
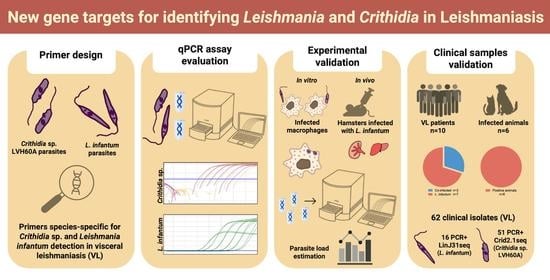
Parasite Detection in Visceral Leishmaniasis Samples by Dye-Based qPCR Using New Gene Targets of Leishmania infantum and Crithidia
 , , , , , , ,
, , , , , , ,
Abstract
:
1. Introduction
2. Material and Methods
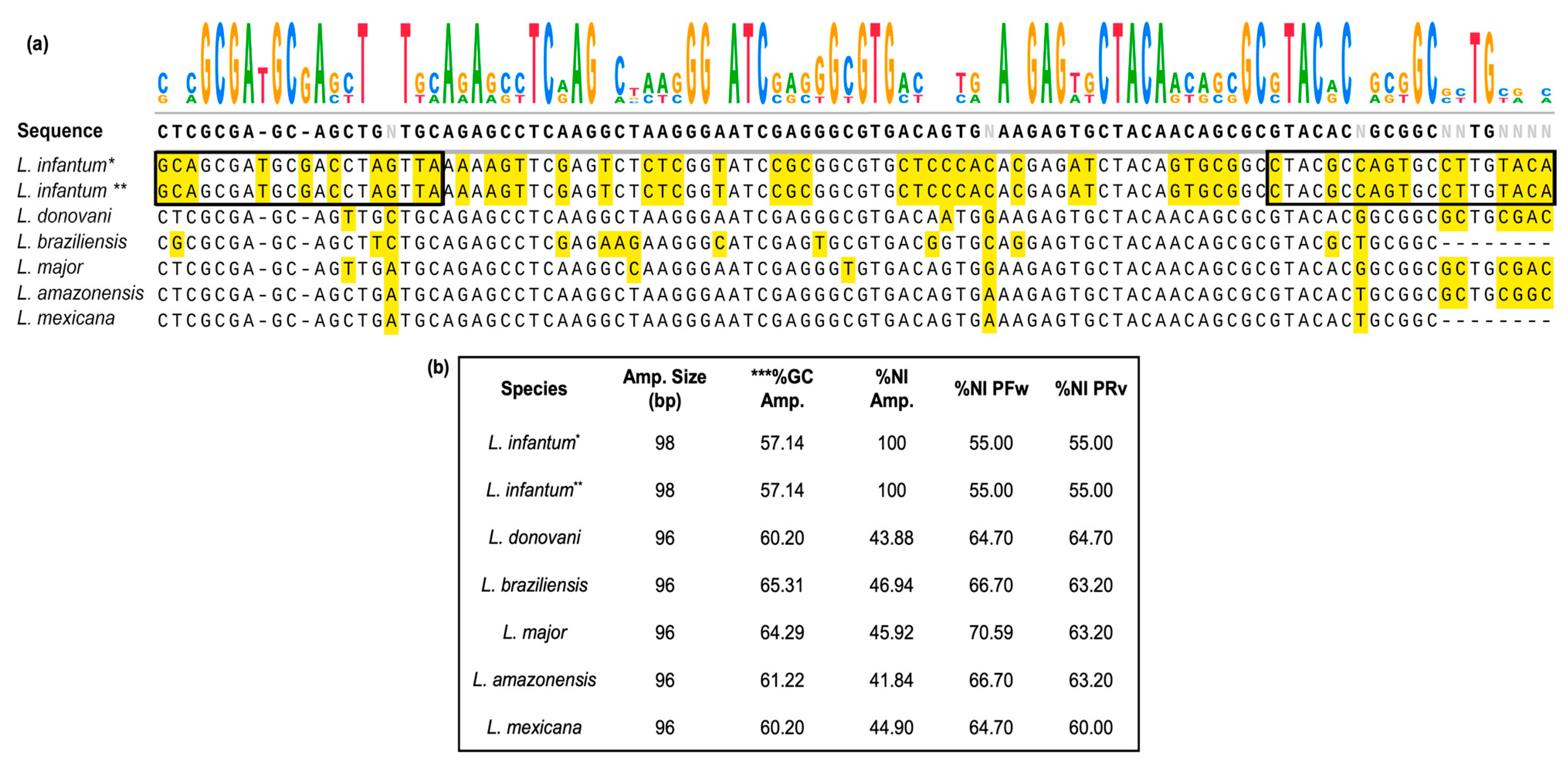
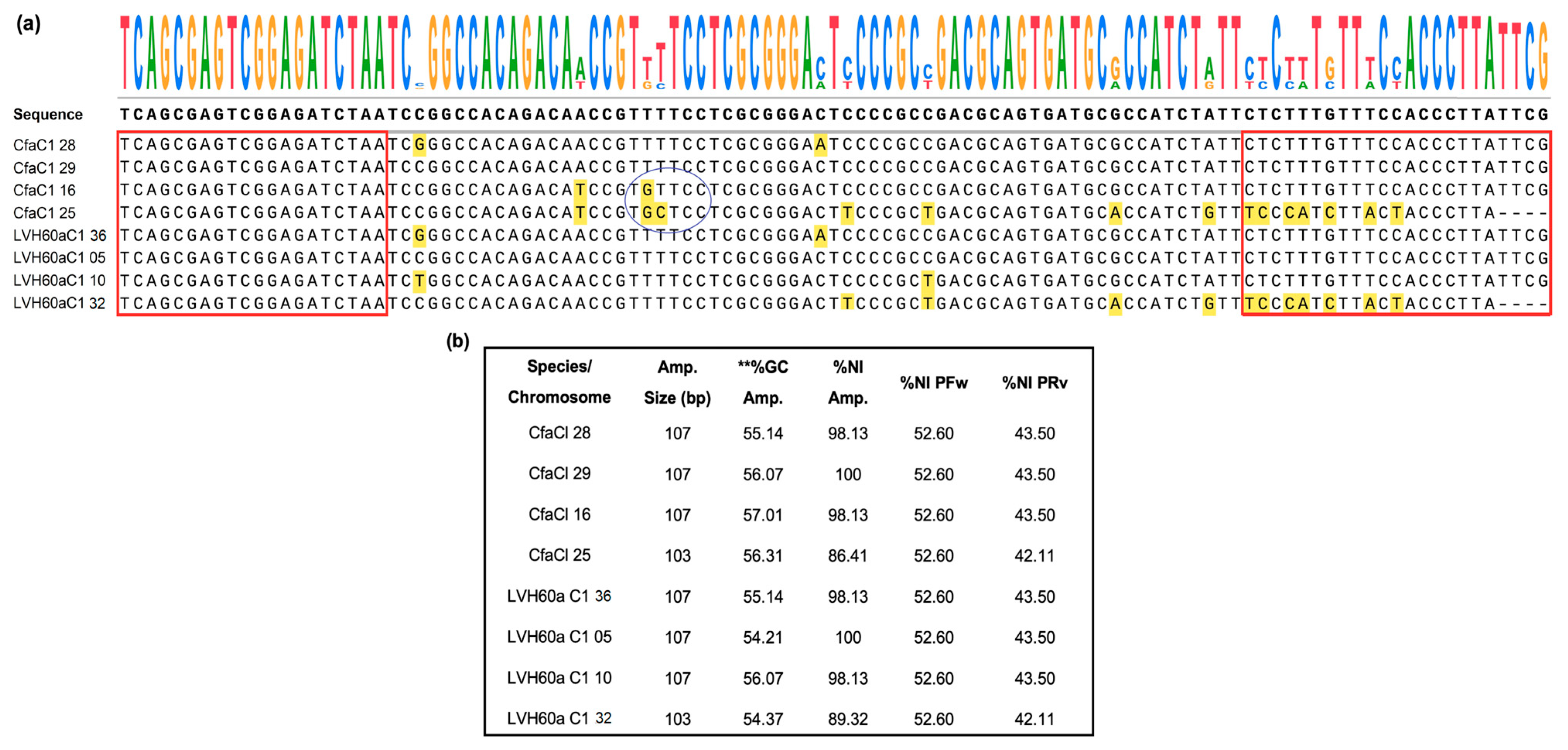
2.1. Search Strategy for New Gene Targets and Primer Design
2.2. DNA Extraction
2.3. Real-Time Quantitative PCR (qPCR) Assays
2.3.1. Standard Curve
2.3.2. qPCR Assay Evaluation
2.4. PCR, Amplicon Sequencing, and Sequence Analysis
2.5. Experimental Validation
2.5.1. In Vitro Infection with THP-1 Macrophages
2.5.2. In Vivo Infection
2.5.3. Sample Collection
2.6. Data Analysis
2.7. Ethics Statement
3. Results
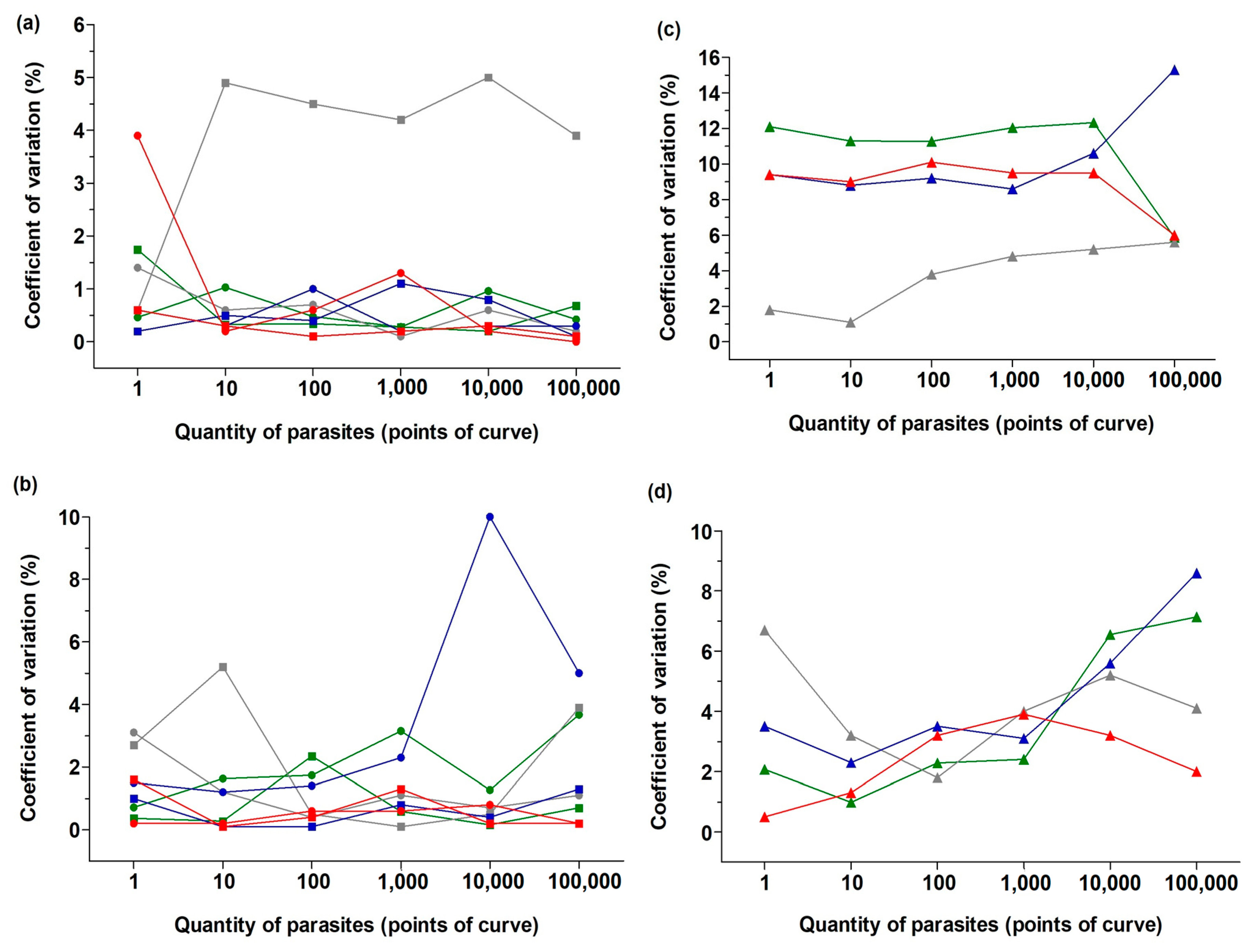
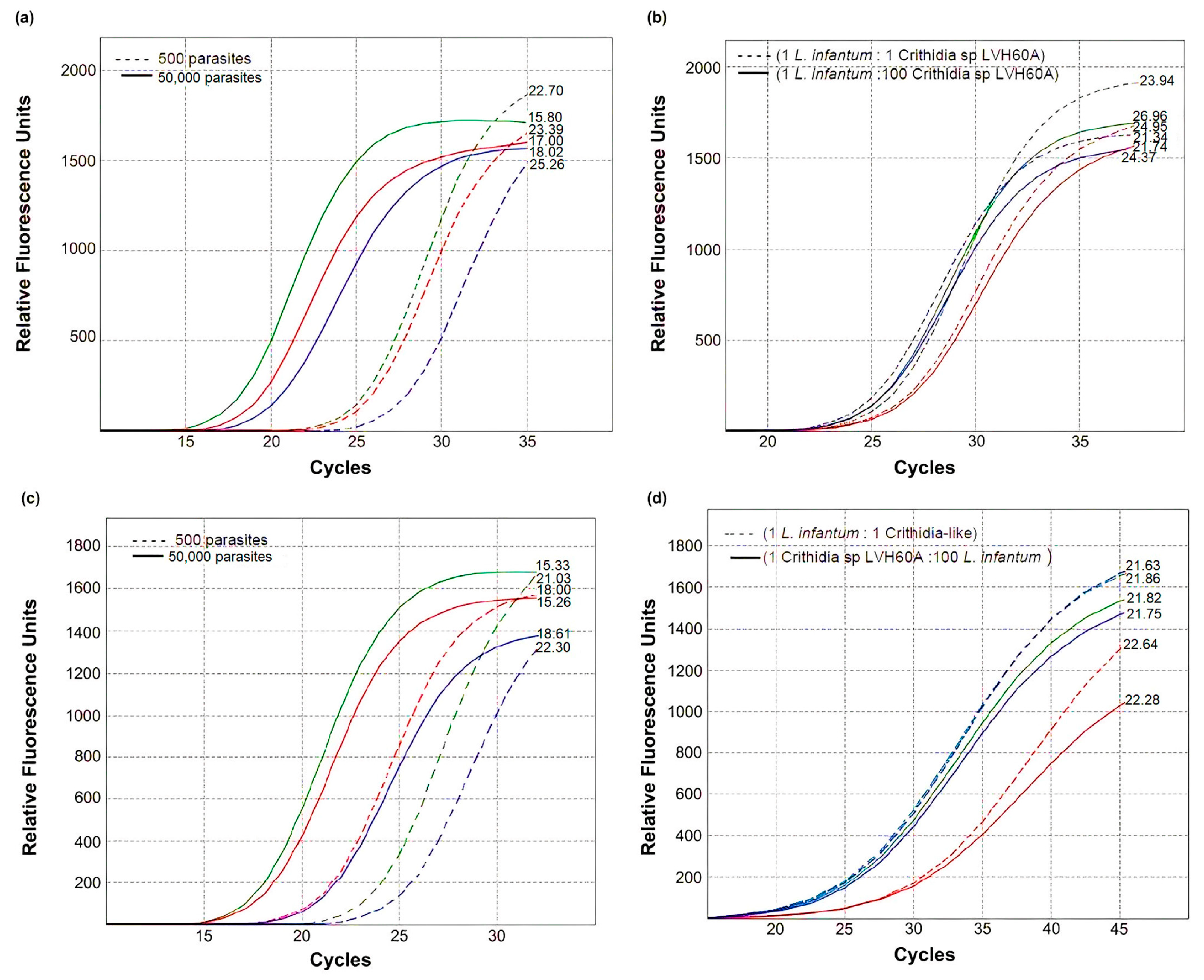
3.1. Real-Time PCR Performance and Validation of the Method
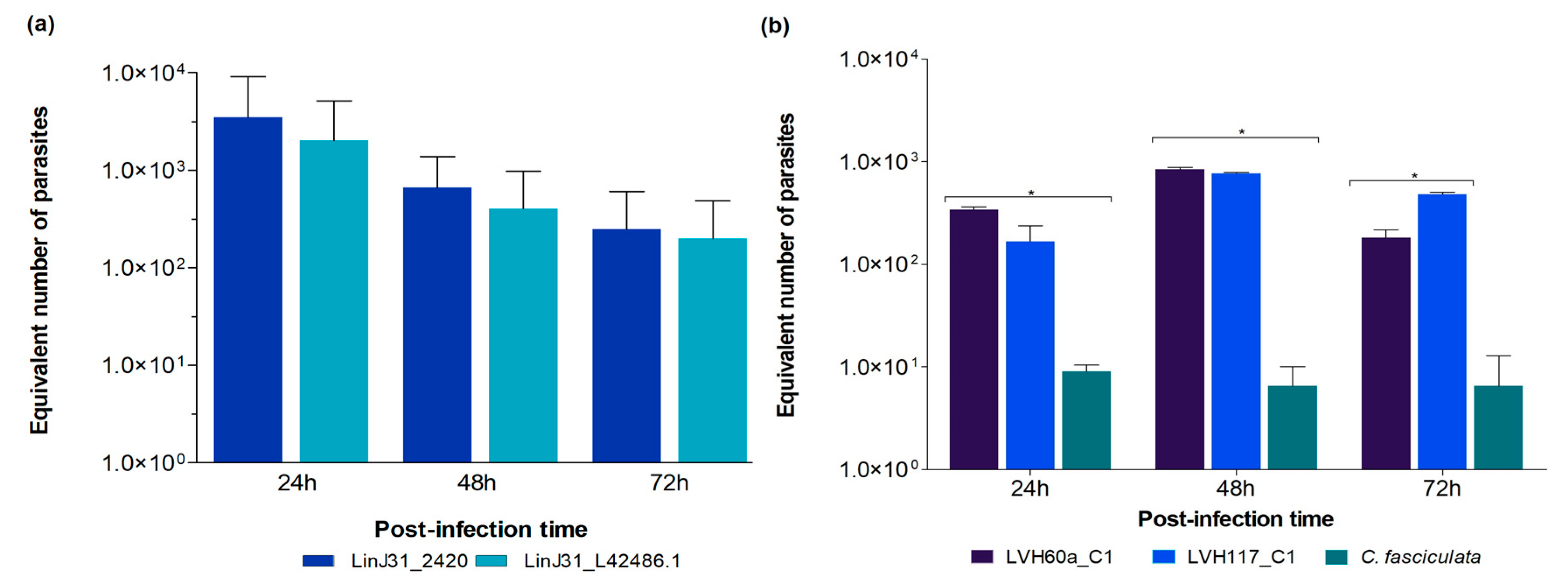
3.2. Validation of the Novel qPCR Assay and Gene Targets in Experimental Conditions
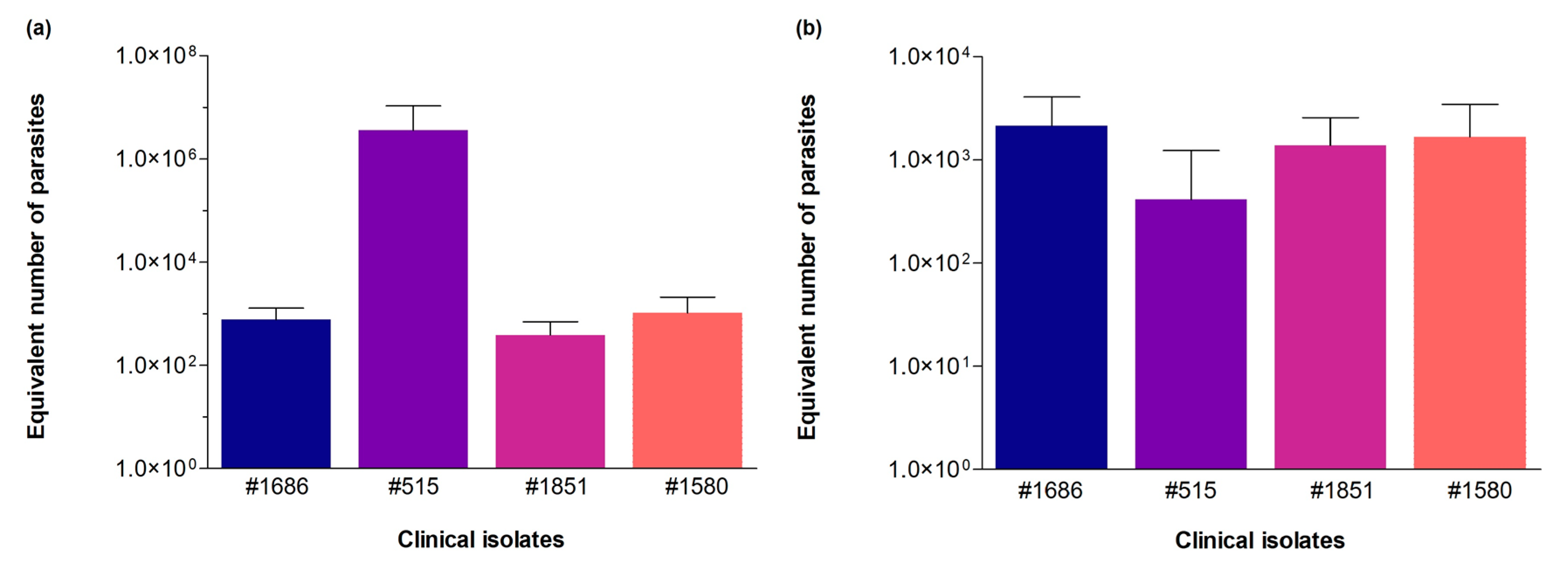
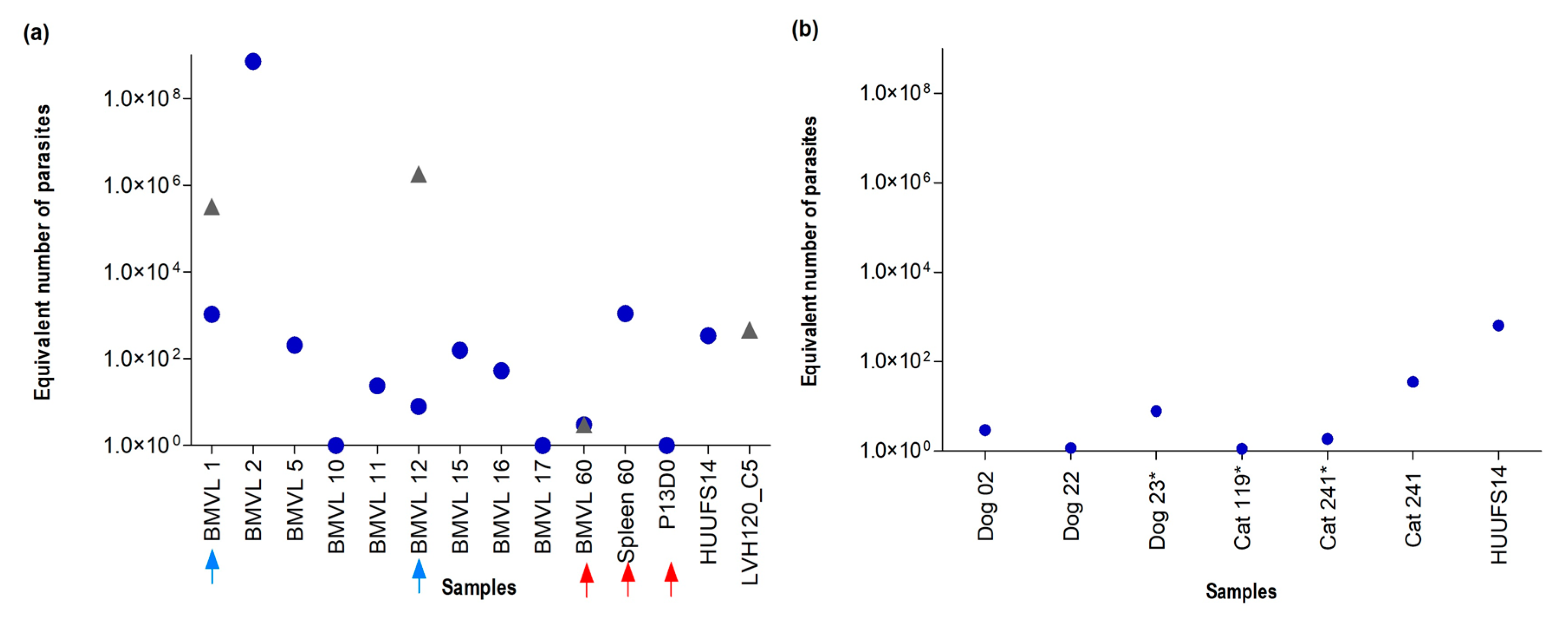
3.3. Validation of the Novel qPCR Assay and Gene Targets through Parasite Detection in Clinical Isolate Cultures and Host Tissue Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azami-Conesa, I.; Gómez-Muñoz, M.T.; Martínez-Díaz, R.A. A Systematic Review (1990–2021) of Wild Animals Infected with Zoonotic Leishmania. Microorganisms 2021, 9, 1101. [Google Scholar] [CrossRef] [PubMed]
- WHO|Leishmaniasis. Available online: http://www.who.int/gho/neglected_diseases/leishmaniasis/en/ (accessed on 23 March 2020).
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; The WHO Leishmaniasis Control Team. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Parasites—Leishmaniasis. Available online: https://www.cdc.gov/parasites/leishmaniasis/index.html (accessed on 26 March 2020).
- Michel, G.; Pomares, C.; Ferrua, B.; Marty, P. Importance of Worldwide Asymptomatic Carriers of Leishmania infantum (L. chagasi) in Human. Acta Trop. 2011, 119, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Gardinassi, L.G.; Garcia, G.R.; Costa, C.H.N.; Silva, V.C.; Santos, I.K.F. de M. Blood Transcriptional Profiling Reveals Immunological Signatures of Distinct States of Infection of Humans with Leishmania infantum. PLoS Negl. Trop. Dis. 2016, 10, e0005123. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, S.R.; Fuzo, C.A.; Oliveira, A.E.R.; Rogerio, L.A.; Takamiya, N.T.; Pessenda, G.; de Melo, E.V.; da Silva, A.M.; Jesus, A.R.; Carregaro, V.; et al. Insight Into the Long Noncoding RNA and MRNA Coexpression Profile in the Human Blood Transcriptome Upon Leishmania infantum Infection. Front. Immunol. 2022, 13, 784463. [Google Scholar] [CrossRef]
- Visceral Leishmaniasis—PAHO/WHO|Pan American Health Organization. Available online: https://www.paho.org/en/topics/leishmaniasis/visceral-leishmaniasis (accessed on 19 January 2022).
- Thakur, S.; Joshi, J.; Kaur, S. Leishmaniasis Diagnosis: An Update on the Use of Parasitological, Immunological and Molecular Methods. J. Parasit. Dis. Off. Organ Indian Soc. Parasitol. 2020, 44, 253–272. [Google Scholar] [CrossRef]
- Reimão, J.Q.; Coser, E.M.; Lee, M.R.; Coelho, A.C. Laboratory Diagnosis of Cutaneous and Visceral Leishmaniasis: Current and Future Methods. Microorganisms 2020, 8, 1632. [Google Scholar] [CrossRef]
- Sudarshan, M.; Singh, T.; Singh, A.K.; Chourasia, A.; Singh, B.; Wilson, M.E.; Chakravarty, J.; Sundar, S. Quantitative PCR in Epidemiology for Early Detection of Visceral Leishmaniasis Cases in India. PLoS Negl. Trop. Dis. 2014, 8, e3366. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Chowdhury, R.; Maruf, S.; Picado, A.; Hossain, F.; Owen, S.I.; Nath, R.; Baker, J.; Hasnain, M.G.; Shomik, M.S.; et al. Gauging the Skin Resident Leishmania Parasites through a Loop Mediated Isothermal Amplification (LAMP) Assay in Post-Kala-Azar Dermal Leishmaniasis. Sci. Rep. 2022, 12, 18069. [Google Scholar] [CrossRef]
- Adams, E.R.; Schoone, G.; Versteeg, I.; Gomez, M.A.; Diro, E.; Mori, Y.; Perlee, D.; Downing, T.; Saravia, N.; Assaye, A.; et al. Development and Evaluation of a Novel Loop-Mediated Isothermal Amplification Assay for Diagnosis of Cutaneous and Visceral Leishmaniasis. J. Clin. Microbiol. 2018, 56, e00386-18. [Google Scholar] [CrossRef] [Green Version]
- Nzelu, C.O.; Kato, H.; Peters, N.C. Loop-Mediated Isothermal Amplification (LAMP): An Advanced Molecular Point-of-Care Technique for the Detection of Leishmania Infection. PLoS Negl. Trop. Dis. 2019, 13, e0007698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Paiva-Cavalcanti, M.; de Morais, R.C.; Pessoa-e-Silva, R.; Trajano-Silva, L.A.; Goncalves-de-Albuquerque, S.D.; Tavares, D.D.; Brelaz-de-Castro, M.C.; Silva, R.D.; Pereira, V.R. Leishmaniases Diagnosis: An Update on the Use of Immunological and Molecular Tools. Cell Biosci. 2015, 5, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcino, G.N.; Carvalho, K.S.S.; Braz, D.C.; Silva, V.C.; Costa, C.H.N.; de Miranda Santos, I.K.F. Evaluation of Methods for Detection of Asymptomatic Individuals Infected with Leishmania infantum in the State of Piauí, Brazil. PLoS Negl. Trop. Dis. 2019, 13, e0007493. [Google Scholar] [CrossRef] [Green Version]
- Sudarshan, M.; Sundar, S. Parasite Load Estimation by QPCR Differentiates between Asymptomatic and Symptomatic Infection in Indian Visceral Leishmaniasis. Diagn. Microbiol. Infect. Dis. 2014, 80, 40–42. [Google Scholar] [CrossRef] [Green Version]
- Kalantari, M.; Motazedian, M.H.; Asgari, Q.; Mohammadpour, I.; Soltani, A.; Azizi, K. Co-Detection and Isolation of Leishmania and Crithidia among Naturally Infected Tatera Indica (Rodentia: Muridae) in Fars Province, Southern Iran. Asian Pac. J. Trop. Biomed. 2018, 8, 279. [Google Scholar] [CrossRef]
- Boucinha, C.; Andrade-Neto, V.V.; Ennes-Vidal, V.; Branquinha, M.H.; dos Santos, A.L.S.; Torres-Santos, E.C.; d’Avila-Levy, C.M. A Stroll Through the History of Monoxenous Trypanosomatids Infection in Vertebrate Hosts. Front. Cell. Infect. Microbiol. 2022, 12, 804707. [Google Scholar] [CrossRef] [PubMed]
- Ghobakhloo, N.; Motazedian, M.H.; Naderi, S.; Ebrahimi, S. Isolation of Crithidia Spp. from Lesions of Immunocompetent Patients with Suspected Cutaneous Leishmaniasis in Iran. Trop. Med. Int. Health 2019, 24, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Mirzapour, A.; Badirzadeh, A.; Ashrafmansouri, M.; Behniafar, H.; Norouzi, M.; Azizi, H.; Behravan, M.; Seyyed Tabaei, S.J. Super Infection of Cutaneous Leishmaniasis Caused by Leishmania Major and L. Tropica to Crithidia fasciculata in Shiraz, Iran. Iran. J. Public Health 2019, 48, 2285–2292. [Google Scholar] [CrossRef]
- Doudi, M.; Karami, M.; Eslami, G.; Setorki, M. A Study of Genetic Polymorphism of Crithidia in Isfahan, Iran. Zahedan J. Res. Med. Sci. 2015, 17. [Google Scholar] [CrossRef]
- Kalantari, M.; Motazedian, M.H.; Asgari, Q.; Soltani, A.; Mohammadpour, I.; Azizi, K. DNA-Based Detection of Leishmania and Crithidia Species Isolated from Humans in Cutaneous and Post-Kala-Azar Dermal Leishmaniasis from Shiraz and Kharameh, Southern Iran. J. Vector Borne Dis. 2020, 57, 52–57. [Google Scholar] [CrossRef]
- Rogerio, L.A.; Takahashi, T.Y.; Cardoso, L.; Takamiya, N.T.; de Melo, E.V.; de Jesus, A.R.; de Oliveira, F.A.; Forrester, S.; Jeffares, D.C.; da Silva, J.S.; et al. Co-Infection of Leishmania infantum and a Crithidia-Related Species in a Case of Refractory Relapsed Visceral Leishmaniasis with Non-Ulcerated Cutaneous Manifestation in Brazil. Int. J. Infect. Dis. 2023, 133, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Fakhar, M.; Derakhshani-nia, M.; Gohardehi, S.; Karamian, M.; Hezarjaribi, H.Z.; Mohebali, M.; Akhoundi, B.; Sharbatkhori, M. Domestic Dogs Carriers of Leishmania infantum, Leishmania tropica and Crithidia fasciculata as Potential Reservoirs for Human Visceral Leishmaniasis in Northeastern Iran. Vet. Med. Sci. 2022, 8, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Dario, M.A.; Lisboa, C.V.; Silva, M.V.; Herrera, H.M.; Rocha, F.L.; Furtado, M.C.; Moratelli, R.; Rodrigues Roque, A.L.; Jansen, A.M. Crithidia Mellificae Infection in Different Mammalian Species in Brazil. Int. J. Parasitol. Parasites Wildl. 2021, 15, 58–69. [Google Scholar] [CrossRef]
- Songumpai, N.; Promrangsee, C.; Noopetch, P.; Siriyasatien, P.; Preativatanyou, K. First Evidence of Co-Circulation of Emerging Leishmania Martiniquensis, Leishmania Orientalis, and Crithidia Sp. in Culicoides Biting Midges (Diptera: Ceratopogonidae), the Putative Vectors for Autochthonous Transmission in Southern Thailand. Trop. Med. Infect. Dis. 2022, 7, 379. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, S.R.; de Santana, A.K.M.; Takamiya, N.T.; Takahashi, T.Y.; Rogerio, L.A.; Oliveira, C.A.B.; Milanezi, C.M.; Trombela, V.A.; Cruz, A.K.; Jesus, A.R.; et al. Non-Leishmania parasite in fatal visceral leishmaniasis–like disease, Brazil. Emerg. Infect. Dis. 2019, 25, 2088. [Google Scholar] [CrossRef] [Green Version]
- Escalona-Montaño, A.R.; Ortiz-Lozano, D.M.; Rojas-Bernabé, A.; Wilkins-Rodriguez, A.A.; Torres-Guerrero, H.; Mondragón-Flores, R.; Mondragón-Gonzalez, R.; Becker, I.; Gutiérrez-Kobeh, L.; Aguirre-Garcia, M.M. Leishmania Mexicana: Promastigotes and Amastigotes Secrete Protein Phosphatases and This Correlates with the Production of Inflammatory Cytokines in Macrophages. Parasitology 2016, 143, 1409–1420. [Google Scholar] [CrossRef]
- de Almeida-Amaral, E.E.; Belmont-Firpo, R.; Vannier-Santos, M.A.; Meyer-Fernandes, J.R. Leishmania Amazonensis: Characterization of an Ecto-Phosphatase Activity. Exp. Parasitol. 2006, 114, 334–340. [Google Scholar] [CrossRef]
- Horáková, E.; Faktorová, D.; Kraeva, N.; Kaur, B.; Abbeele, J.V.D.; Yurchenko, V.; Lukeš, J. Catalase Compromises the Development of the Insect and Mammalian Stages of Trypanosoma Brucei. FEBS J. 2020, 287, 964–977. [Google Scholar] [CrossRef]
- Kraeva, N.; Horáková, E.; Kostygov, A.Y.; Kořený, L.; Butenko, A.; Yurchenko, V.; Lukeš, J. Catalase in Leishmaniinae: With Me or against Me? Infect. Genet. Evol. 2017, 50, 121–127. [Google Scholar] [CrossRef]
- Thakur, L.; Kushwaha, H.R.; Negi, A.; Jain, A.; Jain, M. Leptomonas Seymouri Co-Infection in Cutaneous Leishmaniasis Cases Caused by Leishmania Donovani From Himachal Pradesh, India. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, P.; Sarkar, A.; Datta, S.; Chatterjee, M. Coinfection of Leptomonas Seymouri and Leishmania Donovani in Indian Leishmaniasis. J. Clin. Microbiol. 2012, 50, 2774–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, P.; Prajapati, V.K.; Vanaerschot, M.; Van der Auwera, G.; Dujardin, J.C.; Sundar, S. Detection of Leptomonas Sp. Parasites in Clinical Isolates of Kala-Azar Patients from India. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2010, 10, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Barazesh, A.; Motazedian, M.H.; Fouladvand, M.; Hatam, G.; Tajbakhsh, S.; Ebrahimi, S.; Purkamal, D. Molecular Identification of Species Caused Cutaneous Leishmaniasis in Southern Zone of Iran. J. Arthropod-Borne Dis. 2019, 13, 198–205. [Google Scholar] [CrossRef]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and Gene Copy Number Variation Allow Major Structural Change between Species and Strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A Functional Genomic Resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinformatics 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Vioti, G.; da Silva, M.D.; Galvis-Ovallos, F.; Alves, M.L.; da Silva, D.T.; Leonel, J.A.F.; Pereira, N.W.B.; Benassi, J.C.; Spada, J.C.P.; Maia, C.; et al. Xenodiagnosis in Four Domestic Cats Naturally Infected by Leishmania infantum. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef]
- Novy, F.G.; McNeal, W.J. On the Cultivation of Trypanosoma Brucei. J. Infect. Dis. 1904, 1, 1–30. [Google Scholar] [CrossRef]
- Luz, Z.M.; Coutinho, M.G.; Cançado, J.R.; Krettli, A.U. Hemoculture: Sensitive technique in the detection of Trypanosoma cruzi in chagasic patients in the chronic phase of Chagas disease. Rev. Soc. Bras. Med. Trop. 1994, 27, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Vallur, A.C.; Duthie, M.S.; Reinhart, C.; Tutterrow, Y.; Hamano, S.; Bhaskar, K.R.H.; Coler, R.N.; Mondal, D.; Reed, S.G. Biomarkers for Intracellular Pathogens: Establishing Tools as Vaccine and Therapeutic Endpoints for Visceral Leishmaniasis. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2014, 20, O374–O383. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Ghosh, P.; Khan, M.A.A.; Duthie, M.S.; Vallur, A.C.; Picone, A.; Howard, R.F.; Reed, S.G.; Mondal, D. Real-Time PCR in Detection and Quantitation of Leishmania Donovani for the Diagnosis of Visceral Leishmaniasis Patients and the Monitoring of Their Response to Treatment. PLoS ONE 2017, 12, e0185606. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Hasnain, M.G.; Hossain, F.; Khan, M.A.A.; Chowdhury, R.; Faisal, K.; Mural, M.A.; Baker, J.; Nath, R.; Ghosh, D.; et al. Evaluation of Real-Time PCR for Diagnosis of Post-Kala-Azar Dermal Leishmaniasis in Endemic Foci of Bangladesh. Open Forum Infect. Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Ghosh, P.; Khan, M.A.A.; Hossain, F.; Faisal, K.; Nath, R.; Baker, J.; Wahed, A.A.E.; Maruf, S.; Nath, P.; et al. Evaluation of Rapid Extraction Methods Coupled with a Recombinase Polymerase Amplification Assay for Point-of-Need Diagnosis of Post-Kala-Azar Dermal Leishmaniasis. Trop. Med. Infect. Dis. 2020, 5. [Google Scholar] [CrossRef]
- Chin Yuan, C.; Miley, W.; Waters, D. A Quantification of Human Cells Using an ERV-3 Real Time PCR Assay. J. Virol. Methods 2001, 91, 109–117. [Google Scholar] [CrossRef]
- Adaui, V.; Verdonck, K.; Best, I.; González, E.; Tipismana, M.; Arévalo, J.; Vanham, G.; Campos, M.; Zimic, M.; Gotuzzo, E. SYBR Green–Based Quantitation of Human T-Lymphotropic Virus Type 1 Proviral Load in Peruvian Patients with Neurological Disease and Asymptomatic Carriers: Influence of Clinical Status, Sex, and Familial Relatedness. J. Neurovirol. 2006, 12, 456–465. [Google Scholar] [CrossRef]
- Barker, E.N.; Tasker, S.; Day, M.J.; Warman, S.M.; Woolley, K.; Birtles, R.; Georges, K.C.; Ezeokoli, C.D.; Newaj-Fyzul, A.; Campbell, M.D.; et al. Development and Use of Real-Time PCR to Detect and Quantify Mycoplasma Haemocanis and “Candidatus Mycoplasma Haematoparvum” in Dogs. Vet. Microbiol. 2010, 140, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.E.S.; Coura-Vital, W.; Roatt, B.M.; Bouillet, L.É.M.; Ker, H.G.; de Brito, R.C.F.; de Melo Resende, D.; Carneiro, M.; Giunchetti, R.C.; Marques, M.J.; et al. Molecular Diagnosis of Canine Visceral Leishmaniasis: A Comparative Study of Three Methods Using Skin and Spleen from Dogs with Natural Leishmania infantum Infection. Vet. Parasitol. 2013, 197, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Lins, R.M.M.A.; Oliveira, S.G.; Souza, N.A.; de Queiroz, R.G.; Justiniano, S.C.B.; Ward, R.D.; Kyriacou, C.P.; Peixoto, A.A. Molecular Evolution of the Cacophony IVS6 Region in Sandflies. Insect Mol. Biol. 2002, 11, 117–122. [Google Scholar] [CrossRef]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative Genomic Analysis of Three Leishmania Species That Cause Diverse Human Disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, T.A.; Englund, P.T. The Structure and Replication of Kinetoplast DNA. Annu. Rev. Microbiol. 1995, 49, 117–143. [Google Scholar] [CrossRef]
- Takahashi, T.Y. Triagem de Isolados Clínicos de Leishmania sp. para Sequenciamento Genômico. Available online: https://repositorio.ufscar.br/handle/ufscar/8195/browse?type=author&value=Takahashi%2C+Talita+Yuri (accessed on 30 April 2021).
- Venkataraman, A.; Parlov, M.; Hu, P.; Schnell, D.; Wei, X.; Tiesman, J.P. Spike-in Genomic DNA for Validating Performance of Metagenomics Workflows. BioTechniques 2018, 65, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Jara, M.; Adaui, V.; Valencia, B.M.; Martinez, D.; Alba, M.; Castrillon, C.; Cruz, M.; Cruz, I.; Van der Auwera, G.; Llanos-Cuentas, A.; et al. Real-Time PCR Assay for Detection and Quantification of Leishmania (Viannia) Organisms in Skin and Mucosal Lesions: Exploratory Study of Parasite Load and Clinical Parameters. J. Clin. Microbiol. 2013, 51, 1826–1833. [Google Scholar] [CrossRef] [Green Version]
- Noyes, H.; Stevens, J.R.; Teixeira, M.; Phelan, J.; Holz, P. A Nested PCR for the SsrRNA Gene Detects Trypanosoma Binneyi in the Platypus and Trypanosoma Sp. in Wombats and Kangaroos in Australia1. Int. J. Parasitol. 1999, 29, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and Characterization of a Human Acute Monocytic Leukemia Cell Line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Ferreira, T.; Timbó, R.V.; Minuzzi-Souza, T.T.C.; de Almeida Rocha, D.; Neiva, M.; de Albuquerque Ribeiro, J.; de Almeida, P.S.; Hecht, M.; Nitz, N.; Gurgel-Gonçalves, R. High Molecular Prevalence of Leishmania in Phlebotomine Sand Flies Fed on Chicken Blood in Brazil. Vet. Parasitol. 2018, 259, 80–84. [Google Scholar] [CrossRef]
- Dantas-Torres, F.; da Silva Sales, K.G.; Gomes da Silva, L.; Otranto, D.; Figueredo, L.A. Leishmania-FAST15: A Rapid, Sensitive and Low-Cost Real-Time PCR Assay for the Detection of Leishmania infantum and Leishmania Braziliensis Kinetoplast DNA in Canine Blood Samples. Mol. Cell. Probes 2017, 31, 65–69. [Google Scholar] [CrossRef]
- Caraguel, C.G.B.; Stryhn, H.; Gagné, N.; Dohoo, I.R.; Hammell, K.L. Selection of a Cutoff Value for Real-Time Polymerase Chain Reaction Results to Fit a Diagnostic Purpose: Analytical and Epidemiologic Approaches. J. Vet. Diagn. Investig. Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc 2011, 23, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Domagalska, M.A.; Dujardin, J.-C. Comment letter: Non-Leishmania Parasite in Fatal Visceral Leishmaniasis–like Disease, Brazi. Emerg. Infect. Dis. 2020, 26, 388. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, O.P. Molecular Diagnosis of Visceral Leishmaniasis. Mol. Diagn. Ther. 2018, 22, 443–457. [Google Scholar] [CrossRef]
- Rezaei, Z.; Azarang, E.; Shahabi, S.; Omidian, M.; Pourabbas, B.; Sarkari, B. Leishmania ITS1 Is Genetically Divergent in Asymptomatic and Symptomatic Visceral Leishmaniasis: Results of a Study in Southern Iran. J. Trop. Med. 2020, 2020, 5351098. [Google Scholar] [CrossRef]
- Yurchenko, V.; Butenko, A.; Kostygov, A.Y. Genomics of Trypanosomatidae: Where We Stand and What Needs to Be Done? Pathogens 2021, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Kraeva, N.; Butenko, A.; Hlaváčová, J.; Kostygov, A.; Myškova, J.; Grybchuk, D.; Leštinová, T.; Votýpka, J.; Volf, P.; Opperdoes, F.; et al. Leptomonas Seymouri: Adaptations to the Dixenous Life Cycle Analyzed by Genome Sequencing, Transcriptome Profiling and Co-Infection with Leishmania Donovani. PLoS Pathog. 2015, 11, e1005127. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.Q.; Ferreira-Silva, M.M.; Ratkevicius, C.M.A.; Gómez-Hérnandez, C.; Vito, F.B.D.; Tanaka, S.C.S.V.; Júnior, V.R.; Moraes-Souza, H. Identification of Leishmania infantum in Blood Donors from Endemic Regions for Visceral Leishmaniasis. Parasitology 2021, 148, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; Jeronimo, S.M.B.; Gautam, S.; Sundar, S.; Kang, M.; Kurtz, M.A.; Haque, R.; Schriefer, A.; Talhari, S.; Carvalho, E.M.; et al. Serial Quantitative PCR Assay for Detection, Species Discrimination, and Quantification of Leishmania spp. in Human Samples. J. Clin. Microbiol. 2011, 49, 3892–3904. [Google Scholar] [CrossRef] [Green Version]
- Pita-Pereira, D.; Lins, R.; Oliveira, M.P.; Lima, R.B.; Pereira, B.A.; Moreira, O.C.; Brazil, R.P.; Britto, C. SYBR Green-Based Real-Time PCR Targeting Kinetoplast DNA Can Be Used to Discriminate between the Main Etiologic Agents of Brazilian Cutaneous and Visceral Leishmaniases. Parasit. Vectors 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, R.A.; Laranjeira-Silva, M.F.; Muxel, S.M.; Stocco de Lima, A.C.; Shaw, J.J.; Floeter-Winter, L.M. High Resolution Melting Analysis Targeting Hsp70 as a Fast and Efficient Method for the Discrimination of Leishmania Species. PLoS Negl. Trop. Dis. 2016, 10, e0004485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Paiva Cavalcanti, M.; Dantas-Torres, F.; da Cunha Gonçalves de Albuquerque, S.; Silva de Morais, R.C.; de Brito, M.E.F.; Otranto, D.; Brandão-Filho, S.P. Quantitative Real Time PCR Assays for the Detection of Leishmania (Viannia) Braziliensis in Animals and Humans. Mol. Cell. Probes 2013, 27, 122–128. [Google Scholar] [CrossRef]
- Filgueira, C.P.B.; Moreira, O.C.; Cantanhêde, L.M.; de Farias, H.M.T.; Porrozzi, R.; Britto, C.; Boité, M.C.; Cupolillo, E. Comparison and Clinical Validation of QPCR Assays Targeting Leishmania 18S RDNA and HSP70 Genes in Patients with American Tegumentary Leishmaniasis. PLoS Negl. Trop. Dis. 2020, 14, e0008750. [Google Scholar] [CrossRef]
- Reina, A.M.; Mewa, J.C.; Calzada, J.E.; Saldaña, A. Characterization of Leishmania Spp. Causing Cutaneous Lesions with a Negative Parasitological Diagnosis in Panama. Trop. Med. Infect. Dis. 2022, 7, 282. [Google Scholar] [CrossRef]
- Singh, N.; Chikara, S.; Sundar, S. SOLiDTM Sequencing of Genomes of Clinical Isolates of Leishmania Donovani from India Confirm Leptomonas Co-Infection and Raise Some Key Questions. PLoS ONE 2013, 8, e55738. [Google Scholar] [CrossRef] [Green Version]
- Dedet, J.-P.; Pratlong, F. Leishmania, Trypanosoma and Monoxenous Trypanosomatids as Emerging Opportunistic Agents1. J. Eukaryot. Microbiol. 2000, 47, 37–39. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer F (5′-3′) | Primer R (5′-3′) | Amplicon Size | Usage |
|---|---|---|---|---|
| LinJ31seq | GTGCACGCCAATGTCTTTGT | GCCCATGGTTGAGCTAGGTT | 444 bp | Detection of L. infantum by PCR |
| LinJ31_2420 | GCAGCGATGCGACCTAGTTA | TGTACAAGGCACTGGCGTAG | 98 bp | Detection and quantification of L. infantum by qPCR |
| Crid2.1seq | TCACTTTGGCGGTATCAGTG | GCATCAGCTGACCCTTTCTC | 502 bp | Detection of Crithidia sp. LVH60A and C. fasciculata by PCR |
| LVH60a_Tig001 | GTTAGAGCGACTAGCCCGTG | GGGTAGAGGAGAGAGGTGGG | 128 bp | Detection of Crithidia sp. LVH60A by PCR |
| Catalase_LVH60-12060_1F | TCAGCGAGTCGGAGTCTAA | CGAATAAGGGTGGAAACAAAGAG | 107 bp | Detection and quantification of Crithidia sp. LVH60A and C. fasciculata by qPCR |
| Primer | Species | Chr. | Gene ID or Access Number * | Amp. ** GC% | Amp. Size | Percent Nucleotide Identity |
|---|---|---|---|---|---|---|
| Crid2.1seq | Crithidia fasciculata (CfCl) | 16 | CFAC1_160022800 | 67.47% | 501 bp | A = 87.0 T = 76.0 C = 162.0 G = 176.0 |
| Crithidia sp. LVH60A (LVH60a) | 10 | CP119667.1 | 67.13% | 502 bp | A = 86.0 T = 79.0 C = 166.0 G = 171.0 | |
| Leptomonas pyrrhocoris (H10) | 15 | LpyrH10_15 a | No match | |||
| 16 | LpyrH10_16 b | |||||
| Leptomonas seymouri (ATCC_30220) | 48 | Lsey_0048 a | No match | |||
| 01 | Lsey_01 b | |||||
| Tissue | Number of Samples | PCR + LinJ31seq | PCR + Crid2.1seq | Nested-PCR TRY927/SSU561 [57] | |
|---|---|---|---|---|---|
| Patient samples | BMVL a | 18 | 13 | 0 c | 18 |
| PB | 14 | 6 | 0 | 7 | |
| Clinical isolates | CI b | 53 | 14 | 48 | 46 |
| Skin (nodule/papule) | 4 | 2 | 2 | 4 c | |
| Spleen | 1 | 0 | 1 | 1 d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takamiya, N.T.; Rogerio, L.A.; Torres, C.; Leonel, J.A.F.; Vioti, G.; de Sousa Oliveira, T.M.F.; Valeriano, K.C.; Porcino, G.N.; de Miranda Santos, I.K.F.; Costa, C.H.N.; et al. Parasite Detection in Visceral Leishmaniasis Samples by Dye-Based qPCR Using New Gene Targets of Leishmania infantum and Crithidia. Trop. Med. Infect. Dis. 2023, 8, 405. https://doi.org/10.3390/tropicalmed8080405
Takamiya NT, Rogerio LA, Torres C, Leonel JAF, Vioti G, de Sousa Oliveira TMF, Valeriano KC, Porcino GN, de Miranda Santos IKF, Costa CHN, et al. Parasite Detection in Visceral Leishmaniasis Samples by Dye-Based qPCR Using New Gene Targets of Leishmania infantum and Crithidia. Tropical Medicine and Infectious Disease. 2023; 8(8):405. https://doi.org/10.3390/tropicalmed8080405
Chicago/Turabian StyleTakamiya, Nayore Tamie, Luana Aparecida Rogerio, Caroline Torres, João Augusto Franco Leonel, Geovanna Vioti, Tricia Maria Ferreira de Sousa Oliveira, Karoline Camila Valeriano, Gabriane Nascimento Porcino, Isabel Kinney Ferreira de Miranda Santos, Carlos H. N. Costa, and et al. 2023. "Parasite Detection in Visceral Leishmaniasis Samples by Dye-Based qPCR Using New Gene Targets of Leishmania infantum and Crithidia" Tropical Medicine and Infectious Disease 8, no. 8: 405. https://doi.org/10.3390/tropicalmed8080405