Lagacé, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Kaplan GG.Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Chien, N.S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef]
- Pranculienė, G.; Steponaitienė, R.; Skiecevičienė, J.; Kučinskienė, R.; Kiudelis, G.; Adamonis, K.; Labanauskas, L.; Kupčinskas, L. Associations between NOD2, IRGM and ORMDL3 polymorphisms and pediatric-onset inflammatory bowel disease in the Lithuanian population. Medicina (Kaunas) 2016, 52, 325–330. [Google Scholar] [CrossRef]
- Zheng, W.; Rosenstiel, P.; Huse, K.; Sina, C.; Valentonyte, R.; Mah, N.; Zeitlmann, L.; Grosse, J.; Ruf, N.; Hampe, J. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. 2006, 7, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Maurel, M.; Obacz, J.; Avril, T.; Ding, Y.P.; Papadodima, O.; Treton, X.; Daniel, F.; Pilalis, E.; Hörberg, J.; Ogier-Denis, E.; et al. Control of anterior GRadient 2 (AGR2) dimerization links endoplasmic reticulum proteostasis to inflammation. EMBO Mol. Med. 2019, 11, e10120. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Blumberg, R.S.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Shkoda, A.; Ruiz, P.A.; Daniel, H.; Kim, S.C.; Rogler, G.; Sartor, R.B.; Haller, D. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: Impact on chronic inflammation. Gastroenterology 2007, 132, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.M.; Barlow, P.G.; Henderson, P.; Stevens, C. Interactions between Autophagy and the Unfolded Protein Response: Implications for Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 661–671. [Google Scholar] [CrossRef]
- Cao, S.S.; Wang, M.; Harrington, J.C.; Chuang, B.M.; Eckmann, L.; Kaufman, R.J. Phosphorylation of eIF2α is dispensable for differentiation but required at a posttranscriptional level for paneth cell function and intestinal homeostasis in mice. Inflamm. Bowel Dis. 2014, 20, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.M.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Ishitsuka, Y.; Hayasaka, M.; Yamada, Y.; Miyata, K.; Endo, M.; Kondo, Y.; Moriuchi, H.; Irikura, M.; Tanaka, K.; et al. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol. Res. 2015, 99, 52–62. [Google Scholar] [CrossRef]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayaub, E.A.; Kolb, P.S.; Mohammed-Ali, Z.; Tat, V.; Murphy, J.; Bellaye, P.S.; Shimbori, C.; Boivin, F.J.; Lai, R.; Lynn, E.G.; et al. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J. Pathol. 2016, 239, 411–425. [Google Scholar] [CrossRef]
- Malhi, H.; Kropp, E.M.; Clavo, V.F.; Kobrossi, C.R.; Han, J.; Mauer, A.S.; Yong, J.; Kaufman, R.J. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 2013, 288, 18624–18642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y.; et al. Chop Deficiency Protects Mice Against Bleomycin-induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol. Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef]
- Bain, C.C.; Mowat, A.M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 2014, 260, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hine, A.M.; Loke, P. Intestinal Macrophages in Resolving Inflammation. J. Immunol. 2019, 203, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Tang, P.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kuemmerle, J.F. The fate of myofibroblasts during the development of fibrosis in Crohn’s disease. J. Dig. Dis. 2020, 21, 326–331. [Google Scholar] [CrossRef]
- Mifflin, R.C.; Pinchuk, I.V.; Saada, J.I.; Powell, D.W. Intestinal myofibroblasts: Targets for stem cell therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G684–G696. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Kuemmerle, J.F. Endoplasmic reticulum stress-mediated macrophage-to-myofibroblast transition contributes to intestinal fibrosis in murine TNBS-induced colitis. In Gastroenterology; W.B. Saunders Co-Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxins: Intracellular trafficking to the ER leading to activation of host cell stress responses. Toxins 2010, 2, 1515–1535. [Google Scholar] [CrossRef] [Green Version]
- Werner, T.; Wagner, S.J.; Martinez, I.; Walter, J.; Chang, J.; Clavel, T.; Kisling, S.; Schuemann, K.; Haller, D. Depletion of luminal iron alters the gut microbiota and prevents Crohn’s disease-like ileitis. Gut 2011, 60, 325–333. [Google Scholar] [CrossRef]
- Li, C.; Kuemmerle, J. Epigenetic regulation of intestinal fibrosis. In Firbrostenotic Inflammatory Bowel Disease, 1st ed.; Rieder, F., Ed.; Springer: Cham, Switzerland, 2018; Chapter 4; pp. 39–58. [Google Scholar]
- Li, C.; Kuemmerle, J.F. Genetic and epigenetic regulation of intestinal fibrosis. United Eur. Gastroenterol. J. 2016, 4, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.; Mann, D.A. Epigenetic regulation of wound healing and fibrosis. Curr. Opin. Rheumatol. 2013, 25, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Llinàs-Arias, P.; Rosselló-Tortella, M.; López-Serra, P.; Pérez-Salvia, M.; Setién, F.; Marin, S.; Muñoz, J.P.; Junza, A.; Capellades, J.; Esteller, M.; et al. Epigenetic loss of the endoplasmic reticulum-associated degradation inhibitor SVIP induces cancer cell metabolic reprogramming. JCI Insight 2019, 5, e125888. [Google Scholar] [CrossRef] [Green Version]
- Sieber, J.; Wieder, N.; Ostrosky-Frid, M.; Dvela-Levitt, M.; Aygün, O.; Udeshi, N.D.; Carr, S.A.; Greka, A. Lysine trimethylation regulates 78-kDa glucose-regulated protein proteostasis during endoplasmic reticulum stress. J. Biol. Chem. 2017, 292, 18878–18885. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Wang, M.; Chen, Z.; Bhatt, K.; Oh, H.J.; Lanting, L.; Deshpande, S.; Jia, Y.; Lai, J.Y.; Natarajan, R.; et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Commun. 2016, 7, 12864. [Google Scholar] [CrossRef]
- Byrd, A.E.; Brewer, J.W. Micro(RNA)managing endoplasmic reticulum stress. IUBMB Life 2013, 65, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C. Intestinal fibrosis in inflammatory bowel disease: The role of microRNAs. Explor. Res. Hypothese Med. 2015, 1, 7–16. [Google Scholar]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Oakes, S.A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.; Venturelli, S.; Zender, L.; Bitzer, M. Cellular senescence in gastrointestinal diseases: From pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pluquet, O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends Biochem. Sci. 2020, 45, 371–374. [Google Scholar] [CrossRef]
- Hosomi, S.; Kaser, A.; Blumberg, R.S. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr. Opin. Gastroenterol. 2015, 31, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Blumberg, R.S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011, 140, 1738–1747. [Google Scholar] [CrossRef] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.; Keita, Å.V.; Saxena, A.; Reyes, J.L.; Mancini, N.L.; Al Rajabi, A.; Wang, A.; Baggio, C.H.; Dicay, M.; McKay, D.M.; et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J. Biol. Chem. 2018, 293, 3073–3087. [Google Scholar] [CrossRef] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling-from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Myers, L.K.; Wooley, P.H.; Corrigall, V.M.; Bodman-Smith, M.D.; Panayi, G.S.; Thompson, S.J. Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4-producing regulatory T cells: A novel function for an ancient protein. Arthritis Rheum. 2006, 54, 854–863. [Google Scholar] [CrossRef]
- Kirkham, B.; Chaabo, K.; Hall, C.; Garrood, T.; Mant, T.; Allen, E.; Vincent, A.; Vasconcelos, J.C.; Prevost, A.T.; Panayi, G.S.; et al. Safety and patient response as indicated by biomarker changes to binding immunoglobulin protein in the phase I/IIA RAGULA clinical trial in rheumatoid arthritis. Rheumatology 2016, 55, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C. Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut. Gastrointest. Disord. 2021, 3, 31-43. https://doi.org/10.3390/gidisord3010004
Li C. Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut. Gastrointestinal Disorders. 2021; 3(1):31-43. https://doi.org/10.3390/gidisord3010004
Chicago/Turabian StyleLi, Chao. 2021. "Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut" Gastrointestinal Disorders 3, no. 1: 31-43. https://doi.org/10.3390/gidisord3010004