Time-Resolved X-ray Absorption Spectroscopy in (Photo)Electrochemistry

 , , ,
, , ,
Abstract
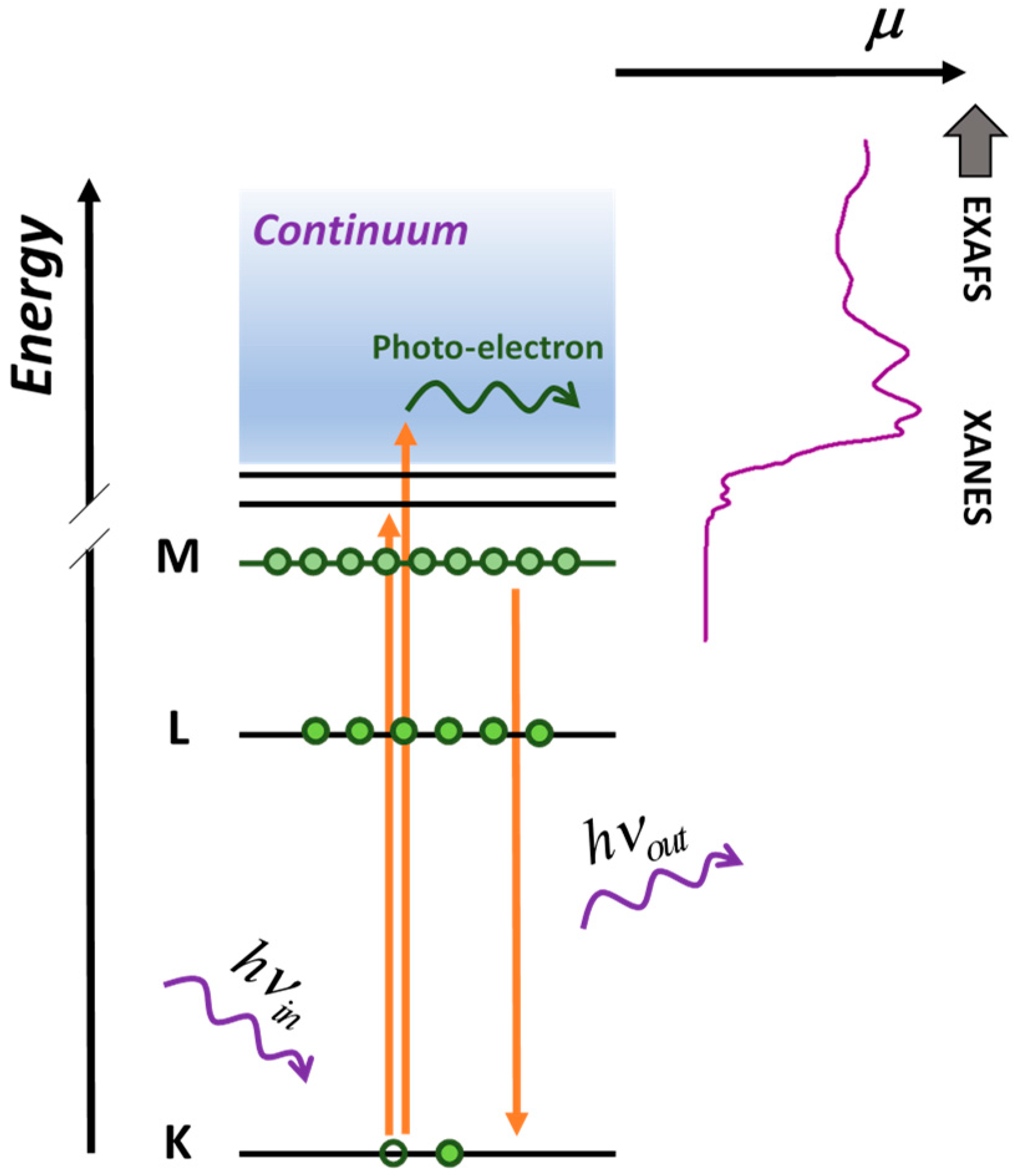
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
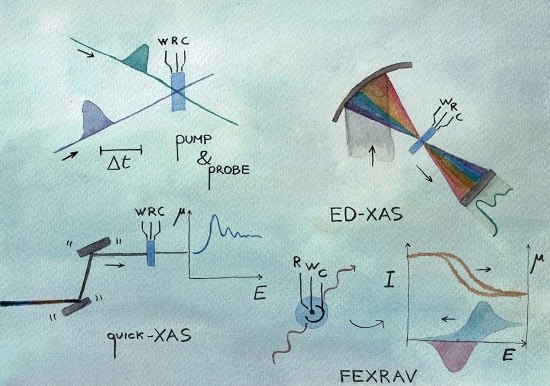
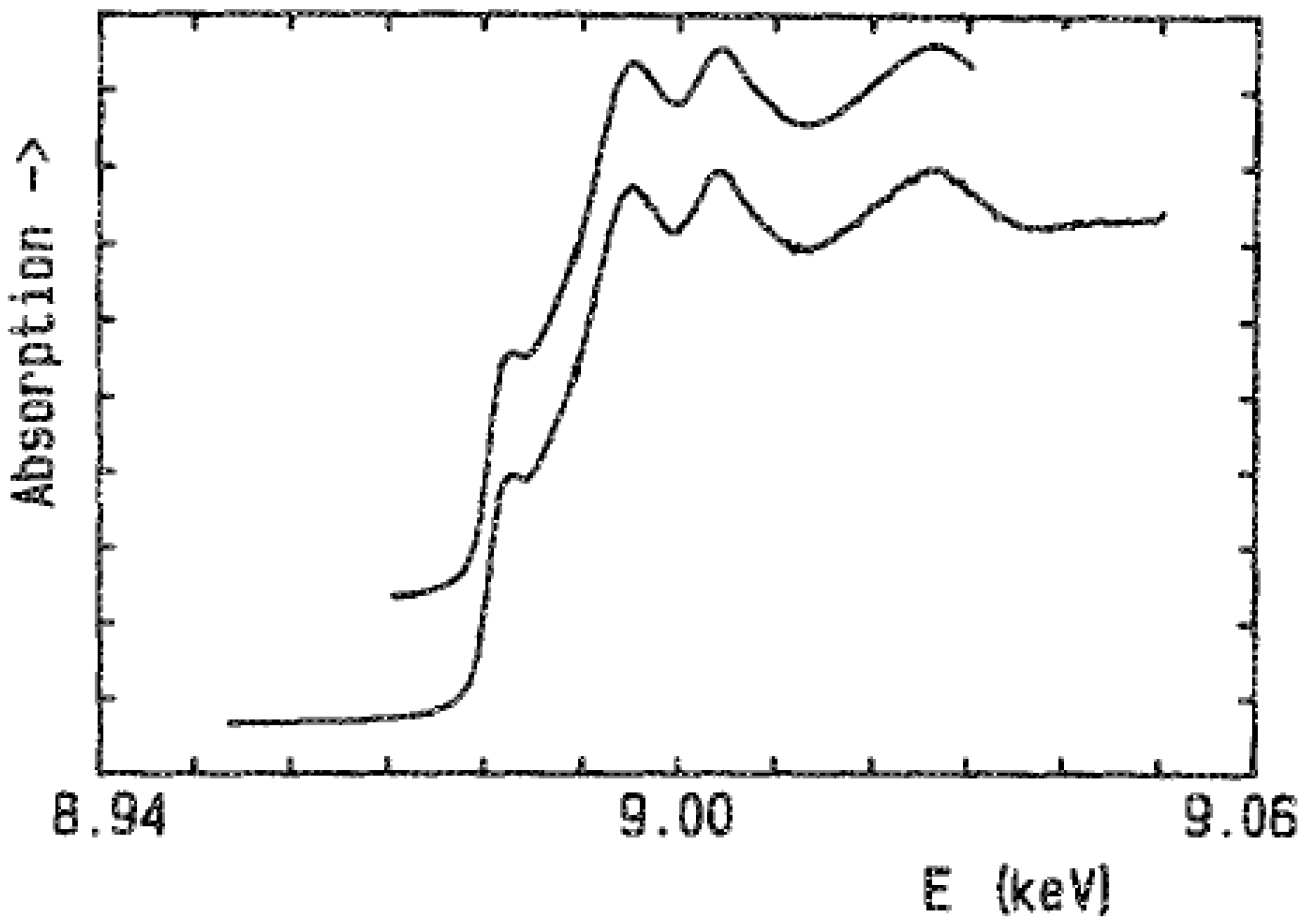
2. Quick-XAS
3. Energy Dispersive XAS
4. Pump & Probe XAS
5. FEXRAV
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Simpson, B.H.; Rodríguez-López, J. Emerging techniques for the in situ analysis of reaction intermediates on photo-electrochemical interfaces. Anal. Methods 2015, 7, 7029–7041. [Google Scholar] [CrossRef]
- Caudillo-Flores, U.; Muñoz-Batista, M.J.; Kubacka, A.; Fernández-García, M. Operando Spectroscopy in Photocatalysis. Chem. Photo Chem. 2018, 777–785. [Google Scholar]
- Minguzzi, A.; Ghigna, P. X-ray absorption spectroscopy in electrochemistry from fundamentals to fixed energy X-ray absorption voltammetry. In Electroanalytical Chemistry: A Series of Advances, Volume 27; Bard, A.J., Zoski, C.G., Eds.; CRC Press–Taylor and Francis Group: Boca Raton, FL, USA, 2017; pp. 119–181. ISBN 9781351981675. [Google Scholar]
- Russell, A.E.; Rose, A. X-ray absorption spectroscopy of low temperature fuel cell catalysts. Chem. Rev. 2004, 104, 4613–4635. [Google Scholar] [CrossRef]
- Abruña, H.D.; Bommarito, G.M.; Yee, H.S. X-ray Standing Waves and Surface EXAFS Studies of Electrochemical Interfaces. Acc. Chem. Res. 1995, 28, 273–279. [Google Scholar] [CrossRef]
- Sharpe, L.R.; Heineman, W.R.; Elder, R.C. EXAFS Spectroelectrochemistry. Chem. Rev. 1990, 90, 705–722. [Google Scholar] [CrossRef]
- Doyle, R.L.; Godwin, I.J.; Brandon, M.P.; Lyons, M.E.G. Redox and electrochemical water splitting catalytic properties of hydrated metal oxide modified electrodes. Phys. Chem. Chem. Phys. 2013, 15, 13737–13783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frahm, R. Quick scanning exafs: First experiments. Nucl. Inst. Methods Phys. Res. A 1988, 270, 578–581. [Google Scholar] [CrossRef]
- Frahm, R. New method for time dependent X-ray absorption studies. Rev. Sci. Instrum. 1989, 60, 2515–2518. [Google Scholar] [CrossRef]
- Stötzel, J.; Lützenkirchen-Hecht, D.; Frahm, R. A new flexible monochromator setup for quick scanning X-ray absorption spectroscopy. Rev. Sci. Instrum. 2010, 81, 073109. [Google Scholar] [CrossRef]
- Müller, O.; Nachtegaal, M.; Just, J.; Lützenkirchen-Hecht, D.; Frahm, R. Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10ms time resolution. J. Synchrotron Radiat. 2016, 23, 260–266. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Q.; Zhou, Y.; Li, H.; Yang, X.Q.; Nam, K.W.; Ehrlich, S.N.; Khalid, S.; Meng, Y.S. High rate delithiation behaviour of LiFePO4studied by quick X-ray absorption spectroscopy. Chem. Commun. 2012, 48, 11537–11539. [Google Scholar] [CrossRef]
- Ishiguro, N.; Saida, T.; Uruga, T.; Nagamatsu, S.I.; Sekizawa, O.; Nitta, K.; Yamamoto, T.; Ohkoshi, S.I.; Iwasawa, Y.; Yokoyama, T.; et al. Operando time-resolved X-ray absorption fine structure study for surface events on a Pt3Co/C cathode catalyst in a polymer electrolyte fuel cell during voltage-operating processes. ACS Catal. 2012, 2, 1319–1330. [Google Scholar] [CrossRef]
- Gorlin, Y.; Lassalle-Kaiser, B.; Benck, J.D.; Gul, S.; Webb, S.M.; Yachandra, V.K.; Yano, J.; Jaramillo, T.F. In situ X-ray absorption spectroscopy investigation of a bifunctional manganese oxide catalyst with high activity for electrochemical water oxidation and oxygen reduction. J. Am. Chem. Soc. 2013, 135, 8525–8534. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.G.; Conradson, S.D.; Wilson, M.S.; Gottesfeld, S.; Raistrick, I.D.; Valerio, J.; Lovato, M. Direct observation of surface oxide formation and reduction on platinum clusters by time-resolved X-ray absorption spectroscopy. J. Electroanal. Chem. 1995, 384, 99–103. [Google Scholar] [CrossRef]
- McBreen, J.; O’Grady, W.E.; Tourillon, G.; Dartyge, E.; Fontaine, A.; Pandya, K.I. In situ time-resolved X-ray absorption near edge structure study of the nickel oxide electrode. J. Phys. Chem. 1989, 93, 6308–6311. [Google Scholar] [CrossRef]
- Dent, A.; Evans, J.; Newton, M.; Corker, J.; Russell, A.; Abdul Rahman, M.B.; Fiddy, S.; Mathew, R.; Farrow, R.; Salvini, G.; et al. High-quality energy-dispersive XAFS on the 1 s timescale applied to electrochemical and catalyst systems. J. Synchrotron Radiat. 1999, 6, 381–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascarelli, S.; Mathon, O.; Muñoz, M.; Mairs, T.; Susini, J. Energy-dispersive absorption spectroscopy for hard-X-ray micro-XAS applications. J. Synchrotron Radiat. 2006, 13, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Labiche, J.-C.; Mathon, O.; Pascarelli, S.; Newton, M.A.; Ferre, G.G.; Curfs, C.; Vaughan, G.B.M.; Homs, A.; Carreiras, D.F. The fast readout low noise camera as a versatile X-ray detector for time resolved dispersive extended X-ray absorption fine structure and diffraction studies of dynamic problems in materials science, chemistry, and catalysis. Rev. Sci. Instrum. 2007, 78, 91301. [Google Scholar] [CrossRef] [PubMed]
- Pascarelli, S.; Mathon, O. Advances in high brilliance energy dispersive X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 5535–5546. [Google Scholar] [CrossRef]
- Rose, A.; South, O.; Harvey, I.; Diaz-Moreno, S.; Owen, J.R.; Russell, A.E. In situ time resolved studies of hydride and deuteride formation in Pd/C electrodes via energy dispersive X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2005, 7, 366–372. [Google Scholar] [CrossRef]
- O’Malley, R.; Vollmer, A.; Lee, J.; Harvey, I.; Headspith, J.; Diaz-Moreno, S.; Rayment, T. Time-resolved studies of diffusion via energy dispersive X-ray absorption spectroscopy. Electrochem. commun. 2003, 5, 1–5. [Google Scholar] [CrossRef]
- Allen, P.G.; Conradson, S.D.; Wilson, M.S.; Gottesfeld, S.; Raistrick, I.D.; Valerio, J.; Lovato, M. In situ structural characterization of a platinum electrocatalyst by dispersive X-ray absorption spectroscopy. Electrochim. Acta 1994, 39, 2415–2418. [Google Scholar] [CrossRef]
- Imai, H.; Izumi, K.; Matsumoto, M.; Kubo, Y.; Kato, K.; Imai, Y. In Situ and Real-Time Monitoring of Oxide Growth in a Few Monolayers at Surfaces of Platinum Nanoparticles in Aqueous Media In Situ and Real-Time Monitoring of Oxide Growth in a Few Monolayers at Surfaces of Platinum Nanoparticles in Aqueous. J. Am. Chem. Soc. 2009, 131, 6293–6300. [Google Scholar] [CrossRef] [PubMed]
- Minguzzi, A.; Montagna, L.; Falqui, A.; Vertova, A.; Rondinini, S.; Ghigna, P. Dynamics of oxide growth on Pt nanoparticles electrodes in the presence of competing halides by operando energy dispersive X-ray absorption spectroscopy. Electrochim. Acta 2018, 270, 378–386. [Google Scholar] [CrossRef]
- Zolfaghari, A.; Conway, B.E.; Jerkiewicz, G. Elucidation of the effects of competitive adsorption of Cl-and Br- ions on the initial stages of Pt surface oxidation by means of electrochemical nanogravimetry. Electrochim. Acta 2002, 47, 1173–1187. [Google Scholar] [CrossRef]
- Rondinini, S.; Minguzzi, A.; Achilli, E.; Locatelli, C.; Agostini, G.; Spinolo, G.; Vertova, A.; Ghigna, P. The dynamics of pseudocapacitive phenomena studied by Energy Dispersive XAS on hydrous iridium oxide electrodes in alkaline media. Electrochim. Acta 2016, 212, 247–253. [Google Scholar] [CrossRef]
- Achilli, E.; Vertova, A.; Visibile, A.; Locatelli, C.; Minguzzi, A.; Rondinini, S.; Ghigna, P. Structure and Stability of a Copper(II) Lactate Complex in Alkaline Solution: A Case Study by Energy-Dispersive X-ray Absorption Spectroscopy. Inorg. Chem. 2017, 56. [Google Scholar] [CrossRef] [PubMed]
- Bressler, C.; Milne, C.; Pham, V.-T.; Elnahhas, A.; van der Veen, R.M.; Gawelda, W.; Johnson, S.; Beaud, P.; Grolimund, D.; Kaiser, M.; Borca, C.N.; et al. Femtosecond XANES study of the light-induced spin crossover dynamics in an iron(II) complex. Science 2009, 323, 489–492. [Google Scholar] [CrossRef]
- Rittmann-Frank, M.H.; Milne, C.J.; Rittmann, J.; Reinhard, M.; Penfold, T.J.; Chergui, M. Mapping of the photoinduced electron traps in TiO2 by picosecond X-ray absorption spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 5858–5862. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Moré, R.; Schilling, M.; Smolentsev, G.; Azzaroli, N.; Fox, T.; Luber, S.; Patzke, G.R. {Co4O4} and {CoxNi4-xO4} Cubane Water Oxidation Catalysts as Surface Cut-Outs of Cobalt Oxides. J. Am. Chem. Soc. 2017, 139, 14198–14208. [Google Scholar] [CrossRef]
- Moonshiram, D.; Gimbert-Suriñach, C.; Guda, A.; Picon, A.; Lehmann, C.S.; Zhang, X.; Doumy, G.; March, A.M.; Benet-Buchholz, J.; Soldatov, A.; et al. Tracking the Structural and Electronic Configurations of a Cobalt Proton Reduction Catalyst in Water. J. Am. Chem. Soc. 2016, 138, 10586–10596. [Google Scholar] [CrossRef] [PubMed]
- Canton, S.E.; Zhang, X.; Liu, Y.; Zhang, J.; Pápai, M.; Corani, A.; Smeigh, A.L.; Smolentsev, G.; Attenkofer, K.; Jennings, G.; et al. Watching the dynamics of electrons and atoms at work in solar energy conversion. Faraday Discuss. 2015, 185, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Chergui, M. Emerging photon technologies for chemical dynamics. Faraday Discuss. 2014, 171, 11–40. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.D.; Abela, R. Novel opportunities for time-resolved absorption spectroscopy at the X-ray free electron laser. Phys. Chem. Chem. Phys. 2010, 12, 5647–5652. [Google Scholar] [CrossRef] [PubMed]
- Lemke, H.T.; Bressler, C.; Chen, L.X.; Fritz, D.M.; Gaffney, K.J.; Galler, A.; Gawelda, W.; Haldrup, K.; Hartsock, R.W.; Ihee, H.; et al. Femtosecond X-ray absorption spectroscopy at a hard X-ray free electron laser: Application to spin crossover dynamics. J. Phys. Chem. A 2013, 117, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Oloff, L.P.; Chainani, A.; Matsunami, M.; Takahashi, K.; Togashi, T.; Osawa, H.; Hanff, K.; Quer, A.; Matsushita, R.; Shiraishi, R.; et al. Time-resolved HAXPES using a microfocused XFEL beam: From vacuum space-charge effects to intrinsic charge-carrier recombination dynamics. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Uemura, Y.; Kido, D.; Wakisaka, Y.; Uehara, H.; Ohba, T.; Niwa, Y.; Nozawa, S.; Sato, T.; Ichiyanagi, K.; Fukaya, R.; et al. Dynamics of Photoelectrons and Structural Changes of Tungsten Trioxide Observed by Femtosecond Transient XAFS. Angew. Chemie Int. Ed. 2016, 55, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- Picón, A.; Lehmann, C.S.; Bostedt, C.; Rudenko, A.; Marinelli, A.; Osipov, T.; Rolles, D.; Berrah, N.; Bomme, C.; Bucher, M.; et al. Hetero-site-specific X-ray pump-probe spectroscopy for femtosecond intramolecular dynamics. Nat. Commun. 2016, 7, 5–10. [Google Scholar] [CrossRef]
- Santomauro, F.G.; Lübcke, A.; Rittmann, J.; Baldini, E.; Ferrer, A.; Silatani, M.; Zimmermann, P.; Grübel, S.; Johnson, J.A.; Mariager, S.O.; et al. Femtosecond X-ray absorption study of electron localization in photoexcited anatase TiO2. Sci. Rep. 2015, 5, 14834. [Google Scholar] [CrossRef] [Green Version]
- Baran, T.; Fracchia, M.; Vertova, A.; Achilli, E.; Naldoni, A.; Malara, F.; Rossi, G.; Rondinini, S.; Ghigna, P.; Minguzzi, A.; et al. Operando and Time-Resolved X-ray Absorption Spectroscopy for the Study of Photoelectrode Architectures. Electrochim. Acta 2016, 207, 16–21. [Google Scholar] [CrossRef]
- Filipponi, A.; Borowski, M.; Loeffen, P.W.; De Panfilis, S.; Di Cicco, A.; Sperandini, F.; Minicucci, M.; Giorgetti, M. Single-energy X-ray absorption detection: A combined electronic and structural local probe for phase transitions in condensed matter. J. Phys. Condens. Matter 1998, 10, 235–253. [Google Scholar] [CrossRef]
- Minguzzi, A.; Lugaresi, O.; Locatelli, C.; Rondinini, S.; D’Acapito, F.; Achilli, E.; Ghigna, P. Fixed energy X-ray absorption voltammetry. Anal. Chem. 2013, 85, 7009–7013. [Google Scholar] [CrossRef] [PubMed]
- Rondinini, S.; Lugaresi, O.; Achilli, E.; Locatelli, C.; Vertova, A.; Ghigna, P.; Minguzzi, A.; Comninellis, C. Fixed Energy X-ray Absorption Voltammetry and Extended X-ray Absorption Fine Structure of Ag Nanoparticle Electrodes. J. Electroanal. Chem. 2016, 766, 71–77. [Google Scholar] [CrossRef]
- Baran, T.; Wojtyła, S.; Lenardi, C.; Vertova, A.; Ghigna, P.; Achilli, E.; Fracchia, M.; Rondinini, S.; Minguzzi, A. An Efficient CuxO Photocathode for Hydrogen Production at Neutral pH: New Insights from Combined Spectroscopy and Electrochemistry. ACS Appl. Mater. Interfaces 2016, 8, 21250–21260. [Google Scholar] [CrossRef] [PubMed]
- Fracchia, M.; Visibile, A.; Ahlberg, E.; Vertova, A.; Minguzzi, A.; Ghigna, P.; Rondinini, S. α- and γ-FeOOH: Stability, Reversibility, and Nature of the Active Phase under Hydrogen Evolution. ACS Appl. Energy Mater. 2018, 1, 1716–1725. [Google Scholar] [CrossRef]
- Montegrossi, G.; Giaccherini, A.; Berretti, E.; Di Benedetto, F.; Innocenti, M.; d’Acapito, F.; Lavacchi, A. Computational Speciation Models: A Tool for the Interpretation of Spectroelectrochemistry for Catalytic Layers under Operative Conditions. J. Electrochem. Soc. 2017, 164, E3690–E3695. [Google Scholar] [CrossRef] [Green Version]
- Achilli, E.; Minguzzi, A.; Visibile, A.; Locatelli, C.; Vertova, A.; Naldoni, A.; Rondinini, S.; Auricchio, F.; Marconi, S.; Fracchia, M.; et al. 3D-printed photo-spectroelectrochemical devices for in situ and in operando X-ray absorption spectroscopy investigation. J. Synchrotron Radiat. 2016, 23, 622–628. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fracchia, M.; Ghigna, P.; Vertova, A.; Rondinini, S.; Minguzzi, A. Time-Resolved X-ray Absorption Spectroscopy in (Photo)Electrochemistry. Surfaces 2018, 1, 138-150. https://doi.org/10.3390/surfaces1010011
Fracchia M, Ghigna P, Vertova A, Rondinini S, Minguzzi A. Time-Resolved X-ray Absorption Spectroscopy in (Photo)Electrochemistry. Surfaces. 2018; 1(1):138-150. https://doi.org/10.3390/surfaces1010011
Chicago/Turabian StyleFracchia, Martina, Paolo Ghigna, Alberto Vertova, Sandra Rondinini, and Alessandro Minguzzi. 2018. "Time-Resolved X-ray Absorption Spectroscopy in (Photo)Electrochemistry" Surfaces 1, no. 1: 138-150. https://doi.org/10.3390/surfaces1010011