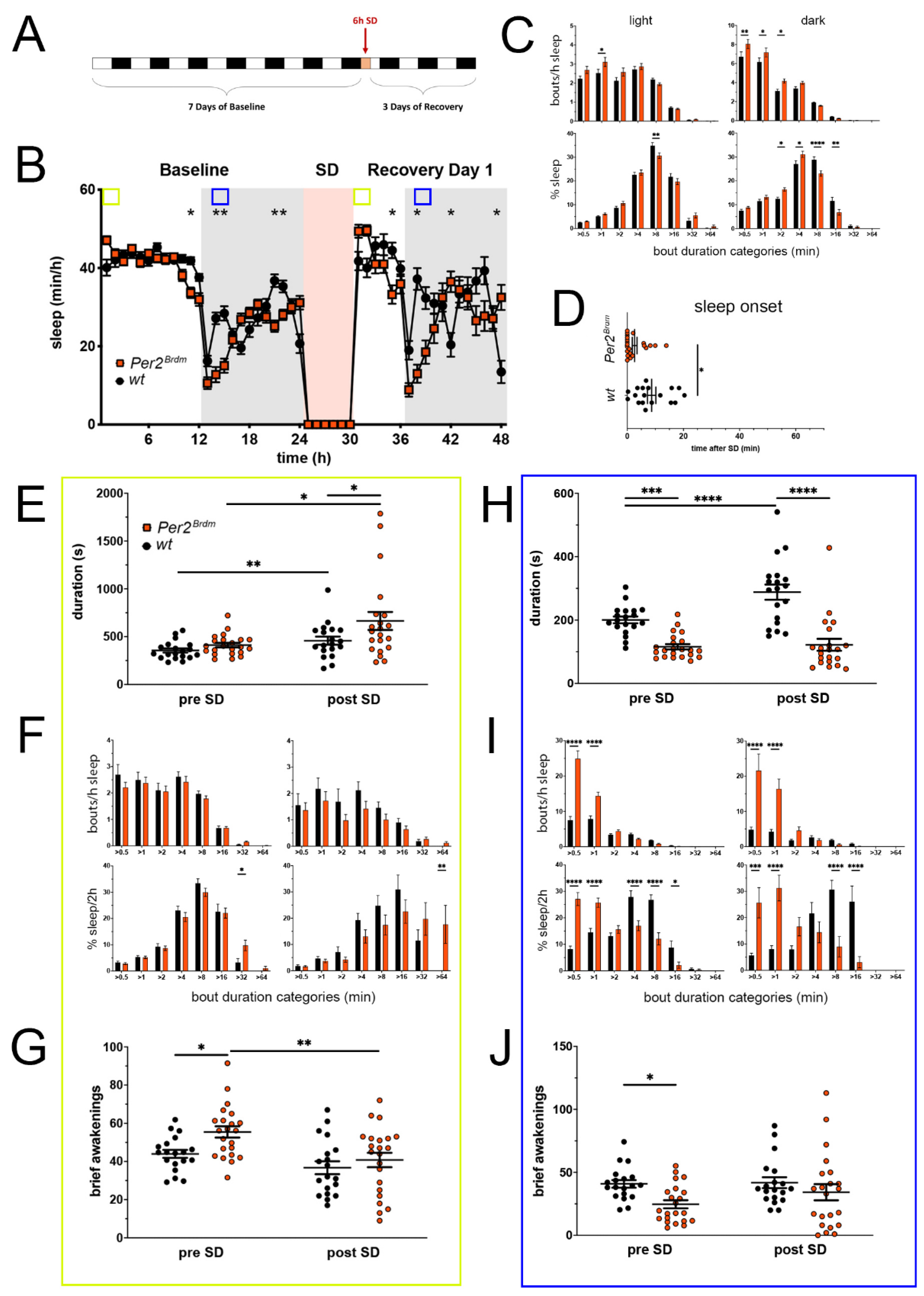
Figure 1.
Sleep–wake analysis of Per2Brdm mutant mice (red) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). Statistically significant differences of sleep between the two genotypes by two-way ANOVA with Šídák’s post-test is depicted with an asterisk (* p < 0.05, ** p < 0.01, n = 19 wt, n = 22 Per2Brdm); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed statistically significant differences between the two genotypes with * p < 0.05, ** p < 0.01, **** p < 0.0001, n = 19 wt, n = 22 Per2Brdm; (D) sleep onset after SD. The Per2Brdm mutant mice show a significantly earlier onset compared to wild type controls. Mann–Whitney test with * p < 0.05, n = 18 wt, n = 22 Per2Brdm; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is significantly increased in Per2Brdm mutant mice post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05, n = 19 wt, n = 21 Per2Brdm; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). Per2Brdm mice sleep more than controls pre- as well as post-SD in the >32 min and >64 min sleep bout categories. Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.01, n = 19 wt, n = 22 Per2Brdm; (G) brief awakenings are increased in Per2Brdm mice pre-SD but not post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05; ** p < 0.01. (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is significantly decreased in Per2Brdm mutant mice pre-SD and post-SD. Two-way ANOVA with Šídák’s post-test with *** p < 0.001, **** p < 0.0001, n = 19 wt, n = 21 Per2Brdm; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Per2Brdm mice sleep had more short sleep bouts in pre-SD as well as post-SD, * p < 0.05, *** p < 0.001, **** p < 0.0001, but wild-type controls showed a higher sleep percentage in mid and long sleep bouts in pre-SD as well as post-SD. Two-way ANOVA with Šídák’s post-test with *** p < 0.001, n = 19 wt, n = 22 Per2Brdm; (J) brief awakenings are decreased in Per2Brdm mice pre-SD, but not post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05, n = 19 wt, n = 22 Per2Brdm.
Figure 1.
Sleep–wake analysis of Per2Brdm mutant mice (red) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). Statistically significant differences of sleep between the two genotypes by two-way ANOVA with Šídák’s post-test is depicted with an asterisk (* p < 0.05, ** p < 0.01, n = 19 wt, n = 22 Per2Brdm); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed statistically significant differences between the two genotypes with * p < 0.05, ** p < 0.01, **** p < 0.0001, n = 19 wt, n = 22 Per2Brdm; (D) sleep onset after SD. The Per2Brdm mutant mice show a significantly earlier onset compared to wild type controls. Mann–Whitney test with * p < 0.05, n = 18 wt, n = 22 Per2Brdm; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is significantly increased in Per2Brdm mutant mice post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05, n = 19 wt, n = 21 Per2Brdm; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). Per2Brdm mice sleep more than controls pre- as well as post-SD in the >32 min and >64 min sleep bout categories. Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.01, n = 19 wt, n = 22 Per2Brdm; (G) brief awakenings are increased in Per2Brdm mice pre-SD but not post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05; ** p < 0.01. (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is significantly decreased in Per2Brdm mutant mice pre-SD and post-SD. Two-way ANOVA with Šídák’s post-test with *** p < 0.001, **** p < 0.0001, n = 19 wt, n = 21 Per2Brdm; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Per2Brdm mice sleep had more short sleep bouts in pre-SD as well as post-SD, * p < 0.05, *** p < 0.001, **** p < 0.0001, but wild-type controls showed a higher sleep percentage in mid and long sleep bouts in pre-SD as well as post-SD. Two-way ANOVA with Šídák’s post-test with *** p < 0.001, n = 19 wt, n = 22 Per2Brdm; (J) brief awakenings are decreased in Per2Brdm mice pre-SD, but not post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05, n = 19 wt, n = 22 Per2Brdm.
![Clockssleep 05 00017 g001]()
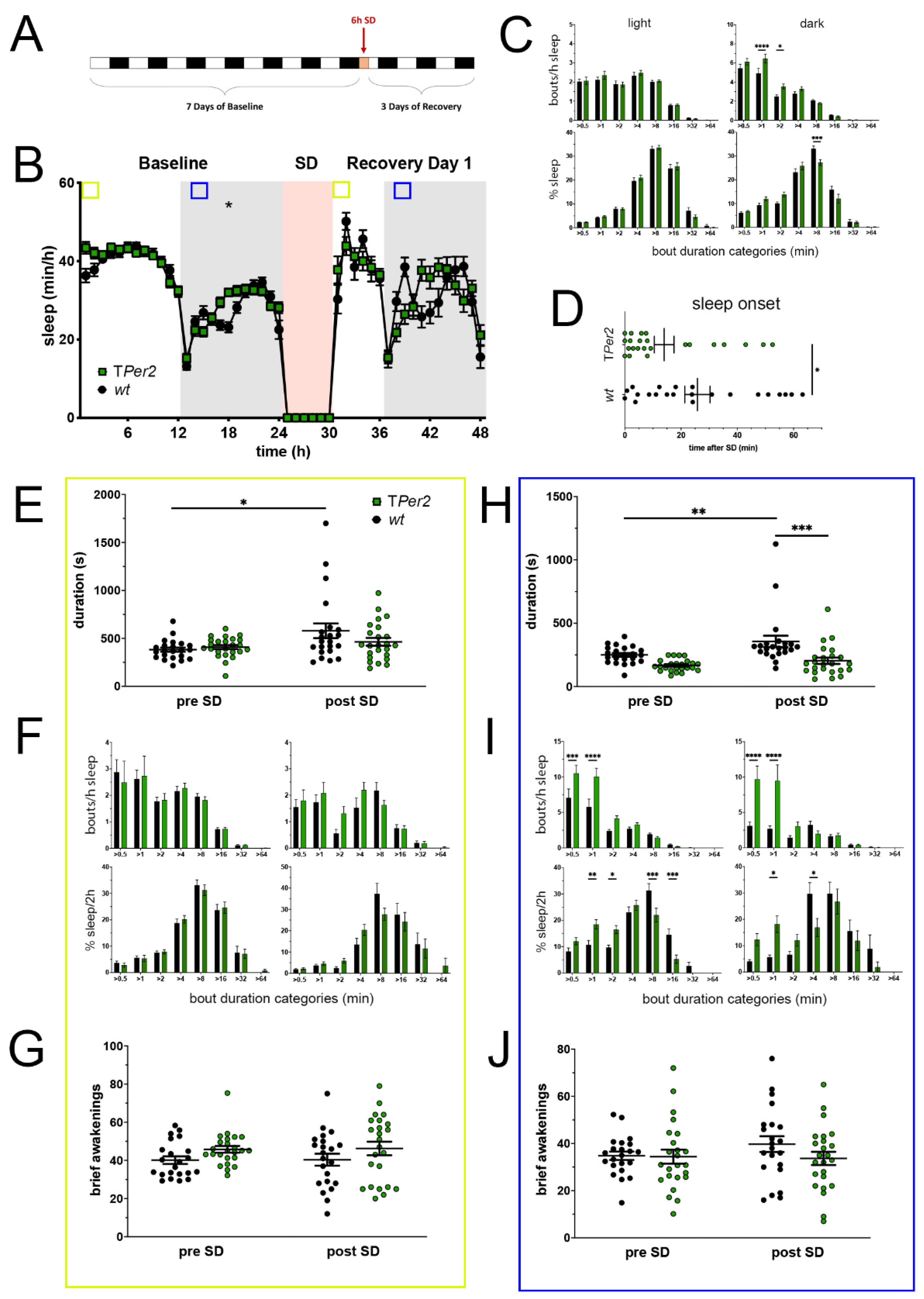
Figure 2.
Sleep–wake analysis of total Per2 knock-out (TPer2 KO) mice (green) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). Statistically significant differences of sleep between the two genotypes by two-way ANOVA with Šídák’s post-test is depicted with an asterisk (* p < 0.05, n = 22 wt, n = 24 TPer2); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed statistically significant differences between the two genotypes with * p < 0.05, *** p < 0.001, **** p < 0.0001, n = 22 wt, n = 24 TPer2; (D) sleep onset after SD. The TPer2 KO mice show a significantly earlier onset compared to wild type controls. Mann–Whitney test with * p < 0.05, n = 22 wt, n = 23 TPer2; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is comparable in both genotypes. Two-way ANOVA with Šídák’s post-test, n = 22 wt, n = 24 TPer2; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). TPer2 KO mice do not sleep more than controls pre- as well as post-SD. Two-way ANOVA with Šídák’s post-test, n = 22 wt, n = 24 TPer2; (G) brief awakenings in TPer2 mice are comparable to control animals. Two-way ANOVA with Šídák’s post-test. (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is significantly decreased in TPer2 KO mice postTwoSD. 2-way ANOVA with Šídák’s post-test with ** p < 0.01, *** p < 0.001, n = 22 wt, n = 24 TPer2; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). TPer2 mice had more short sleep bouts in pre-SD as well as post-SD, *** p < 0.001, **** p < 0.0001, but wild-type controls showed a higher sleep percentage in mid and long sleep bouts in pre-SD as well as post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.01, *** p < 0.001, n = 22 wt, n = 24 TPer2; (J) brief awakenings are comparable to controls in TPer2 mice pre-SD and post-SD. Two-way ANOVA with Šídák’s post-test, n = 22 wt, n = 24 TPer2.
Figure 2.
Sleep–wake analysis of total Per2 knock-out (TPer2 KO) mice (green) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). Statistically significant differences of sleep between the two genotypes by two-way ANOVA with Šídák’s post-test is depicted with an asterisk (* p < 0.05, n = 22 wt, n = 24 TPer2); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed statistically significant differences between the two genotypes with * p < 0.05, *** p < 0.001, **** p < 0.0001, n = 22 wt, n = 24 TPer2; (D) sleep onset after SD. The TPer2 KO mice show a significantly earlier onset compared to wild type controls. Mann–Whitney test with * p < 0.05, n = 22 wt, n = 23 TPer2; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is comparable in both genotypes. Two-way ANOVA with Šídák’s post-test, n = 22 wt, n = 24 TPer2; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). TPer2 KO mice do not sleep more than controls pre- as well as post-SD. Two-way ANOVA with Šídák’s post-test, n = 22 wt, n = 24 TPer2; (G) brief awakenings in TPer2 mice are comparable to control animals. Two-way ANOVA with Šídák’s post-test. (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is significantly decreased in TPer2 KO mice postTwoSD. 2-way ANOVA with Šídák’s post-test with ** p < 0.01, *** p < 0.001, n = 22 wt, n = 24 TPer2; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). TPer2 mice had more short sleep bouts in pre-SD as well as post-SD, *** p < 0.001, **** p < 0.0001, but wild-type controls showed a higher sleep percentage in mid and long sleep bouts in pre-SD as well as post-SD. Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.01, *** p < 0.001, n = 22 wt, n = 24 TPer2; (J) brief awakenings are comparable to controls in TPer2 mice pre-SD and post-SD. Two-way ANOVA with Šídák’s post-test, n = 22 wt, n = 24 TPer2.
![Clockssleep 05 00017 g002]()
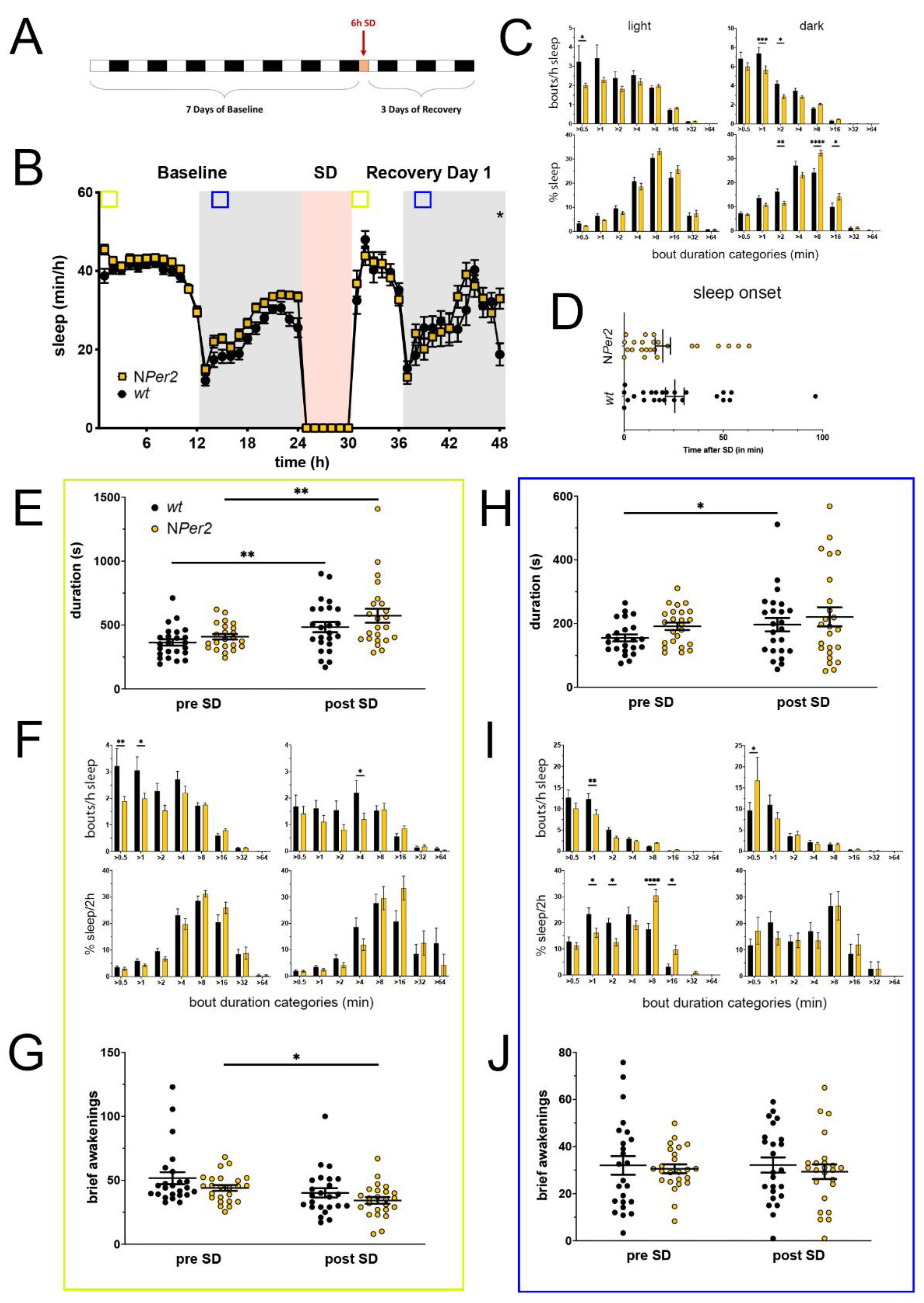
Figure 3.
Sleep–wake analysis of neuronal Per2 knock-out (NPer2 KO) mice (yellow) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). Statistically significant differences of sleep between the two genotypes by two-way ANOVA with Šídák’s post-test is depicted with an asterisk (* p < 0.05, n = 24 wt, n = 24 NPer2); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed statistically significant differences between the two genotypes with * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, n = 24 wt, n = 24 NPer2; (D) sleep onset after SD. The NPer2 KO mice show a similar onset compared to wild type controls. Mann–Whitney test, n = 24 wt, n = 24 NPer2; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is comparable between NPer2 KO and control mice. Two-way ANOVA with Šídák’s post-test, n = 24 wt, n = 23 NPer2; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). NPer2 mice have less short bouts pre-SD and less medium bouts post-SD compared to controls. Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.01, n = 24 wt, n = 24 NPer2; (G) brief awakenings are comparable between NPer2 mice and controls. Two-way ANOVA with Šídák’s post-test, * p < 0.05, n = 24 wt, n = 24 NPer2; (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is comparable between NPer2 KO mice and controls. Two-way ANOVA with Šídák’s post-test, * p < 0.05, n = 24 wt, n = 24 NPer2; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). NPer2 mice had less short sleep bouts in pre-SD but more in post-SD. NPer2 mice showed a higher sleep percentage in mid and long sleep bouts in pre-SD, but not post-SD Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.001, **** p < 0.0001, n = 24 wt, n = 24 NPer2; (J) brief awakenings are comparable between NPer2 and control mice. Two-way ANOVA with Šídák’s post-test, n = 24 wt, n = 24 NPer2.
Figure 3.
Sleep–wake analysis of neuronal Per2 knock-out (NPer2 KO) mice (yellow) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). Statistically significant differences of sleep between the two genotypes by two-way ANOVA with Šídák’s post-test is depicted with an asterisk (* p < 0.05, n = 24 wt, n = 24 NPer2); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed statistically significant differences between the two genotypes with * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, n = 24 wt, n = 24 NPer2; (D) sleep onset after SD. The NPer2 KO mice show a similar onset compared to wild type controls. Mann–Whitney test, n = 24 wt, n = 24 NPer2; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is comparable between NPer2 KO and control mice. Two-way ANOVA with Šídák’s post-test, n = 24 wt, n = 23 NPer2; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). NPer2 mice have less short bouts pre-SD and less medium bouts post-SD compared to controls. Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.01, n = 24 wt, n = 24 NPer2; (G) brief awakenings are comparable between NPer2 mice and controls. Two-way ANOVA with Šídák’s post-test, * p < 0.05, n = 24 wt, n = 24 NPer2; (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is comparable between NPer2 KO mice and controls. Two-way ANOVA with Šídák’s post-test, * p < 0.05, n = 24 wt, n = 24 NPer2; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). NPer2 mice had less short sleep bouts in pre-SD but more in post-SD. NPer2 mice showed a higher sleep percentage in mid and long sleep bouts in pre-SD, but not post-SD Two-way ANOVA with Šídák’s post-test with * p < 0.05, ** p < 0.001, **** p < 0.0001, n = 24 wt, n = 24 NPer2; (J) brief awakenings are comparable between NPer2 and control mice. Two-way ANOVA with Šídák’s post-test, n = 24 wt, n = 24 NPer2.
![Clockssleep 05 00017 g003]()
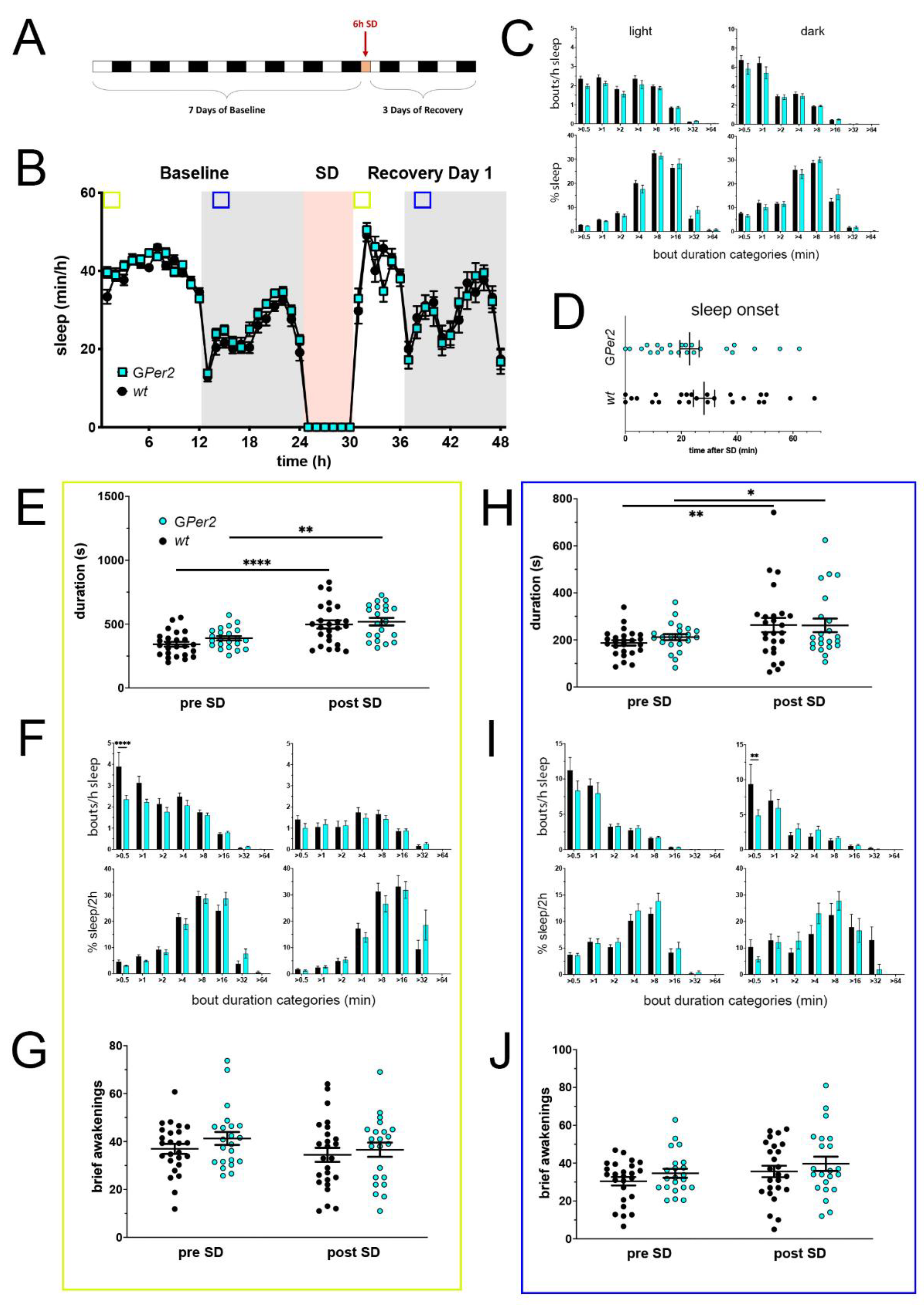
Figure 4.
Sleep–wake analysis of glial Per2 knock-out (GPer2 KO) mice (blue) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). There is no statistically significant difference of sleep between the two genotypes. Two-way ANOVA with Šídák’s post-test (n = 25 wt, n = 22 GPer2); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed no statistically significant differences between the two genotypes, n = 25 wt, n = 22 GPer2; (D) sleep onset after SD. The GPer2 KO mice show a similar onset compared to wild type controls. Mann–Whitney test with n = 25 wt, n = 23 GPer2; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is comparable between GPer2 KO and control mice. Two-way ANOVA with Šídák’s post-test, ** p < 0.01, **** p < 0.0001, n = 25 wt, n = 22 TPer2; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). GPer2 KO mice show less short bouts pre-SD, but have an increased sleep percentage in longer bouts pre- and post-SD compared to controls. Two-way ANOVA with Šídák’s post-test, n = 25 wt, n = 22 GPer2; (G) brief awakenings in GPer2 mice are comparable to control animals. Two-way ANOVA with Šídák’s post-test (n = 25 wt, n = 22 GPer2); (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is comparable between GPer2 KO mice and controls. Two-way ANOVA with Šídák’s post-test, * p < 0.05, ** p < 0.01, n = 25 wt, n = 22 GPer2; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). GPer2 mice had less short sleep bouts post-SD. Two-way ANOVA with Šídák’s post-test with ** p < 0.01, n = 25 wt, n = 22 GPer2; (J) brief awakenings are comparable to controls in GPer2 mice pre-SD and post-SD. Two-way ANOVA with Šídák’s post-test, n = 25 wt, n = 22 GPer2.
Figure 4.
Sleep–wake analysis of glial Per2 knock-out (GPer2 KO) mice (blue) compared to wild type controls (black). (A) Scheme of sleep–wake recordings during undisturbed conditions (baseline = BL), sleep deprivation (SD), and sleep–wake recordings during recovery from SD; (B) time course of mean (±SEM) hourly values of sleep over 7 days summarized over 24 h baseline. White areas depict 12 h of light and gray shaded areas 12 h of darkness. The rose shaded area depicts 6 h of sleep deprivation (SD) in the light portion. The green and blue squares depict pre- and post-SD time windows being analyzed in detail in E-G (green square, 2 h light phase) and H-J (blue square, 2 h dark phase). There is no statistically significant difference of sleep between the two genotypes. Two-way ANOVA with Šídák’s post-test (n = 25 wt, n = 22 GPer2); (C) distribution of sleep bout durations in the light (left panels) and dark phase (right panels) of baseline sleep (hours 0–24 in (B)). The top panels show the number of bouts per hour of sleep in each sleep bout category in the light and dark phase. The bottom panels show the percentage of sleep in each of the categories. Values are shown as mean (±SEM). Two-way ANOVA with Šídák’s post-test revealed no statistically significant differences between the two genotypes, n = 25 wt, n = 22 GPer2; (D) sleep onset after SD. The GPer2 KO mice show a similar onset compared to wild type controls. Mann–Whitney test with n = 25 wt, n = 23 GPer2; (E) average sleep bout duration pre-SD (2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (2 h during recovery after sleep onset). Sleep bout duration is comparable between GPer2 KO and control mice. Two-way ANOVA with Šídák’s post-test, ** p < 0.01, **** p < 0.0001, n = 25 wt, n = 22 TPer2; (F) sleep bout durations pre-SD (left panels, 2 h in BL from ZT0 to ZT2, hours 1–2 in (B)) and post-SD (right panels, 2 h during recovery after sleep onset). GPer2 KO mice show less short bouts pre-SD, but have an increased sleep percentage in longer bouts pre- and post-SD compared to controls. Two-way ANOVA with Šídák’s post-test, n = 25 wt, n = 22 GPer2; (G) brief awakenings in GPer2 mice are comparable to control animals. Two-way ANOVA with Šídák’s post-test (n = 25 wt, n = 22 GPer2); (H) average sleep bout duration pre-SD (2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). Sleep bout duration is comparable between GPer2 KO mice and controls. Two-way ANOVA with Šídák’s post-test, * p < 0.05, ** p < 0.01, n = 25 wt, n = 22 GPer2; (I) sleep bout durations pre-SD (left panels, 2 h in BL from ZT13 to ZT15, hours 13–14 in (B)) and post-SD (right panels, 2 h during recovery from ZT13 to ZT15, hours 38–39 in (B)). GPer2 mice had less short sleep bouts post-SD. Two-way ANOVA with Šídák’s post-test with ** p < 0.01, n = 25 wt, n = 22 GPer2; (J) brief awakenings are comparable to controls in GPer2 mice pre-SD and post-SD. Two-way ANOVA with Šídák’s post-test, n = 25 wt, n = 22 GPer2.
![Clockssleep 05 00017 g004]()
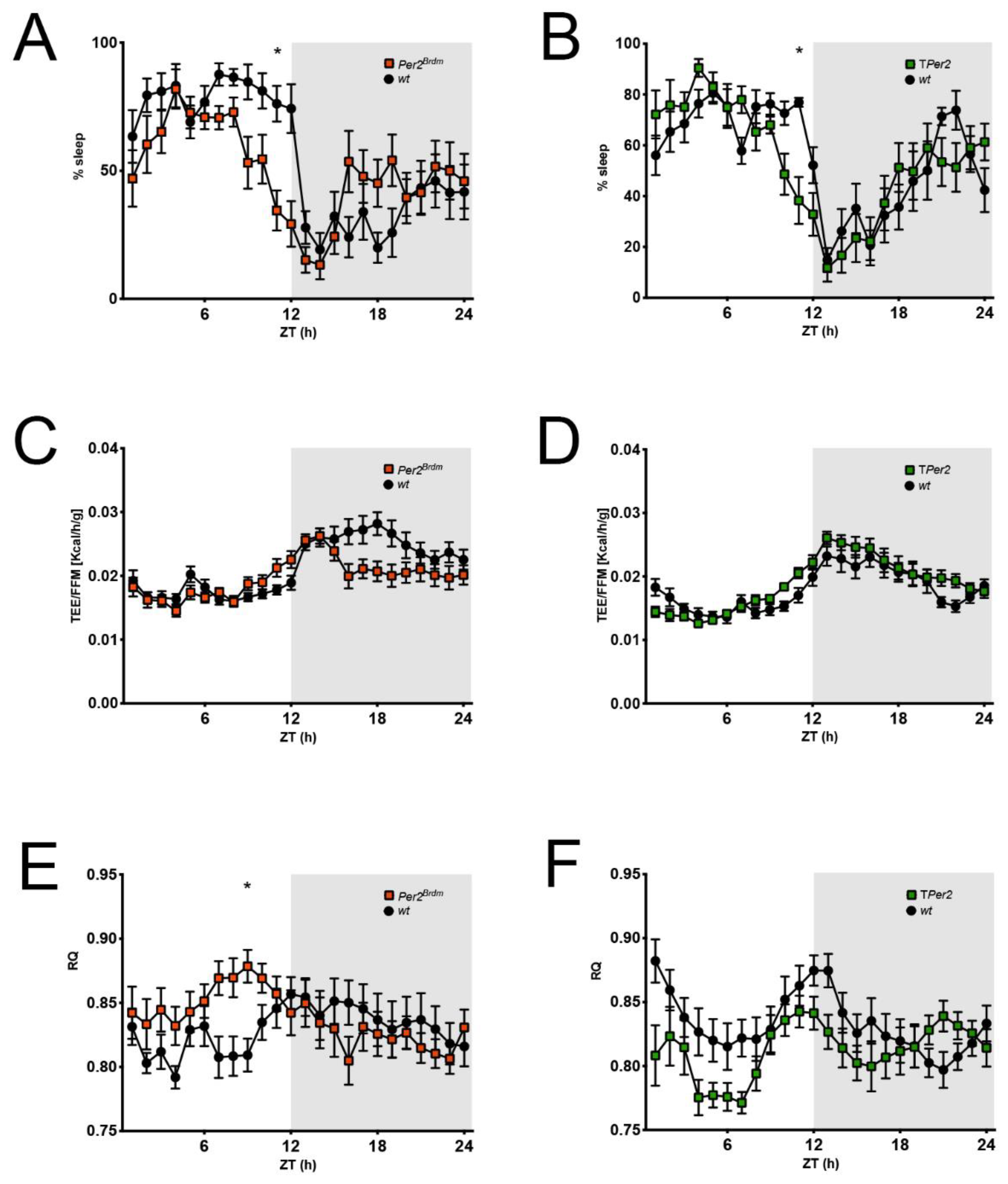
Figure 5.
Comparison of sleep and metabolic data derived from the metabolic cages. (A,B) Percent of sleep per 1 h observation interval defined as absence of any movement sensor activation. Per2Brdm and TPer2 mice have an earlier wake onset than controls; (C,D) total energy expenditure (TEE) per 1 h normalized to the fat-free mass (FFM) of each mouse; (E,F) respiratory quotient (RQ) per 1 h defined as CO2 produced/O2 consumed. Corresponding to an earlier wake onset, RQ increases earlier in Per2Brdm and TPer2 mice. For all panels: n = 12, repeated measures two-way ANOVA with Šídák’s correction for multiple testing, * p < 0.05.
Figure 5.
Comparison of sleep and metabolic data derived from the metabolic cages. (A,B) Percent of sleep per 1 h observation interval defined as absence of any movement sensor activation. Per2Brdm and TPer2 mice have an earlier wake onset than controls; (C,D) total energy expenditure (TEE) per 1 h normalized to the fat-free mass (FFM) of each mouse; (E,F) respiratory quotient (RQ) per 1 h defined as CO2 produced/O2 consumed. Corresponding to an earlier wake onset, RQ increases earlier in Per2Brdm and TPer2 mice. For all panels: n = 12, repeated measures two-way ANOVA with Šídák’s correction for multiple testing, * p < 0.05.
Table 1.
Metabolic Phenotyping. Total energy expenditure (TEE), food intake, and water intake are normalized to FFM. Total ambulatory activity represents all directed ambulatory locomotion (at least 1 cm/s) within the beam break system. t-tests were used to compare knock-out animals to their respective controls; values are means ±SEM ** p < 0.01, * p < 0.05, n = 12 per genotype.
Table 1.
Metabolic Phenotyping. Total energy expenditure (TEE), food intake, and water intake are normalized to FFM. Total ambulatory activity represents all directed ambulatory locomotion (at least 1 cm/s) within the beam break system. t-tests were used to compare knock-out animals to their respective controls; values are means ±SEM ** p < 0.01, * p < 0.05, n = 12 per genotype.
| | Wt | Per2Brdm | Wt (fl/fl) | TPer2 (D/D) |
|---|
| Body weight (g) | 23.7 ± 0.7 ** | 34.9 ± 1.1 | 31.9 ± 0.7 | 31.7 ± 0.7 |
| Fat mass (FM—g) | 2.4 ± 0.8 | 2.8 ± 0.3 | 4.3 ± 0.7 | 5.7 ± 0.5 |
| Fat free mass (FFM—g) | 20.3 ± 0.5 ** | 30.2 ± 0.8 | 25.8 ± 0.8 | 24.3 ± 0.5 |
| Total energy expenditure (TEE/FFM)—kcal/24 h/g) | 0.47 ± 0.02 | 0.51 ± 0.02 | 0.43 ± 0.02 | 0.45 ± 0.01 |
| Respiratory Quotient (RQ) | 0.83 ± 0.01 | 0.84 ± 0.01 | 0.83 ± 0.01 | 0.81 ± 0.01 |
| Respiratory Quotient (RQ light) | 0.82 ± 0.02 ** | 0.85 ± 0.02 | 0.84 ± 0.01 * | 0.81 ± 0.01 |
| Respiratory Quotient (RQ dark) | 0.84 ± 0.02 | 0.82 ± 0.01 | 0.82 ± 0.01 | 0.82 ± 0.01 |
| Food intake/FFM (Kcal/24 h/g) | 0.54 ± 0.04 | 0.51 ± 0.04 | 0.39 ± 0.04 | 0.44 ± 0.05 |
| Food intake (light phase—kcal/12 h) | 3.3 ± 0.7 ** | 7.4 ± 0.4 | 3.6 ± 0.6 | 3.3 ± 0.5 |
| Food intake (dark phase—kcal/12 h) | 7.6 ± 0.8 | 7.8 ± 0.9 | 6.2 ± 0.5 | 7.5 ± 1.1 |
| Water intake/FFM (g/24 h/g) | 0.16 ± 0.06 | 0.19 ± 0.04 | 0.13 ± 0.03 | 0.14 ± 0.02 |
| Water intake (light phase—g) | 0.6 ± 0.1 ** | 2.7 + 0.2 | 1.1 ± 0.2 | 1.2 ± 0.3 |
| Water intake (dark phase—g) | 2.7 ± 0.2 | 3.1 ± 0.3 | 2.2 ± 0.3 | 2.1 ± 0.3 |
| Total Ambulatory Activity (meters) | 123.5 ± 9.7 ** | 89.2 ± 8.3 | 105.1 ± 8.2 | 116.83 ± 9.7 |
| Ambulatory Activity Light (meters) | 38.2 ± 5.4 | 35.2 ± 4.1 | 43.8 ± 3.0 | 40.1 ± 4.9 |
| Ambulatory Activity Dark (meters) | 85.3 ± 7.5 ** | 54.1 ± 5.0 | 61.3 ± 6.8 | 76.7 ± 7.3 |

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}