
Investigation of the Microstructure of Fine-Grained YPO4:Gd Ceramics with Xenotime Structure after Xe Irradiation


 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
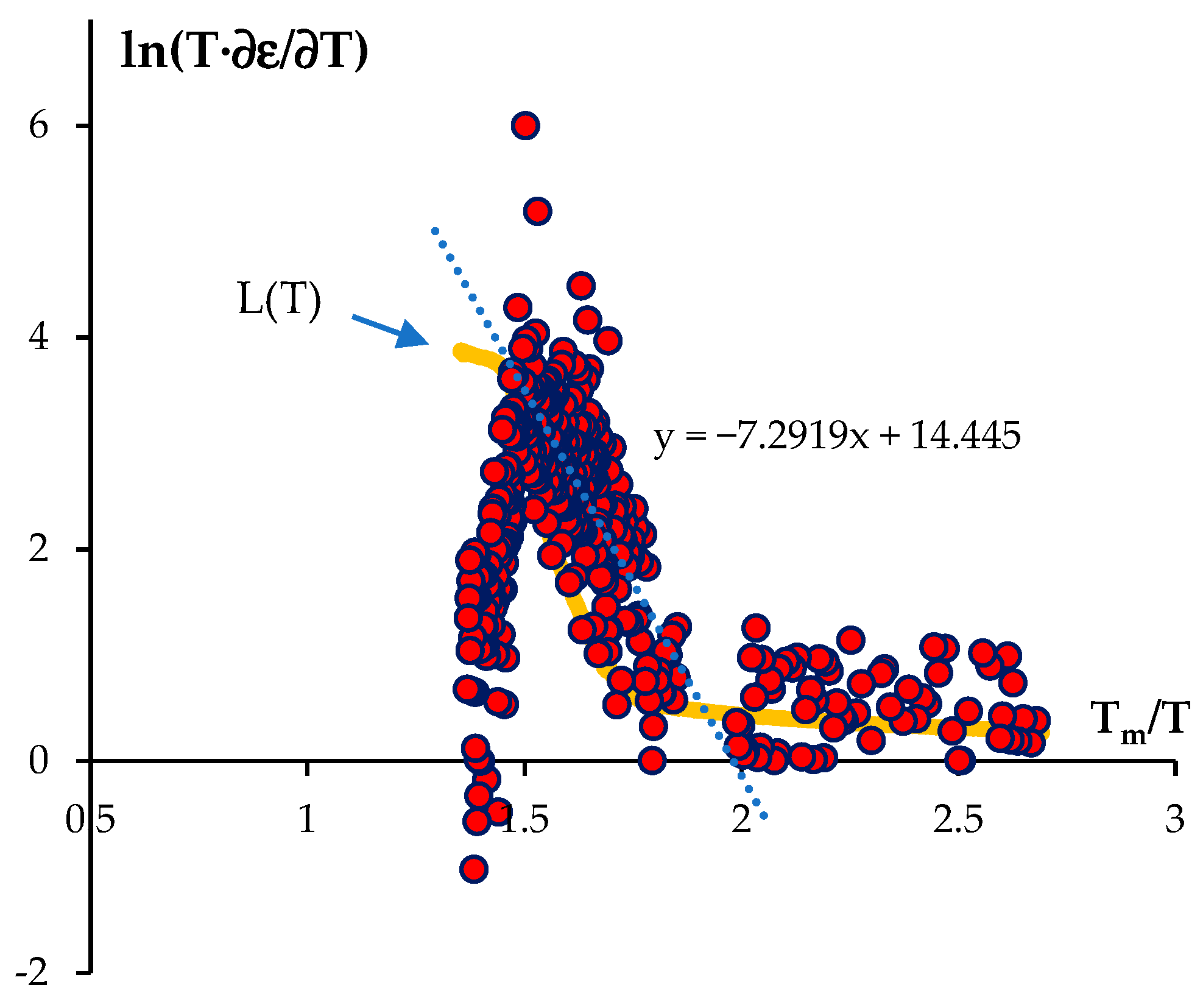
- The phosphate powders Y0.95Gd0.05PO4, where Gd acted as a simulator of Cm, were obtained with the sol-gel method. Ceramic samples with a high relative density (>98%) were obtained with spark plasma sintering (SPS). The sintering temperature was 1400 °C, and the whole sintering process duration was ~18 min. The SPS activation energy for the YPO4:Gd fine-grained ceramic was ~22 kTm (~210 kJ/mol).
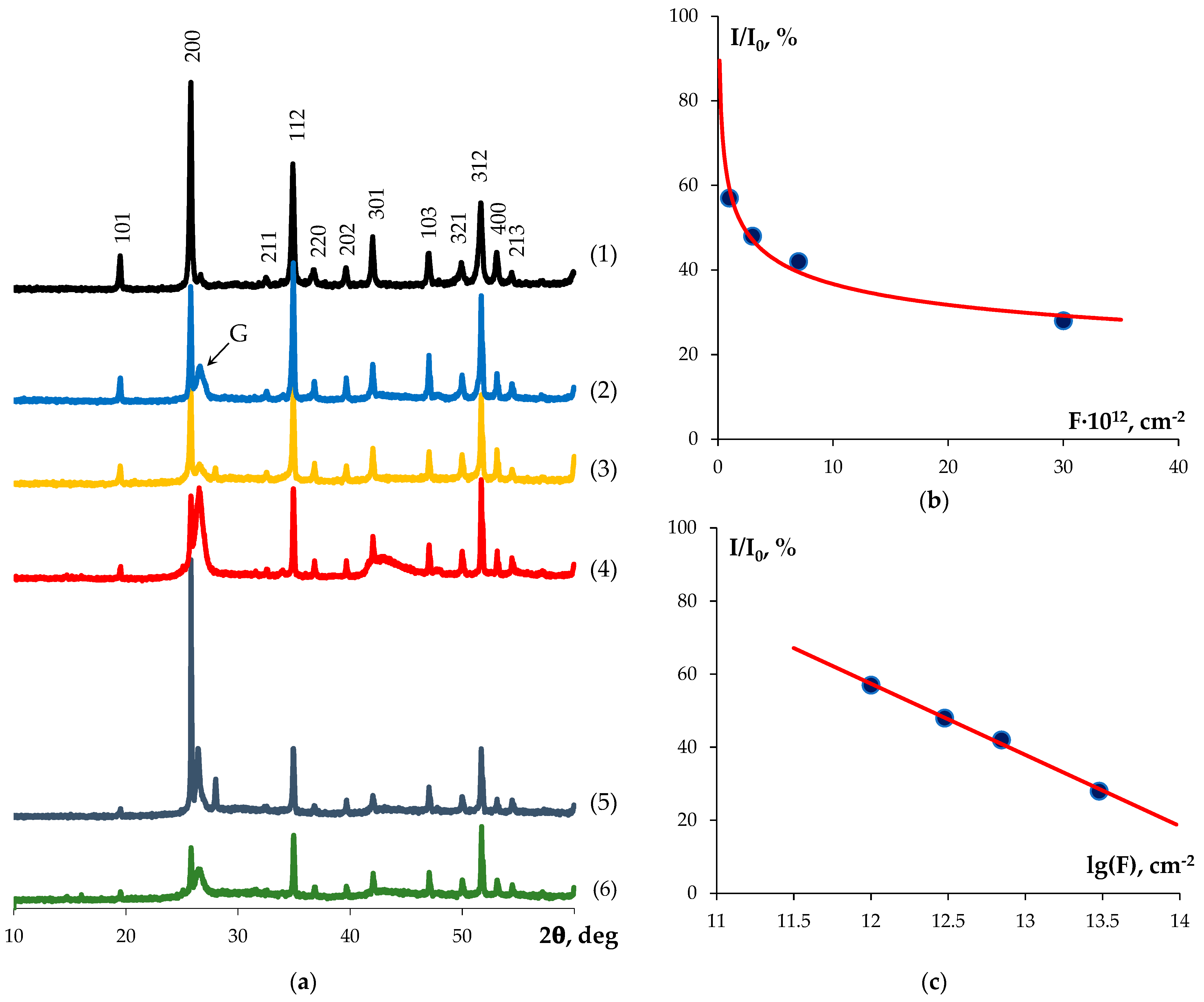
- The ceramic samples demonstrated a high resistance to irradiation from Xe ions with an energy of 167 MeV. At a maximum irradiation fluence of 3 × 1013 cm−2, the surface layers of the ceramic samples partially retained crystallinity. The calculated value of the fluence leading to the complete amorphization of the surface layers was (9.2 ± 0.1) × 1014 cm−2. After annealing at 500 °C, the metamict phase recovered. After heating at 700 °C, the recovery degree reached ~80%.
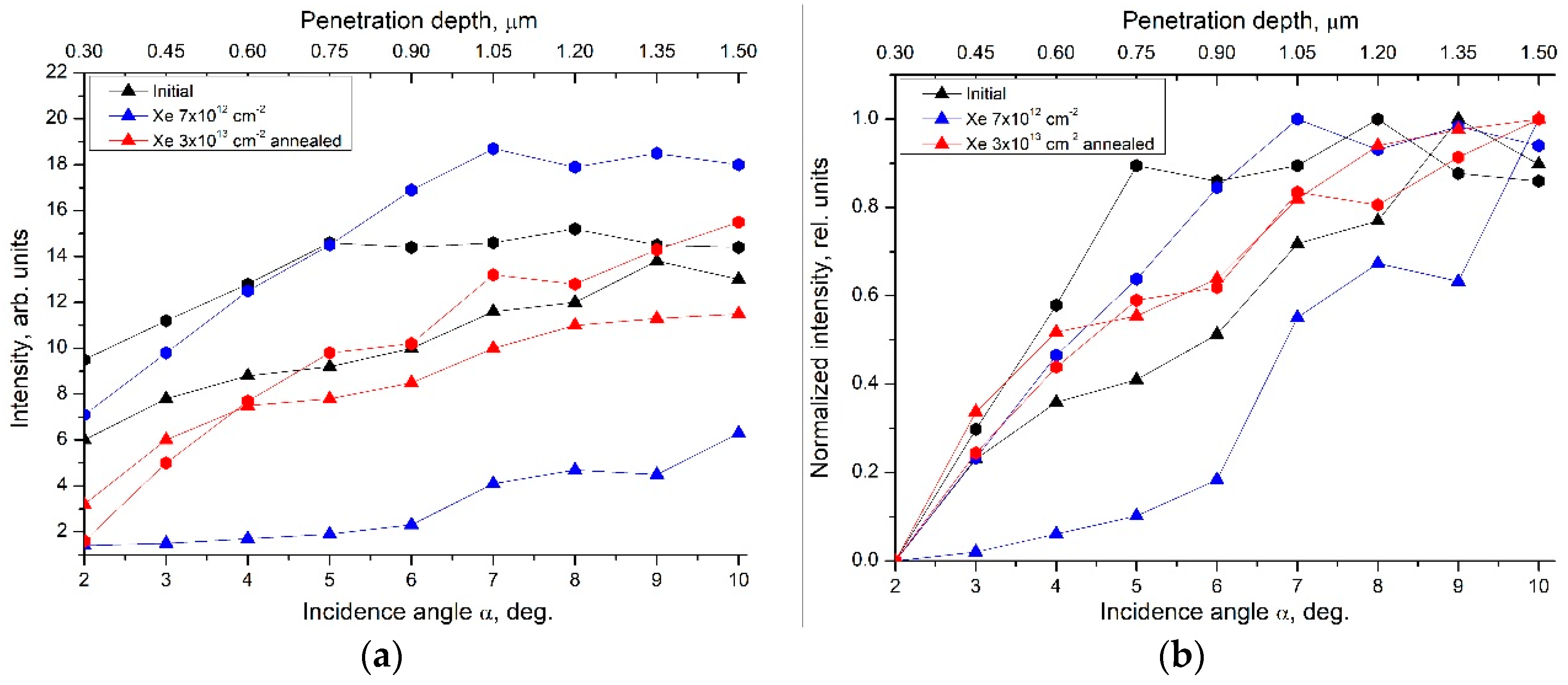
- GIXRD experiments revealed the presence of a graphite phase concentrated mainly near the surfaces of the ceramic samples. Irradiation with high energy ions resulted in the amorphization of the YPO4:Gd phase in the subsurface layers and weakly affected the crystallinity of the graphite phase. Increasing the irradiation fluence resulted in an increase in the amorphization degree of YPO4:Gd from 20% up to 70%. Subsequent annealing of the samples resulted in a decrease in the amorphization degree, down to the level of 20%, as well as in probable burnout and the diffusion of carbon inside the samples, which manifested as a more uniform depth distribution in the graphite phase of the annealed sample.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krivovichev, S.V.; Burns, P.C.; Tananaev, I.G. (Eds.) Structural Chemistry of Inorganic Actinide Compounds; Elsevier: Amsterdam, The Netherlands, 2007; p. 494. [Google Scholar] [CrossRef]
- Emden, B.; Thornber, M.R.; Graham, J.; Lincoln, F.J. The Incorporation of Actinides in Monazite and Xenotime from Placer Deposits in Western Australia. Can. Mineral. 1997, 35, 95–104. [Google Scholar]
- Orlova, A.I.; Ojovan, M.I. Ceramic mineral waste-forms for nuclear waste immobilization. Materials 2019, 12, 2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Hughes, J.M.; Mariano, A.N. Crystal chemistry of the monazite and xenotime structures. Am. Mineral. 1995, 80, 21–26. [Google Scholar] [CrossRef]
- Burakov, B.E.; Ojovan, M.I.; Lee, W.E. Crystalline Materials for Actinide Immobilization. Materials for Engineering; Imperial College Press: London, UK, 2010; Volume 1, p. 216. [Google Scholar] [CrossRef]
- Liu, G.K.; Lia, S.T.; Beitza, J.V.; Abraham, M. Effects of self-radiation damage on electronic properties of 244Cm3+ in an orthophosphate crystal of YPO4. J. Alloys Compd. 1998, 271–273, 872–875. [Google Scholar] [CrossRef]
- Vance, E.R.; Zhang, Y.; McLeod, T.; Davis, J. Actinide valences in xenotime and monazite. J. Nucl. Mater. 2011, 409, 221–224. [Google Scholar] [CrossRef]
- Zglinicki, K.; Malek, R.; Szamalek, K.; Wolkowics, S. Mining waste as a potential additional source of HREE and U for the European Green Deal: A case study if Bangka Island (Indonesia). Minerals 2022, 12, 44. [Google Scholar] [CrossRef]
- Smolka-Danielowska, D.; Walencik-Lata, A. The occurrence of selected radionuclides and rare earth elements in waste at the mine heap from the Polish Mining Group. Minerals 2021, 11, 504. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Geisler-Wierwille, T. Minerals and Natural Analogues. In Comprehensive Nuclear Materials; Konings, R.J.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 563–600. [Google Scholar]
- Boatner, L.A. Synthesis, structure and properties of monazite, pretulite, and xenotime. Rev. Mineral. Geochem. 2002, 48, 87–121. [Google Scholar] [CrossRef]
- Hikichi, Y.; Nomura, T. Melting temperatures of monazite and xenotime. J. Am. Ceram. Soc. 1987, 70, C-252–C-253. [Google Scholar] [CrossRef]
- Rafiuddin, M.R.; Grosvenor, A.P. An investigation of the chemical durability of hydrous and anhydrous rare-earth phosphates. J. Nucl. Mater. 2018, 509, 631–643. [Google Scholar] [CrossRef]
- Meldrum, A.; Boatner, L.A.; Ewing, R.C. Displacive radiation effects in the monazite- and zircon-structure orthophosphates. Phys. Rev. B 1997, 56, 13805. [Google Scholar] [CrossRef]
- Rafiuddin, M.R.; Grosvenor, A.P. Probing the effect of radiation damage on the structure of rare-earth phosphates. J. Alloys Compd. 2015, 653, 279–289. [Google Scholar] [CrossRef]
- Lenz, C.; Thorogood, G.; Aughterson, R.; Ionescu, M.; Gregg, D.J.; Davis, J.; Lumpkin, G.R. The quantification of radiation damage in orthophosphates using confocal μ-luminescence spectroscopy of Nd3+. Front. Chem. 2019, 7, 13. [Google Scholar] [CrossRef]
- Rafiuddin, M.R.; Seydoux-Guillaume, A.-M.; Deschanels, X.; Mesbah, A.; Baumier, C.; Szenknect, S.; Dacheux, N. An in-situ electron microscopy study of dual ion-beam irradiated xenotime-type ErPO4. J. Nucl. Mater. 2020, 539, 152265. [Google Scholar] [CrossRef]
- Hikichi, Y.; Ota, T.; Daimon, K.; Hattori, T. Thermal, mechanical, and chemical properties of sintered xenotime-type RPO4 (R = Y, Er, Yb, or Lu). J. Am. Ceram. Soc. 1998, 81, 2216–2218. [Google Scholar] [CrossRef]
- Cho, I.-S.; Choi, G.K.; An, J.-S.; Kim, J.R.; Hong, K.S. Sintering, microstructure and microwave dielectric properties of rare earth orthophosphates, RePO4 (Re = La, Ce, Nd, Sm, Tb, Dy, Y, Yb). Mater. Res. Bull. 2009, 44, 173–178. [Google Scholar] [CrossRef]
- Kuo, D.-H.; Kriven, W.M. Characterization of yttrium phosphate and a yttrium phosphate/yttrium aluminate laminate. J. Am. Ceram. Soc. 1995, 78, 3121–3124. [Google Scholar] [CrossRef]
- Bregiroux, D.; Lucas, S.; Champion, E.; Audubert, F.; Bernache-Assollant, D. Sintering and microstructure of rare earth phosphate ceramics REPO4 with Re = La, Ce or Y. J. Eur. Ceram. Soc. 2006, 26, 279–287. [Google Scholar] [CrossRef]
- Tokita, M. Progress of Spark Plasma Sintering (SPS) Method, Systems, Ceramics Application and Industrialization. Ceramics 2021, 4, 160–198. [Google Scholar] [CrossRef]
- Munir, Z.A.; Anselmi-Tamburini, U.; Ohyanagi, M. The effect of electric field and pressure on the synthesis and consolidation of materials: A review of the spark plasma sintering method. J. Mater. Sci. 2006, 41, 763–777. [Google Scholar] [CrossRef]
- Hu, Z.-Y.; Zhang, Z.-H.; Cheng, X.-W.; Wang, F.-C.; Zhang, Y.-F.; Li, S.-L. A review of multi-physical fields induced phenomena and effects in spark plasma sintering: Fundamentals and applications. Mater. Des. 2020, 191, 108662. [Google Scholar] [CrossRef]
- Orlova, A.I. Crystalline phosphates for HLW immobilization—Composition, structure, properties and production of ceramics. Spark Plasma Sintering as a promising sintering technology. J. Nucl. Mater. 2022, 559, 153407. [Google Scholar] [CrossRef]
- Olevsky, E.; Dudina, D. Field-Assisted Sintering: Science and Applications; Springer Nature: Cham, Switzerland, 2018; p. 425. [Google Scholar] [CrossRef]
- Papynov, E.K.; Shichalin, O.O.; Mironenko, A.Y.; Ryakov, A.V.; Manakov, I.V.; Makhrov, P.V.; Buravlev, I.G.; Tananaev, I.G.; Avramenko, V.A.; Sergienko, V.I. Synthesis of high-density pellets of uranium dioxide by spark plasma sintering in dies of different types. Radiochemistry 2018, 40, 362–370. [Google Scholar] [CrossRef]
- Papynov, E.K.; Shichalin, O.O.; Medkov, M.A.; Grishchenko, D.N.; Tkachenko, I.A.; Fedorets, A.N.; Pechnikov, V.S.; Golub, A.V.; Buravlev, I.Y.; Tananaev, I.G.; et al. Spark plasma sintering of special-purpose functional ceramics based on UO2, ZrO2, Fe3O4/α-Fe2O3. Glass Phys. Chem. 2018, 44, 632–640. [Google Scholar] [CrossRef]
- Shichalin, O.O.; Papynov, E.K.; Nepomnyushchaya, V.A.; Ivanets, A.I.; Belov, A.A.; Dran’kov, A.N.; Yarusova, S.B.; Buravlev, I.Y.; Tarabanova, A.E.; Fedorets, A.N.; et al. Hydrothermal synthesis and spark plasma sintering of NaY zeolite as solid-state matrices for cesium-137 immobilization. J. Eur. Ceram. Soc. 2022, 42, 3004–3014. [Google Scholar] [CrossRef]
- Papynov, E.K.; Belov, A.A.; Shichalin, O.O.; Buravlev, I.Y.; Azon, S.A.; Gridasova, E.A.; Parotkina, Y.A.; Yagofarov, V.Y.; Drankov, A.N.; Golub, A.V.; et al. Synthesis of perovskite-like SrTiO3 ceramics for radioactive strontium immobilization by Spark Plasma Sintering-Reactive Synthesis. Russ. J. Inorg. Chem. 2021, 66, 645–653. [Google Scholar] [CrossRef]
- Alekseeva, L.; Nokhrin, A.; Boldin, M.; Lantsev, E.; Murashov, A.; Orlova, A.; Chuvil’deev, V. Study of the hydrolytic stability of fine-grained ceramics based on Y2.5Nd0.5Al5O12 oxide with a garnet structure under hydrothermal conditions. Materials 2021, 14, 2152. [Google Scholar] [CrossRef]
- Orlova, A.I.; Volgutov, V.Y.; Mikhailov, D.A.; Bykov, D.M.; Skuratov, V.A.; Chuvil’deev, V.N.; Nokhrin, A.V.; Boldin, M.S.; Sakharov, N.V. Phosphate Ca1/4Sr1/4Zr2(PO4)3 of the NaZr2(PO4)3 structure type: Synthesis of a dense ceramic material and its radiation testing. J. Nucl. Mater. 2014, 446, 232–239. [Google Scholar] [CrossRef]
- Orlova, A.I.; Troshin, A.N.; Mikhailov, D.A.; Chuvil’deev, V.N.; Boldin, M.S.; Sakharov, N.V.; Nokhrin, A.V.; Skuratov, V.A.; Kirilkin, N.S. Phosphorous-containing cesium compounds of pollucite structure. Preparation of high-density ceramic and its radiation test. Radiochemistry 2014, 56, 98–104. [Google Scholar] [CrossRef]
- Orlova, A.I.; Malanina, N.V.; Chuvil’deev, V.N.; Boldin, M.S.; Sakharov, N.V.; Nokhrin, A.V. Praseodymium and neodymium phosphates Ca9Ln(PO4)7 of witlockite structure. Preparation of a ceramic with a high relative density. Radiochemistry 2014, 56, 380–384. [Google Scholar] [CrossRef]
- Potanina, E.A.; Orlova, A.I.; Nokhrin, A.V.; Boldin, M.S.; Sakharov, N.V.; Belkin, O.A.; Chuvil’deev, V.N.; Tokarev, M.G.; Shotin, S.V.; Zelenov, A.Y. Characterization of Nax(Ca/Sr)1-2xNdxWO4 complex tungstates fine-grained ceramics obtained by Spark Plasma Sintering. Ceram. Int. 2018, 44, 4033–4044. [Google Scholar] [CrossRef]
- Mikhailov, D.A.; Orlova, A.I.; Malanina, N.V.; Nokhrin, A.V.; Potanina, E.A.; Chuvil’deev, V.N.; Boldin, M.S.; Sakharov, N.V.; Belkin, O.A.; Kalenova, M.Y.; et al. A study of fine-grained ceramics based on complex oxides ZrO2-Ln2O3 (Ln = Sm, Yb) obtained by Spark Plasma Sintering for inert matrix fuel. Ceram. Int. 2018, 44, 18595–18608. [Google Scholar] [CrossRef]
- Potanina, E.A.; Orlova, A.I.; Mikhailov, D.A.; Nokhrin, A.V.; Chuvil’deev, V.N.; Boldin, M.S.; Sakharov, N.V.; Lantcev, E.A.; Tokarev, M.G.; Murashov, A.A. Spark Plasma Sintering of fine-grained SrWO4 and NaNd(WO4)2 tungstates ceramics with the scheelite structure for nuclear waste immobilization. J. Alloys Compd. 2019, 774, 182–190. [Google Scholar] [CrossRef]
- Shichalin, O.O.; Papynov, E.K.; Maiorov, V.Y.; Belov, A.A.; Modin, E.B.; Buravlev, I.Y.; Azarova, Y.A.; Golub, A.V.; Gridasova, E.A.; Sukhorada, A.E.; et al. Spark plasms sintering of aluminosilicate ceramic matrices for immobilization of cesium radionuclides. Radiochemistry 2019, 61, 185–191. [Google Scholar] [CrossRef]
- Papynov, E.K.; Shichalin, O.O.; Mayorov, V.Y.; Kuryavyi, V.G.; Kaidalova, T.A.; Teplukhina, L.V.; Portnyagin, A.S.; Slobodyuk, A.B.; Belov, A.A.; Tananaev, I.G.; et al. SPS technique for ionizing radiation source fabrication based on dense cesium-containing core. J. Hazard. Mater. 2019, 369, 25–30. [Google Scholar] [CrossRef]
- Boldin, M.S.; Popov, A.A.; Lantsev, E.A.; Nokhrin, A.V.; Chuvil’deev, V.N. Investigation of the densification behavior of alumina during Spark Plasma Sintering. Materials 2022, 15, 2167. [Google Scholar] [CrossRef]
- Lantsev, E.; Nokhrin, A.; Malekhonova, N.; Boldin, M.; Chuvil’deev, V.; Blagoveshchenskiy, Y.; Isaeva, N.; Andreev, P.; Smetanina, K.; Murashov, A. A study of the impact of graphite on the kinetics of SPS in nano- and submicron WC-10% Co powder composition. Ceramics 2021, 4, 331–363. [Google Scholar] [CrossRef]
- Lantsev, E.A.; Malekhonova, N.V.; Nokhrin, A.V.; Chuvil’deev, V.N.; Boldin, M.S.; Andreev, P.V.; Smetanina, K.E.; Balgoveshchenskiy, Y.V.; Isaeva, N.V.; Murashov, A.A. Spark plasma sintering of fine-grained WC hard alloys with ultra-low cobalt content. J. Alloys Compd. 2021, 857, 157535. [Google Scholar] [CrossRef]
- Frost, H.J.; Ashby, M.F. Deformation Mechanism Maps; Elsevier: Amsterdam, The Netherlands, 1982; p. 328. [Google Scholar]
- Pelleg, J. Diffusion in Ceramics; Springer International Publishing: Cham, Switzerland, 2016; p. 448. [Google Scholar] [CrossRef]
- Chuvil’deev, V.N.; Boldin, M.S.; Dyatlova, Y.G.; Rumyantsev, V.I.; Ordan’yan, S.S. Comparative study of hot pressing and Spark Plasma Sintering of Al2O3/ZrO2/Ti(C,N) powders. Russ. J. Inorg. Chem. 2015, 60, 987–993. [Google Scholar] [CrossRef]
- Dudina, D.V.; Bokhonov, B.B.; Ukhina, A.V.; Anisimov, A.G.; Mali, V.I.; Esikov, M.A.; Batraev, I.S.; Kuznechik, O.O.; Pilinevich, L.P. Reactivity of materials towards carbon of graphite foil during Spark Plasma Sintering: A case study using Ni-W powders. Mater. Lett. 2016, 168, 62–67. [Google Scholar] [CrossRef]
- Nokhrin, A.; Andreev, P.; Boldin, M.; Chuvil’deev, V.; Chegurov, M.; Smetanina, K.; Gryaznov, M.; Shotin, S.; Nazarov, A.; Shcherbak, G.; et al. Investigation of microstructure and corrosion resistance of Ti-Al-V titanium alloys obtained by Spark Plasma Sintering. Metals 2021, 11, 945. [Google Scholar] [CrossRef]
- Bernard-Granger, G.; Benameur, N.; Guizard, C.; Nygren, M. Influence of graphite contamination on the optical properties of transparent spinel obtained by spark plasma sintering. Scr. Mater. 2009, 60, 164–167. [Google Scholar] [CrossRef]
- Wang, P.; Yang, M.; Zhang, S.; Tu, R.; Goto, T.; Zhang, L. Suppression of carbon contamination in SPSed CaF2 transparent ceramics by Mo foil. J. Eur. Ceram. Soc. 2017, 37, 4103–4107. [Google Scholar] [CrossRef]
- Kosyanov, D.Y.; Vornovskikh, A.A.; Zakharenko, A.M.; Gridasova, E.A.; Yavetskiy, R.P.; Dobrotvorskaya, M.V.; Tolmacheva, A.V.; Shichalin, O.O.; Papynov, E.K.; Ustinov, A.Y.; et al. Influence of sintering parameters on transparency of reactive SPSed Nd3+:YAG ceramics. Opt. Mater. 2021, 112, 110760. [Google Scholar] [CrossRef]
- Wang, P.; Huang, Z.; Morita, K.; Li, Q.; Yang, M.; Zhang, S.; Goto, T.; Tu, R. Influence of spark plasma sintering conditions on microstructure, carbon contamination, and transmittance of CaF2 ceramics. J. Eur. Ceram. Soc. 2022, 42, 245–257. [Google Scholar] [CrossRef]
- Yong, S.-K.; Choi, D.H.; Lee, K.; Ko, S.-Y.; Cheong, D.-I.; Park, Y.-J.; Go, S.-I. Study of the carbon contamination and carboxylate group formation in Y2O3-MgO nanocomposites fabricated by spark plasma sintering. J. Eur. Ceram. Soc. 2020, 40, 847–851. [Google Scholar] [CrossRef]
- Hammoud, H.; Garnier, V.; Fantozzi, G.; Lachaud, E.; Taider, S. Mechanism of carbon contamination in transparent MgAl2O4 and Y3Al5O12 ceramics sintered by Spark Plasma Sintering. Ceramics 2019, 2, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Kim, B.-N.; Yoshida, H.; Higara, K.; Sakka, Y. Distribution of carbon contamination in oxide ceramics occurring during spark-plasma-sintering (SPS) processing: II—Effect of SPS and loading temperatures. J. Eur. Ceram. Soc. 2018, 38, 2596–2604. [Google Scholar] [CrossRef]
- Nečina, V.; Pabst, W. Reduction of temperature gradient and carbon contamination in electric current assisted sintering (ECAS/SPS) using a “saw-tooth” heating schedule. Ceram. Int. 2019, 45, 22987–22990. [Google Scholar] [CrossRef]
- Henke, B.L.; Gullikson, E.M.; Davis, J.C. X-ray interactions: Photoabsorption, scattering, transmission, and reflection at E=50-30000 eV, Z=1-92. At. Data Nucl. Data Tables 1993, 54, 181–342. [Google Scholar] [CrossRef] [Green Version]
- Young, W.S.; Cutler, I.B. Initial sintering with constant rates of heating. J. Am. Ceram. Soc. 1970, 53, 659–663. [Google Scholar] [CrossRef]
- Golovkina, L.S.; Orlova, A.I.; Nokhrin, A.V.; Boldin, M.S.; Chuvil’deev, V.N.; Sakharov, N.V.; Belkin, O.A.; Shotin, S.V.; Zelenov, A.Y. Spark Plasma Sintering of fine-grained ceramic-metal composites based on garnet-structure oxide Y2.5Nd0.5Al5O12 for inert matrix fuel. Mater. Chem. Phys. 2018, 214, 516–526. [Google Scholar] [CrossRef]
- Golovkina, L.S.; Orlova, A.I.; Boldin, M.S.; Sakharov, N.V.; Chuvil’deev, V.N.; Nokhrin, A.V.; Konings, R.; Staicu, D. Development of composite ceramic materials with improved thermal conductivity and plasticity based on garnet-type oxide. J. Nucl. Mater. 2017, 489, 158–163. [Google Scholar] [CrossRef]
- Golovkina, L.S.; Orlova, A.I.; Nokhrin, A.V.; Boldin, M.S.; Lantsev, E.A.; Chuvil’deev, V.N.; Sakharov, N.V.; Shotin, S.V.; Zelenov, A.Y. Spark Plasma Sintering of fine-grained ceramic-metal composites YAG:Nd-(W,Mo) based on garnet-type oxide Y2.5Nd0.5Al5O12 for inert matrix fuel. J. Nucl. Mater. 2018, 511, 109–121. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhaylov, D.A.; Potanina, E.A.; Nokhrin, A.V.; Orlova, A.I.; Yunin, P.A.; Sakharov, N.V.; Boldin, M.S.; Belkin, O.A.; Skuratov, V.A.; Issatov, A.T.; et al. Investigation of the Microstructure of Fine-Grained YPO4:Gd Ceramics with Xenotime Structure after Xe Irradiation. Ceramics 2022, 5, 237-252. https://doi.org/10.3390/ceramics5020019
Mikhaylov DA, Potanina EA, Nokhrin AV, Orlova AI, Yunin PA, Sakharov NV, Boldin MS, Belkin OA, Skuratov VA, Issatov AT, et al. Investigation of the Microstructure of Fine-Grained YPO4:Gd Ceramics with Xenotime Structure after Xe Irradiation. Ceramics. 2022; 5(2):237-252. https://doi.org/10.3390/ceramics5020019
Chicago/Turabian StyleMikhaylov, Dmitriy A., Ekaterina A. Potanina, Aleksey V. Nokhrin, Albina I. Orlova, Pavel A. Yunin, Nikita V. Sakharov, Maksim S. Boldin, Oleg A. Belkin, Vladimir A. Skuratov, Askar T. Issatov, and et al. 2022. "Investigation of the Microstructure of Fine-Grained YPO4:Gd Ceramics with Xenotime Structure after Xe Irradiation" Ceramics 5, no. 2: 237-252. https://doi.org/10.3390/ceramics5020019