
Temperature-Dependent Structure–Function Properties of Bacterial Xylose Isomerase Enzyme for Food Applications: An In Silico Study
 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of the Experimental Sequences
2.2. Physicochemical Characterization
2.3. Structural Analysis
2.3.1. Primary Structure Prediction
2.3.2. Secondary Structure Analysis
2.3.3. Tertiary Structure Analysis
2.4. Homology Modeling and Structural Validation
2.5. Functional Analysis
2.6. Molecular Docking Analysis
2.6.1. Preparation of Amino-2-Hydroxymethyl-Propane-1,3-Diol and (4R)-2-Methylpentane-2,4- Diol Ligands
2.6.2. Molecular Docking to Investigate Protein–Ligand Interaction
3. Results and Discussions
3.1. Retrieval of the Experimental Sequences
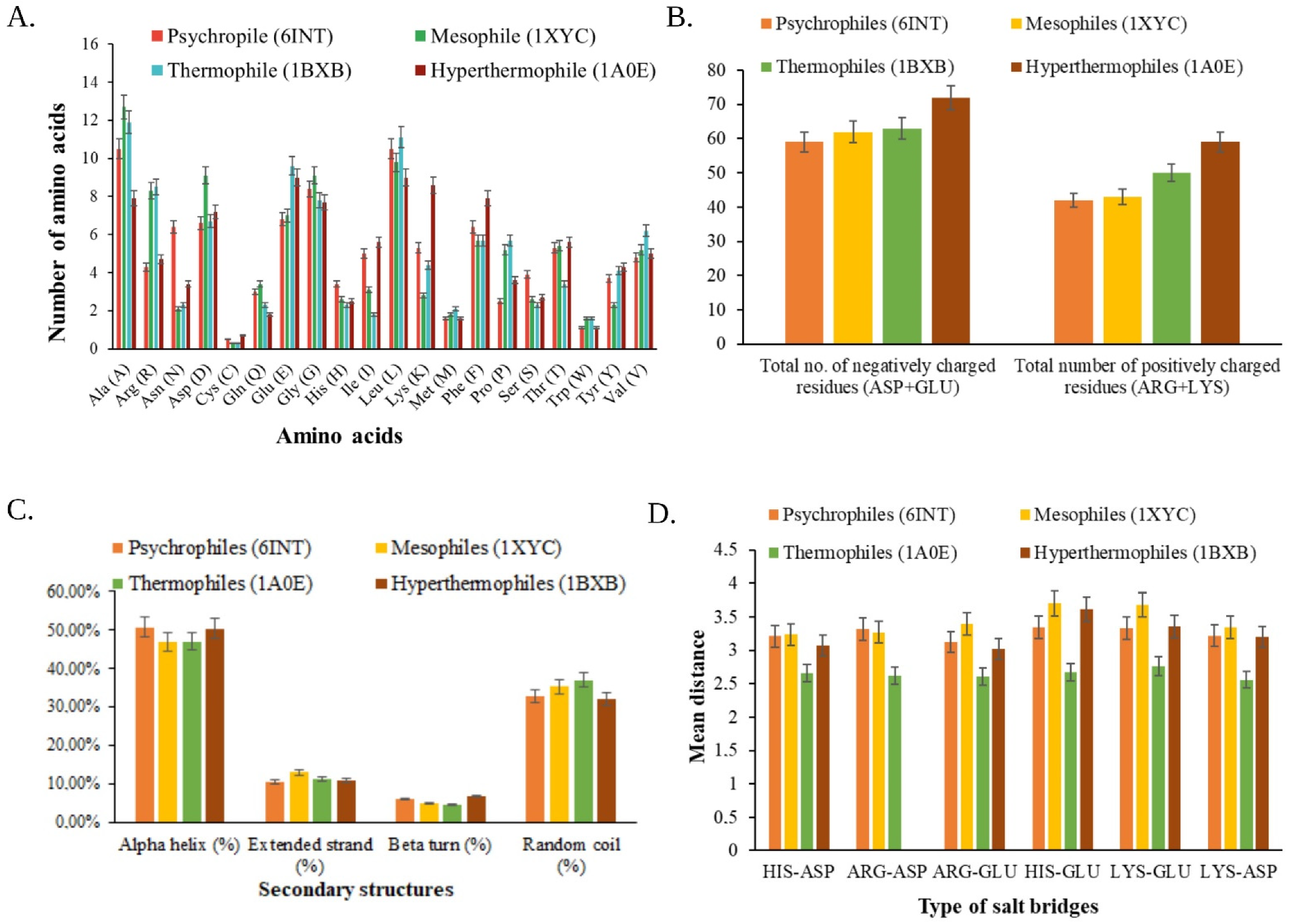
3.2. Physicochemical Characterization
3.3. Structural Analysis
3.3.1. Primary Structure Prediction
3.3.2. Secondary Structure Prediction
3.3.3. Analysis of Tertiary Structure
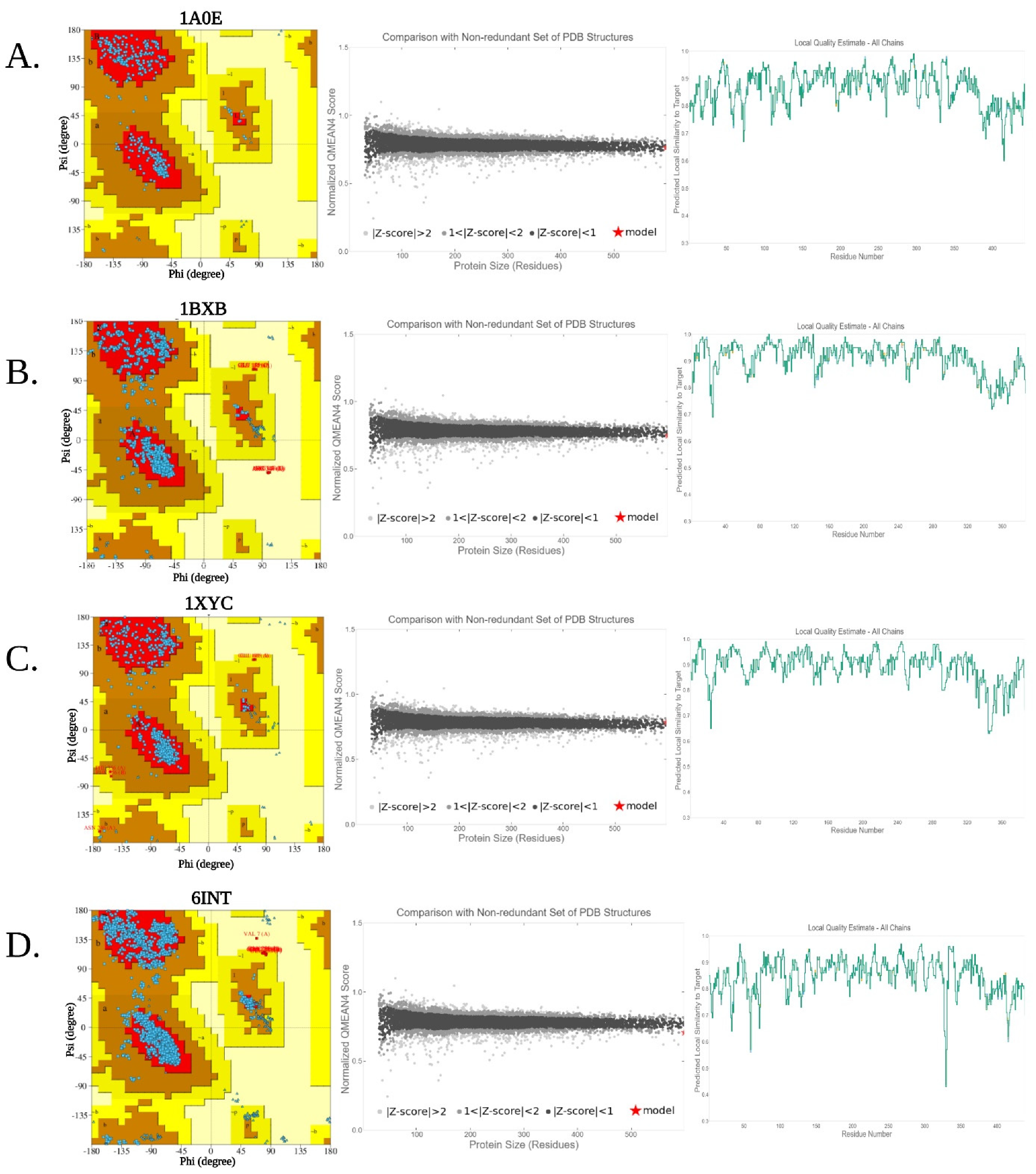
3.4. Homology Modeling and Structural Validation
3.5. Functional Analysis
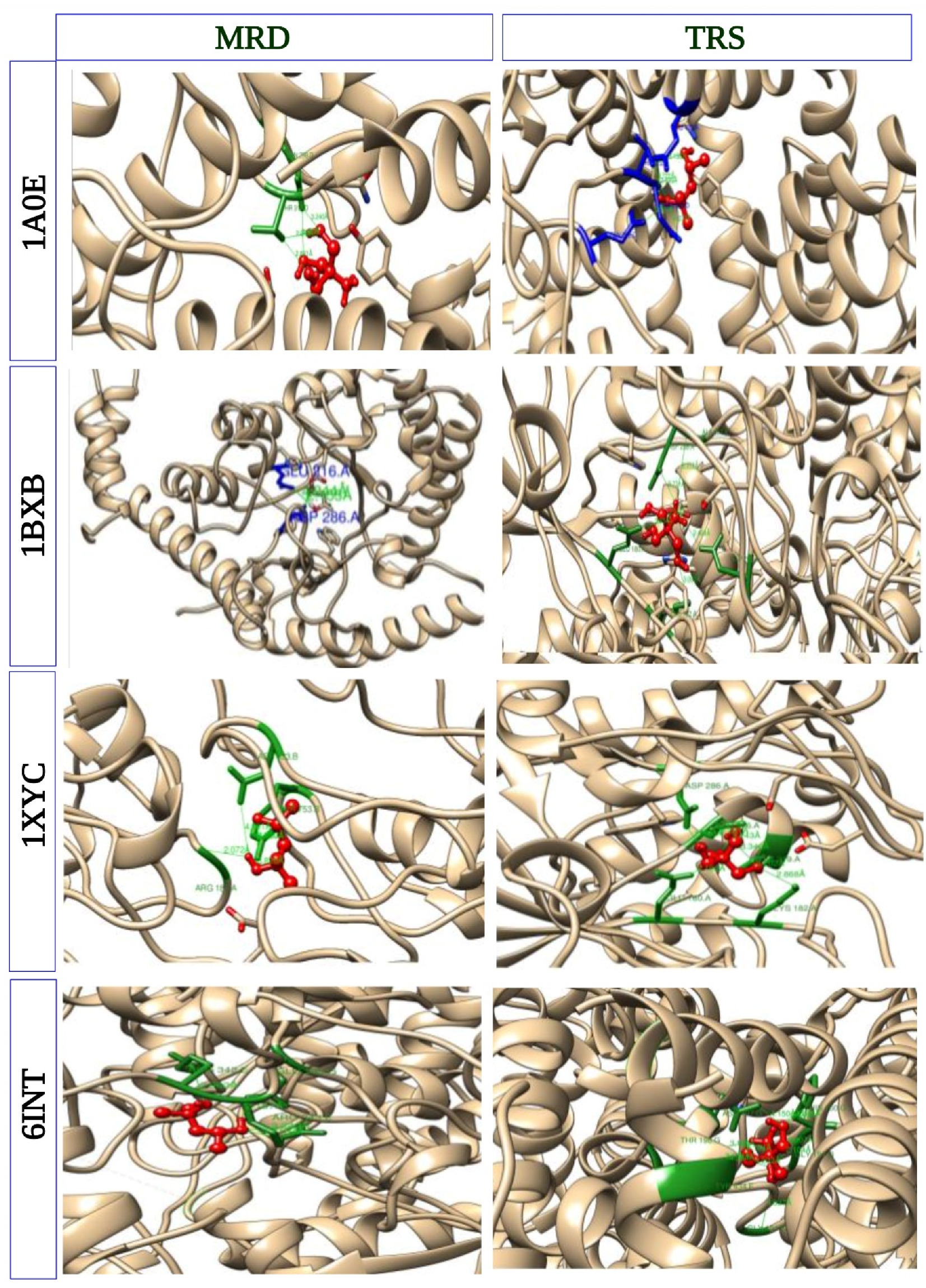
3.6. Molecular Docking to Investigate Protein–Ligand Interaction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, R.; Kumar, M.; Mittal, A.; Mehta, P.K. Microbial enzymes: Industrial progress in 21st century. 3 Biotech 2016, 6, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, V.; Rao, M. Glucose Isomerase. In Enzyme Technology; Springer: New York, NY, USA, 2006; pp. 239–252. [Google Scholar] [CrossRef]
- Bhosale, S.H.; Rao, M.B.; Deshpande, V.V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 1996, 60, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhabi, N.A.; Esmail, G.A.; Ghilan, A.K.M.; Arasu, M.V. Isolation and screening of Streptomyces sp. Al-Dhabi-49 from the environment of Saudi Arabia with concomitant production of lipase and protease in submerged fermentation. Saudi J. Biol. Sci. 2020, 27, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Glucose isomerase: Functions, structures, and applications. Appl. Sci. 2022, 12, 428. [Google Scholar] [CrossRef]
- Kilara, A.; Shahani, K.M.; Shukla, T.P. The use of immobilized enzymes in the food industry: A review. Crit. Rev. Food Sci. Nutr. 1979, 12, 161–198. [Google Scholar] [CrossRef] [PubMed]
- Staudigl, P.; Haltrich, D.; Peterbauer, C.K. L-Arabinose isomerase and D-xylose isomerase from Lactobacillus reuteri: Characterization, coexpression in the food grade host Lactobacillus plantarum, and application in the conversion of D-galactose and D-glucose. J. Agric. Food Chem. 2014, 62, 1617–1624. [Google Scholar] [CrossRef]
- Singh, R.S.; Singh, T.; Pandey, A. Microbial Enzymes—An Overview. In Advances in Enzyme Technology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–40. [Google Scholar]
- Rozanov, A.S.; Zagrebel’nyi, S.N.; Beklemishchev, A.B. Cloning of Escherichia coli K12 xylose isomerase (glucose isomerase) gene and studying the enzymatic properties of its expression product. Prikl. Biokhim. Mikrobiol. 2009, 45, 38–44. [Google Scholar] [CrossRef]
- Haldrup, A.; Noerremark, M.; Okkels, F.T. Plant selection principle based on xylose isomerase. Vitr. Cell. Dev. Biol.–Plant 2001, 37, 114–119. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; John, M.W., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Flores-Castañón, N.; Sarkar, S.; Banerjee, A. Structural, functional, and molecular docking analyses of microbial cutinase enzymes against polyurethane monomers. J. Hazard. Mater. Lett. 2022, 3, 100063. [Google Scholar] [CrossRef]
- Sarkar, S.; Banerjee, A.; Chakraborty, N.; Soren, K.; Chakraborty, P.; Bandopadhyay, R. Structural-functional analyses of textile dye degrading azoreductase, laccase and peroxidase: A comparative in silico study. Electron. J. Biotechnol. 2020, 43, 48–54. [Google Scholar] [CrossRef]
- Pramanik, K.; Kundu, S.; Banerjee, S.; Ghosh, P.K.; Maití, T.K. Computational-based structural, functional and phylogenetic analysis of Enterobacter phytases. 3 Biotech 2018, 8, 262. [Google Scholar] [CrossRef]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Costantini, S.; Colonna, G.; Facchiano, A.M. ESBRI: A web server for evaluating salt bridges in proteins. Bioinformation 2008, 3, 137. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; Von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the local structural quality of transmembrane protein models using statistical potentials (QMEANBrane). Bioinformatics 2014, 30, i505–i511. [Google Scholar] [CrossRef] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Caimano, M.J.; Sivasankaran, S.K.; Allard, A.; Hurley, D.; Hokamp, K.; Grassmann, A.A.; Hinton, J.C.D.; Nally, J.E. A model system for studying the transcriptomic and physiological changes associated with mammalian host-adaptation by Leptospira interrogans serovar Copenhageni. PLoS Pathog. 2014, 10, e1004004. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.-X.; Cao, Y. CB-Dock2: Improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Gan, J.; Xiao, Z.-X.; Cao, Y. FitDock: Protein–ligand docking by template fitting. Brief. Bioinform. 2022, 23, bbac087. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Grimm, M.; Dai, W.-T.; Hou, M.-C.; Xiao, Z.-X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Hasan, R.; Rony, M.N.H.; Ahmed, R. In silico characterization and structural modeling of bacterial metalloprotease of family M4. J. Genet. Eng. Biotechnol. 2021, 19, 25. [Google Scholar] [CrossRef]
- Gamage, D.G.; Gunaratne, A.; Periyannan, G.R.; Russell, T.G. Applicability of Instability Index for In vitro Protein Stability Prediction. Protein Pept. Lett. 2019, 26, 339–347. [Google Scholar] [CrossRef]
- Pramanik, K.; Soren, T.; Mitra, S.; Maiti, T.K. In silico structural and functional analysis of Mesorhizobium ACC deaminase. Comput. Biol. Chem. 2017, 68, 12–21. [Google Scholar] [CrossRef]
- Petukhov, M.; Kil, Y.; Kuramitsu, S.; Lanzov, V. Insights into thermal resistance of proteins from the intrinsic stability of their α-helices. Proteins 1997, 29, 309–320. [Google Scholar] [CrossRef]
- Kumar, S.; Tsai, C.-J.; Nussinov, R. Factors enhancing protein thermostability. Protein Eng. Des. Sel. 2000, 13, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side chain to main chain hydrogen bonds stabilize apolyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [Green Version]
- Behbahani, M.; Rabiei, P.; Mohabatkar, H. A Comparative Analysis of Allergen Proteins between Plants and Animals Using Several Computational Tools and Chou’s pseaac Concept. Int. Arch. Allergy Immunol. 2020, 181, 813–821. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Abdelhedi, O.; Nasri, R.; Mora, L.; Jridi, M.; Toldrá, F.; Nasri, M. In silico analysis and molecular docking study of angiotensin I-converting enzyme inhibitory peptides from smooth-hound viscera protein hydrolysates fractionated by ultrafiltration. Food Chem. 2018, 239, 453–463. [Google Scholar] [CrossRef]
- Gezegen, H.; Gürdere, M.B.; Dinçer, A.; Özbek, O.; Koçyiğit, Ü.M.; Taslimi, P.; Tüzün, B.; Budak, Y.; Ceylan, M. Synthesis, molecular docking, and biological activities of new cyanopyridine derivatives containing phenylurea. Arch. Pharm. 2021, 354, 2000334. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent advances and applications of molecular docking to G protein-coupled receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Shimshi, M.; Raz, K.; Eliaz, N.N.; Mhashal, A.R.; Ansbacher, T.; Major, D.T. Enzydock: Protein–ligand docking of multiple reactive states along a reaction coordinate in enzymes. J. Chem. Theory Comput. 2019, 15, 5116–5134. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, L. Improved protein–ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics 2014, 30, 1674–1680. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Serial No. | Physicochemical Characters | |||||||
|---|---|---|---|---|---|---|---|---|
| PDB ID | Bacterial Isolates | Number of AA | Theoretical PI | MW (Da) | II | AI | GRAVY | |
| 1. | Psychrophile (6INT) | Paenibacillus sp. R4 | 438 | 5.34 | 48,880.99 | 30.77 | 84.95 | −0.272 |
| 2. | Mesophile (1XYC) | Streptomyces olivochromogenes | 386 | 4.98 | 42,791.95 | 32.95 | 78.24 | −0.381 |
| 3. | Thermophile (1BXB) | Thermus thermophilus HB8 | 387 | 5.33 | 43,906.75 | 30.01 | 80.26 | −0.411 |
| 4. | Hyperthermophile (1A0E) | Thermotoga neapolitana | 443 | 5.47 | 50,761.77 | 29.61 | 79.53 | −0.377 |
| Serial No. | Quality Assesment Scores | |||||
|---|---|---|---|---|---|---|
| PDB ID | Bacterial Isolates | 3D-1D Score (%) | ERRAT Quality Factor | QMEAN Z-Score | AA in FR of Ramamchandran Plot (%) | |
| 1. | Psychrophile (6INT) | Paenibacillus sp. R4 | 88.70 | 96.15 | −0.30 | 91.4 |
| 2. | Mesophile (1XYC) | Streptomyces olivochromogenes | 91.06 | 98.95 | 1.25 | 92.6 |
| 3. | Thermophile (1BXB) | Thermus thermophilus HB8 | 87.47 | 97.62 | 0.43 | 89.9 |
| 4. | Hyperthermophile (1A0E) | Thermotoga neapolitana | 88.71 | 96.49 | −0.28 | 92.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Mehta, N.; Suravajhala, R.; Meza, C.; Sarkar, S.; Banerjee, A. Temperature-Dependent Structure–Function Properties of Bacterial Xylose Isomerase Enzyme for Food Applications: An In Silico Study. Clean Technol. 2022, 4, 1317-1329. https://doi.org/10.3390/cleantechnol4040081
Sharma M, Mehta N, Suravajhala R, Meza C, Sarkar S, Banerjee A. Temperature-Dependent Structure–Function Properties of Bacterial Xylose Isomerase Enzyme for Food Applications: An In Silico Study. Clean Technologies. 2022; 4(4):1317-1329. https://doi.org/10.3390/cleantechnol4040081
Chicago/Turabian StyleSharma, Maurya, Naayaa Mehta, Renuka Suravajhala, Cynthia Meza, Shrabana Sarkar, and Aparna Banerjee. 2022. "Temperature-Dependent Structure–Function Properties of Bacterial Xylose Isomerase Enzyme for Food Applications: An In Silico Study" Clean Technologies 4, no. 4: 1317-1329. https://doi.org/10.3390/cleantechnol4040081