
Systemic Levels of Pro-Inflammatory Cytokines and Post-Treatment Modulation in Tuberculous Lymphadenitis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statement of Ethics
2.2. Research Participants
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Analysis
3. Results
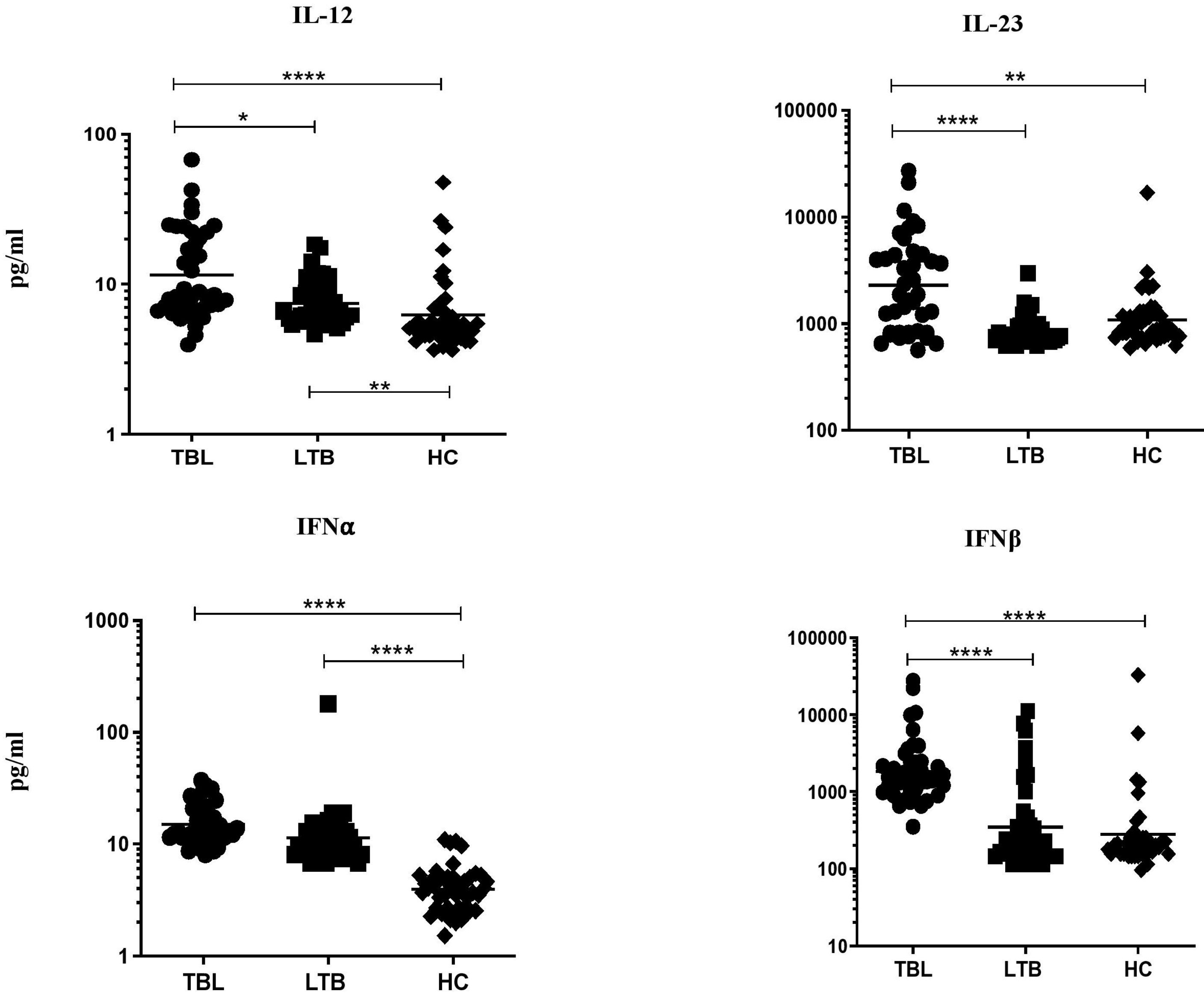
3.1. TBL Group Has Increased Pro-Inflammatory Cytokine Levels
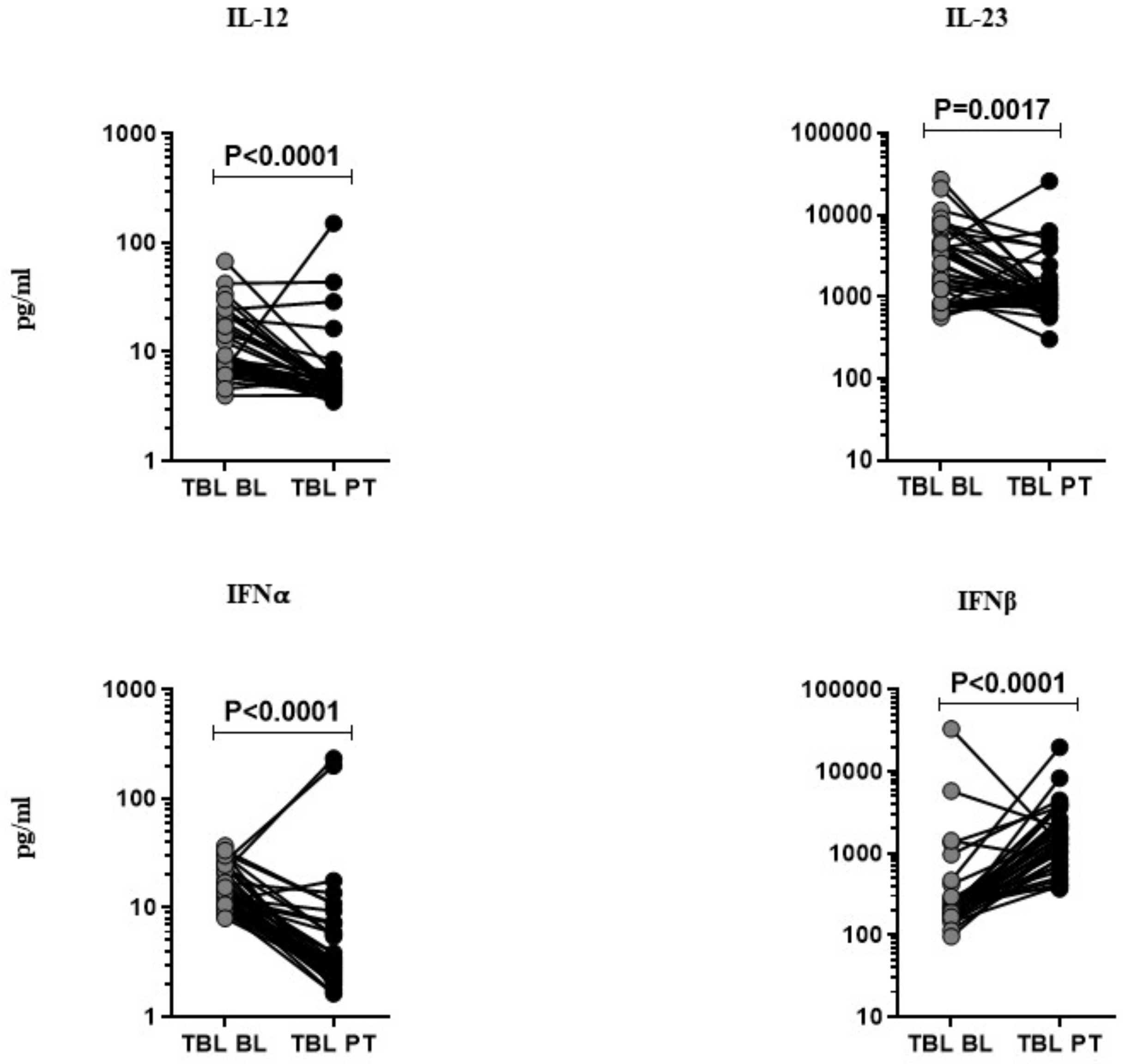
3.2. Anti-Tuberculosis Treatment Effect on Pro-Inflammatory Cytokines
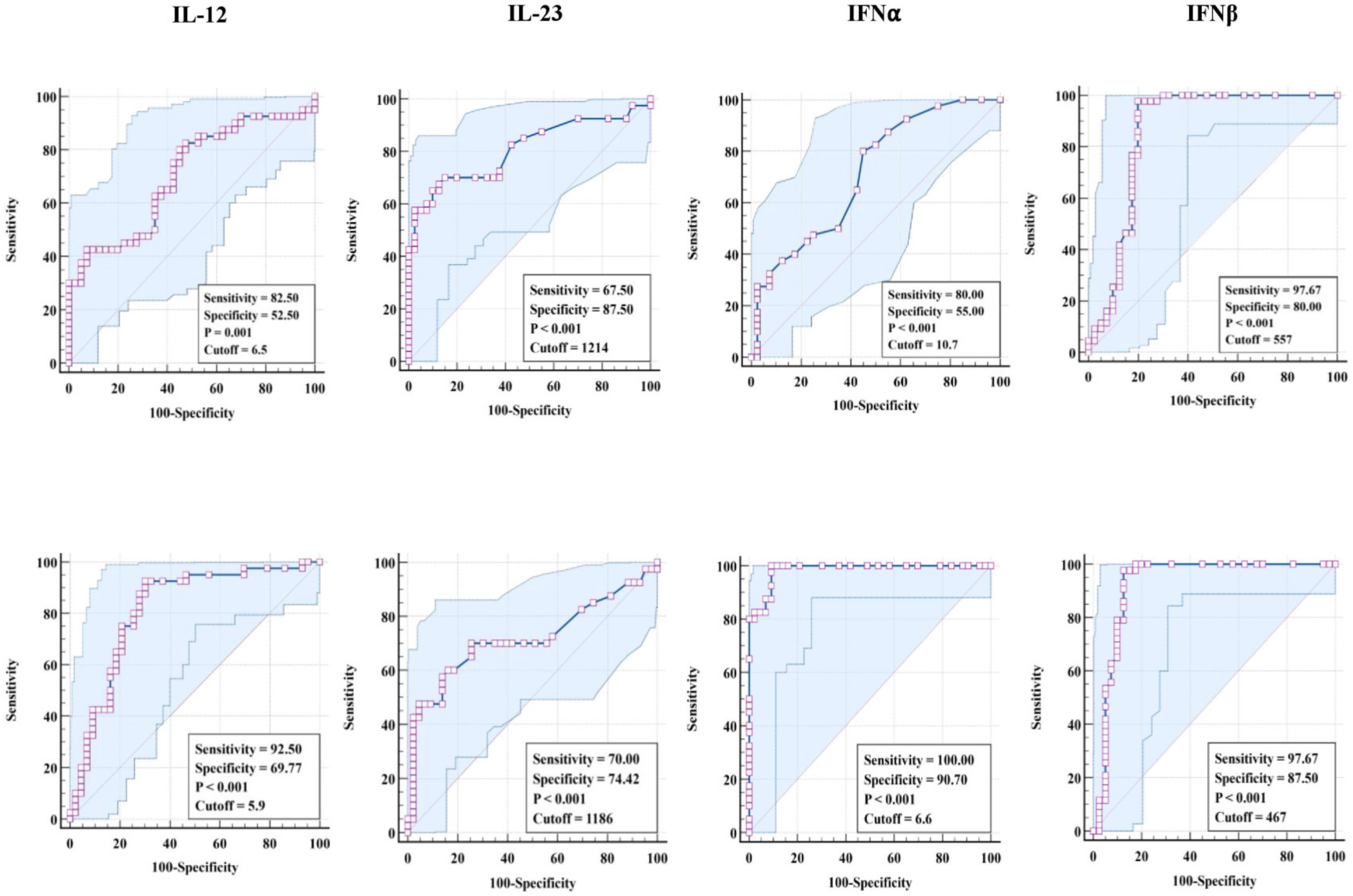
3.3. ROC Analysis of Pro-Inflammatory Cytokines in TBL Disease
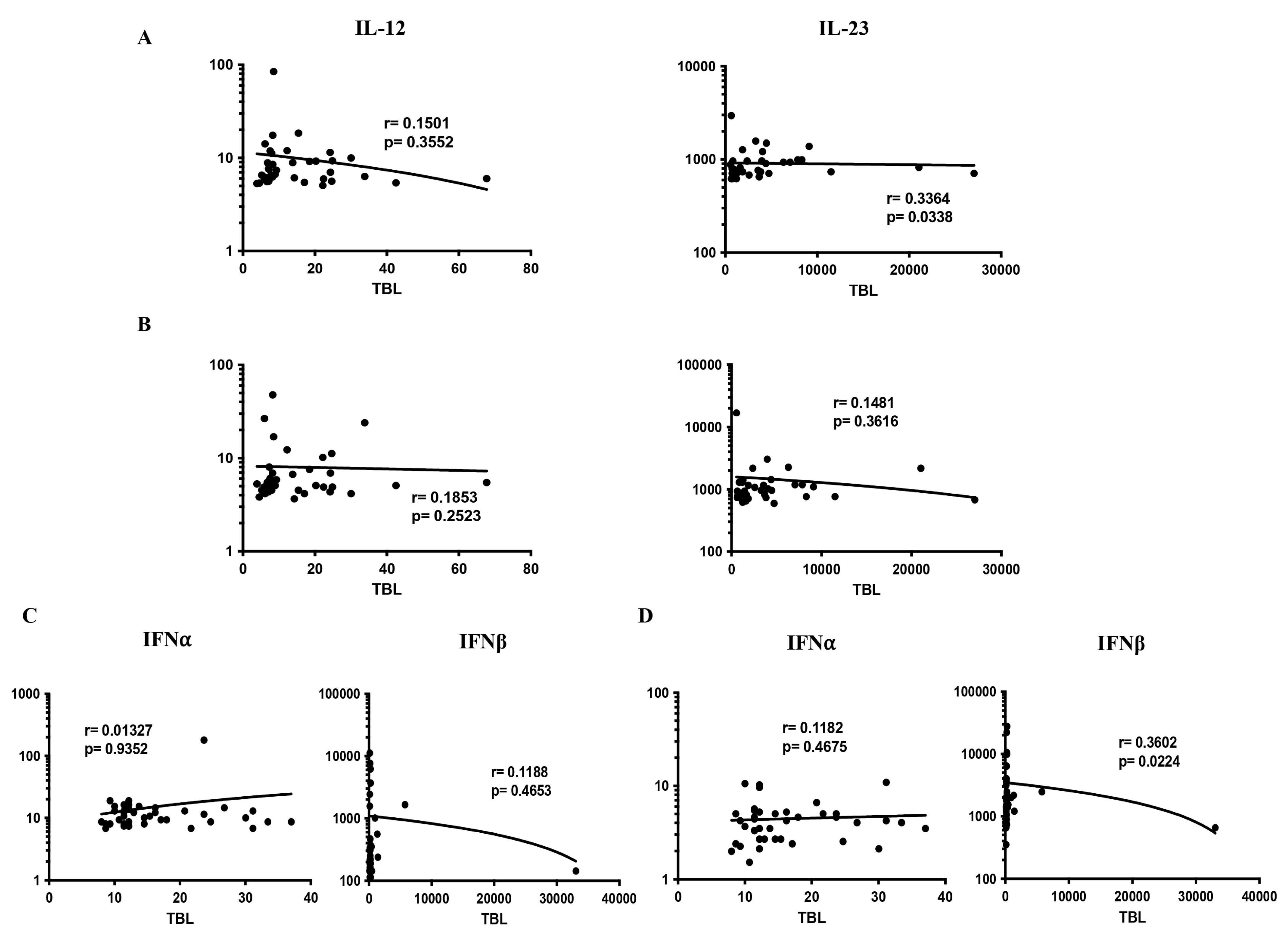
3.4. Correlation Analysis of Pro-Inflammatory Cytokines in TBL Disease
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Publication. Global Tuberculosis Report 2022. 2022. Available online: https://www.who.int/health-topics/tuberculosis#tab=tab_1 (accessed on 10 January 2023).
- Sharma, S.K.; Ryan, H.; Khaparde, S.; Sachdeva, K.S.; Singh, A.D.; Mohan, A.; Sarin, R.; Paramasivan, C.N.; Kumar, P.; Nischal, N.; et al. Index—TB guidelines: Guidelines on extrapulmonary tuberculosis for India. Indian J. Med. Res. 2017, 145, 448–463. [Google Scholar] [PubMed]
- Fontanilla, J.M.; Barnes, A.; von Reyn, C.F. Current diagnosis and management of peripheral tuberculous lymphadenitis. Clin. Infect. Dis. 2011, 53, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Handa, U.; Mundi, I.; Mohan, S. Nodal tuberculosis revisited: A review. J. Infect. Dev. Ctries. 2012, 6, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Kong, Y.; Wilson, F.; Foxman, B.; Fowler, A.H.; Marrs, C.F.; Cave, M.D.; Bates, J.H. Identification of risk factors for extrapulmonary tuberculosis. Clin. Infect. Dis. 2004, 38, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Barber, K.D.; Barber, D.L. Innate and adaptive cellular immune responses to mycobacterium tuberculosis infection. Cold Spring Harb. Perspect. Med. 2015, 5, a018424. [Google Scholar]
- O’Garra, A.; Redford, P.S.; McNab, F.W.; Bloom, C.I.; Wilkinson, R.J.; Berry, M.P. The immune response in tuberculosis. Annu. Rev. Immunol. 2013, 31, 475–527. [Google Scholar] [CrossRef]
- Cliff, J.M.; Kaufmann, S.H.; McShane, H.; van Helden, P.; O’Garra, A. The human immune response to tuberculosis and its treatment: A view from the blood. Immunol. Rev. 2015, 264, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Dorhoi, A.; Kaufmann, S.H. Perspectives on host adaptation in response to Mycobacterium tuberculosis: Modulation of inflammation. Semin. Immunol. 2014, 26, 533–542. [Google Scholar] [CrossRef]
- Etna, M.P.; Giacomini, E.; Severa, M.; Coccia, E.M. Pro-and anti-inflammatory cytokines in tuberculosis: A two-edged sword in TB pathogenesis. Semin. Immunol. 2014, 26, 543–551. [Google Scholar] [CrossRef]
- Cooper, A.M. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 2009, 27, 393–422. [Google Scholar] [CrossRef] [Green Version]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Ritter, K.; Rousseau, J.; Hölscher, C. The Role of gp130 cytokines in tuberculosis. Cells 2020, 9, 2695. [Google Scholar] [CrossRef]
- Hunter, C.A. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 2005, 5, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M.; Kipnis, A.; Turner, J.; Magram, J.; Ferrante, J.; Orme, I.M. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J. Immunol. 2002, 168, 1322–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khader, S.A.; Pearl, J.E.; Sakamoto, K.; Gilmartin, L.; Bell, G.K.; Jelley-Gibbs, D.M.; Ghilardi, N.; deSauvage, F.; Cooper, A.M. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen- specific IFN-g responses if IL-12p70 is available. J. Immunol. 2005, 175, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, T.; Ryan, A.; Britton, W. Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J. Immunol. 2006, 177, 8684–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, T.; Ryan, A.; Triccas, J.; Britton, W. Plasmid interleukin-23 (IL-23), but not plasmid IL-27, enhances the protective efficacy of a DNA vaccine against Mycobacterium tuberculosis infection. Infect. Immun. 2006, 74, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, R.; Zhong, S.; Ren, H.; Zou, Y.; Chen, X.; Shi, X.; Wang, M.; Long, H.; Luo, Y. The immunogenicity and protective efficacy of Mtb8.4/hIL-12 chimeric gene vaccine. Vaccine 2006, 24, 1315–1323. [Google Scholar] [CrossRef]
- Yoshida, S.; Tanaka, T.; Kita, Y.; Kuwayama, S.; Kanamaru, N.; Muraki, Y.; Hashimoto, S.; Inoue, Y.; Sakatani, M.; Kobayashi, E.; et al. DNA vaccine using hemagglutinating virus of Japan-liposome encapsulating combination encoding mycobacterial heat shock protein 65 and interleukin-12 confers protection against Mycobacterium tuberculosis by T cell activation. Vaccine 2006, 24, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.L.; Schultz, T.E.; Duke, T.J.; Blumenthal, A. Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front. Immunol. 2017, 8, 1633. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Teixeira, L.; Mayer-Barber, K.; Sher, A.; O’Garra, A. Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med. 2018, 215, 1273–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, A.A.; Ma, X.; Cuomo, P.; Park, K.; Wahl, L.; Wolf, S.F.; Zhou, H.; Trinchieri, G.; Karp, C.L. Type I interferons and IL-12: Convergence and cross-regulation among mediators of cellular immunity. Eur. J. Immunol. 2001, 31, 2026–2034. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Bloom, C.I.; Graham, C.M.; Berry, M.P.; Rozakeas, F.; Redford, P.S.; Wang, Y.; Xu, Z.; Wilkinson, K.A.; Wilkinson, R.J.; Kendrick, Y.; et al. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS ONE 2013, 8, e70630. [Google Scholar] [CrossRef]
- Kathamuthu, G.R.; Moideen, K.; Baskaran, D.; Banurekha, V.V.; Nair, D.; Sekar, G.; Sridhar, R.; Vidyajayanthi, B.; Gajendraraj, G.; Parandhaman, D.K.; et al. Tuberculous lymphadenitis is associated with enhanced baseline and antigen-specific induction of type 1 and type 17 cytokines and reduced interleukin-1β (IL-1β) and IL-18 at the site of infection. Clin. Vaccine Immunol. 2017, 24, e00045-17. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Samperio, P. Role of interleukin-12 family cytokines in the cellular response to mycobacterial disease. Int. J. Infect. Dis. 2010, 14, e366-71. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.J.; Ornelles, D.A.; Mitchell, L.M.; Brzoza-Lewis, K.L.; Hiltbold, E.M. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J. Immunol. 2008, 181, 8576–8584. [Google Scholar] [CrossRef] [Green Version]
- Altare, F.; Durandy, A.; Lammas, D.; Emile, J.F.; Lamhamedi, S.; Le Deist, F.; Drysdale, P.; Jouanguy, E.; Döffinger, R.; Bernaudin, F.; et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 1998, 280, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C.; Atkinson, R.A.; Arendse, B.; Brown, N.; Myburgh, E.; Alber, G.; Brombacher, F. A protective and agonistic function of IL-12p40 in mycobacterial infection. J. Immunol. 2001, 167, 6957–6966. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomini, E.; Iona, E.; Ferroni, L.; Miettinen, M.; Fattorini, L.; Orefici, G.; Julkunen, I.; Coccia, E.M. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J. Immunol. 2001, 166, 7033–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glosson-Byers, N.L.; Sehra, S.; Kaplan, M.H. STAT4 is required for IL-23 responsiveness in Th17 memory cells and NKT cells. Jak-Stat 2014, 3, e955393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Tato, C.; Muul, L.; Laurence, A.; O’Shea, J. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2007, 56, 2936–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.; Boniface, K.; Chan, J.; McKenzie, B.; Blumenschein, W.; Mattson, J.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef]
- Khader, S.A.; Cooper, A.M. IL-23 and IL-17 in tuberculosis. Cytokine 2008, 41, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.P.; Moideen, K.; Banurekha, V.V.; Nair, D.; Babu, S. Plasma Proinflammatory Cytokines Are Markers of Disease Severity and Bacterial Burden in Pulmonary Tuberculosis. Open Forum Infect. Dis. 2019, 6, ofz257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovarik, P.; Castiglia, V.; Ivin, M.; Ebner, F. Type I interferons in bacterial infections: A balancing act. Front. Immunol. 2016, 7, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, D.T.; Hedges, J.F.; Jutila, M.A. Getting “inside” type I IFNs: Type I IFNs in intracellular bacterial infections. J. Immunol. Res. 2017, 2017, 9361802. [Google Scholar] [CrossRef] [Green Version]
- Bolívar, S.; Anfossi, R.; Humeres, C.; Vivar, R.; Boza, P.; Muñoz, C.; Pardo-Jimenez, V.; Olivares-Silva, F.; Díaz-Araya, G. IFN-β plays both pro- and anti-inflammatory roles in the rat cardiac fibroblast through differential STAT protein activation. Front. Pharmacol. 2018, 28, 1368. [Google Scholar] [CrossRef] [PubMed]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E., III; Freedman, V.H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aman, M.J.; Tretter, T.; Eisenbeis, I.; Bug, G.; Decker, T.; Aulitzky, W.E.; Tilg, H.; Huber, C.; Peschel, C. Interferon-a stimulates production of interleukin-10 in activated CD4+ T-cells and monocytes. Blood 1996, 87, 4731–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.; Rubio, M.; Nakajima, T.; Delespesse, G.; Sarfati, M. IFN-⍺ priming of human monocytes differentially regulates gram-positive and gram-negative bacteria-induced IL-10 release and selectively enhances IL-12 p70, CD80, and MHC class I expression. J. Immunol. 1998, 161, 2011–2018. [Google Scholar] [CrossRef]
- Chaussabel, D.; Semnani, R.T.; McDowell, M.A.; Sacks, D.; Sher, A.; Nutman, T.B. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 2003, 102, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Remoli, M.E.; Giacomini, E.; Lutfalla, G.; Dondi, E.; Orefici, G.; Battistini, A.; Uzé, G.; Pellegrini, S.; Coccia, E.M. Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J. Immunol. 2002, 169, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 2007, 179, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat Commun. 2020, 11, 5566. [Google Scholar] [CrossRef] [PubMed]
- Kathamuthu, G.R.; Moideen, K.; Bhaskaran, D.; Sekar, G.; Sridhar, R.; Vidyajayanthi, B.; Gajendraraj, G.; Babu, S. Reduced systemic and mycobacterial antigen-stimulated concentrations of IL-1β and IL-18 in tuberculous lymphadenitis. Cytokine 2017, 90, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Demographics | TBL | LTBI | HC |
|---|---|---|---|
| Number of subjects recruited (n) | 40 | 40 | 43 |
| Gender (M/F) | 13/27 | 15/25 | 12/28 |
| Median age in years (Range) | 28 (18–51) | 32 (21–62) | 39 (23–55) |
| Lymph node culture grade (0/1+/2+/3+) | 8/30/2/0 | Not done | Not done |
| Interferon-gamma release assay | Not done | Positive | Negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kathamuthu, G.R.; Moideen, K.; Sridhar, R.; Baskaran, D.; Babu, S. Systemic Levels of Pro-Inflammatory Cytokines and Post-Treatment Modulation in Tuberculous Lymphadenitis. Trop. Med. Infect. Dis. 2023, 8, 150. https://doi.org/10.3390/tropicalmed8030150
Kathamuthu GR, Moideen K, Sridhar R, Baskaran D, Babu S. Systemic Levels of Pro-Inflammatory Cytokines and Post-Treatment Modulation in Tuberculous Lymphadenitis. Tropical Medicine and Infectious Disease. 2023; 8(3):150. https://doi.org/10.3390/tropicalmed8030150
Chicago/Turabian StyleKathamuthu, Gokul Raj, Kadar Moideen, Rathinam Sridhar, Dhanaraj Baskaran, and Subash Babu. 2023. "Systemic Levels of Pro-Inflammatory Cytokines and Post-Treatment Modulation in Tuberculous Lymphadenitis" Tropical Medicine and Infectious Disease 8, no. 3: 150. https://doi.org/10.3390/tropicalmed8030150