

A Rapid and Sensitive Detection of HIV-1 with a One-Pot Two-Stage Reverse Transcription Recombinase Aided Real-Time PCR Assay
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Nucleic Acid Extraction
2.2. Primer and Probe Design of RT-RAP Assay
2.3. Plasmid Construction
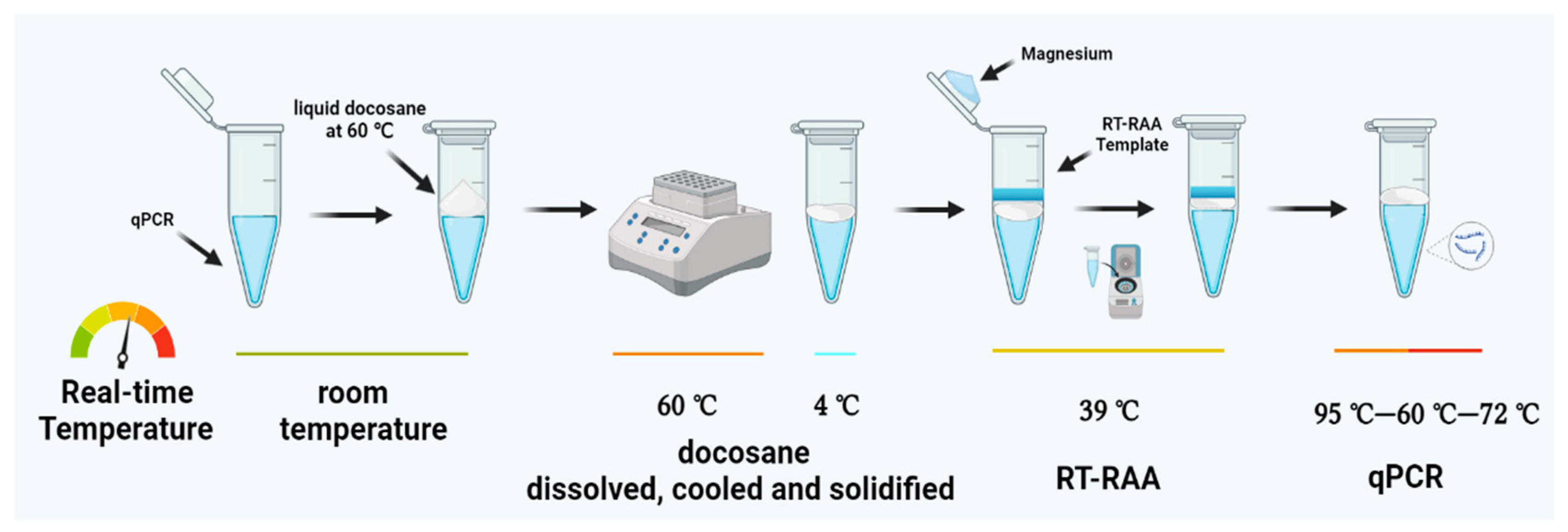
2.4. Solid Docosane Barrier
2.5. One-Pot Two-Stage RT-RAP Assay
2.6. Sensitivity, Specificity, and Reproducibility of the RT-RAP Assay
2.7. RT-RAP Detection of Clinical Samples
2.8. Statistical Analysis
3. Results
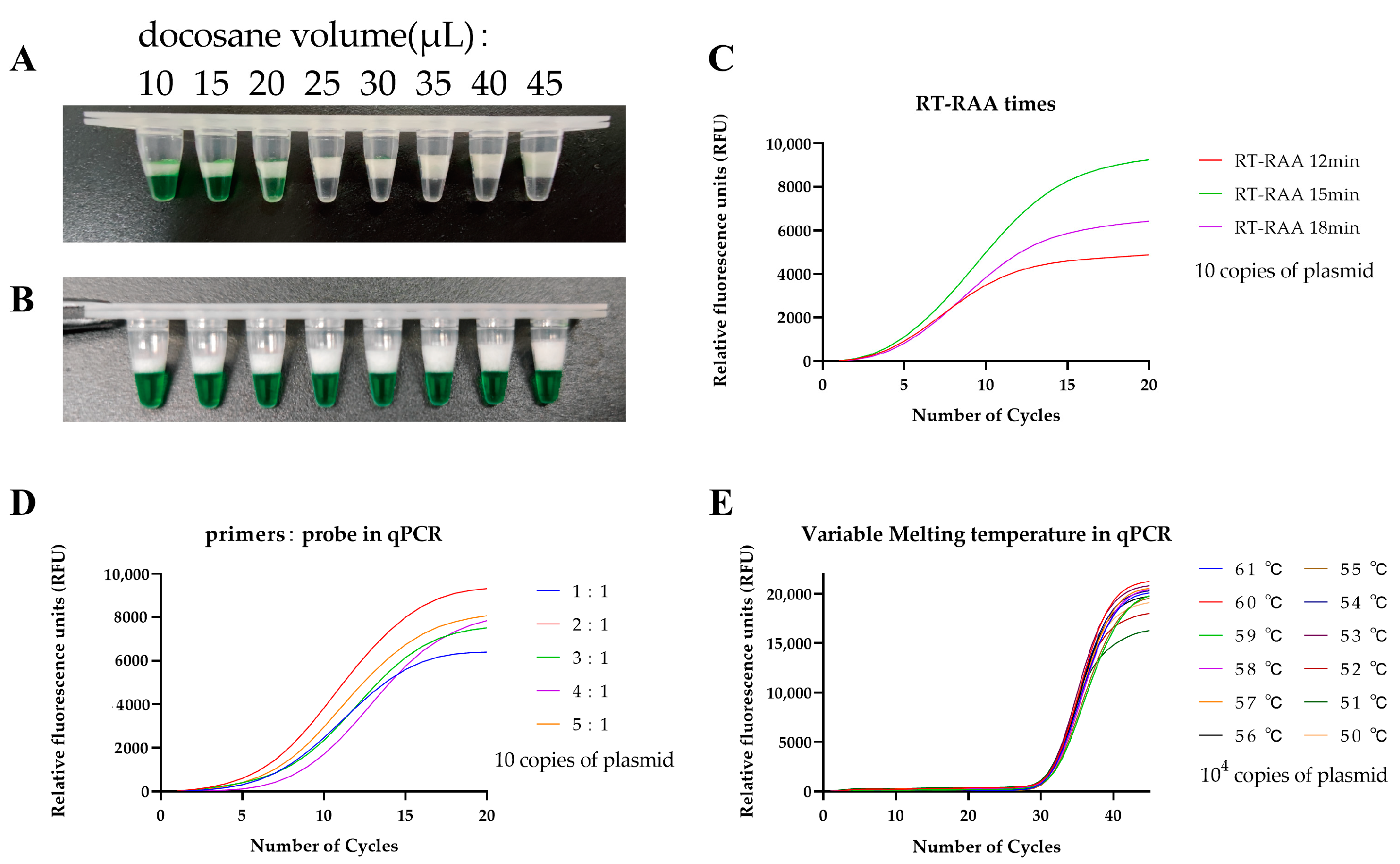
3.1. Optimization of the One-Pot Two-Stage RT-RAP Assays
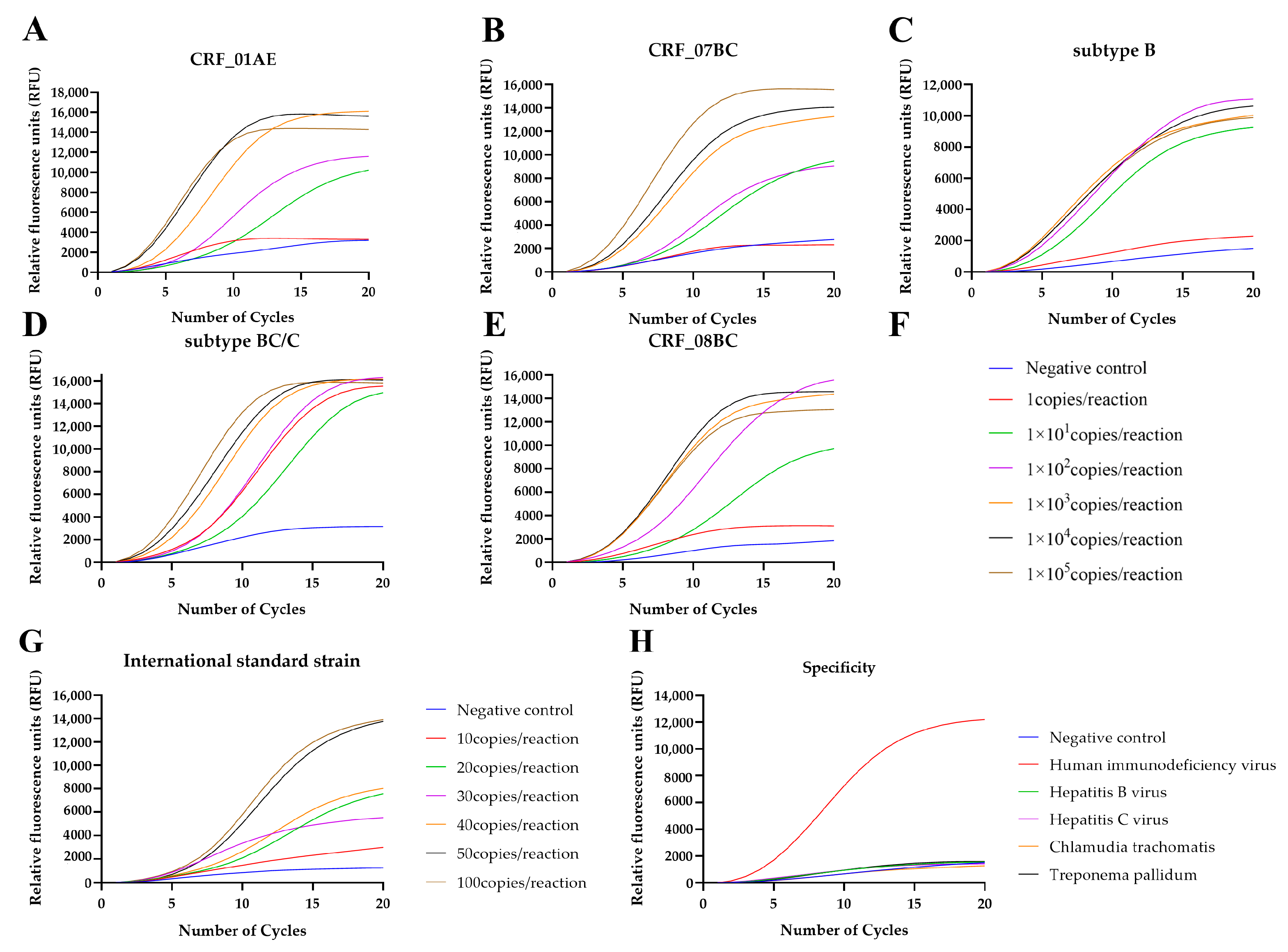
3.2. Sensitivity, Specificity, and Reproducibility of the RT-RAP Assay
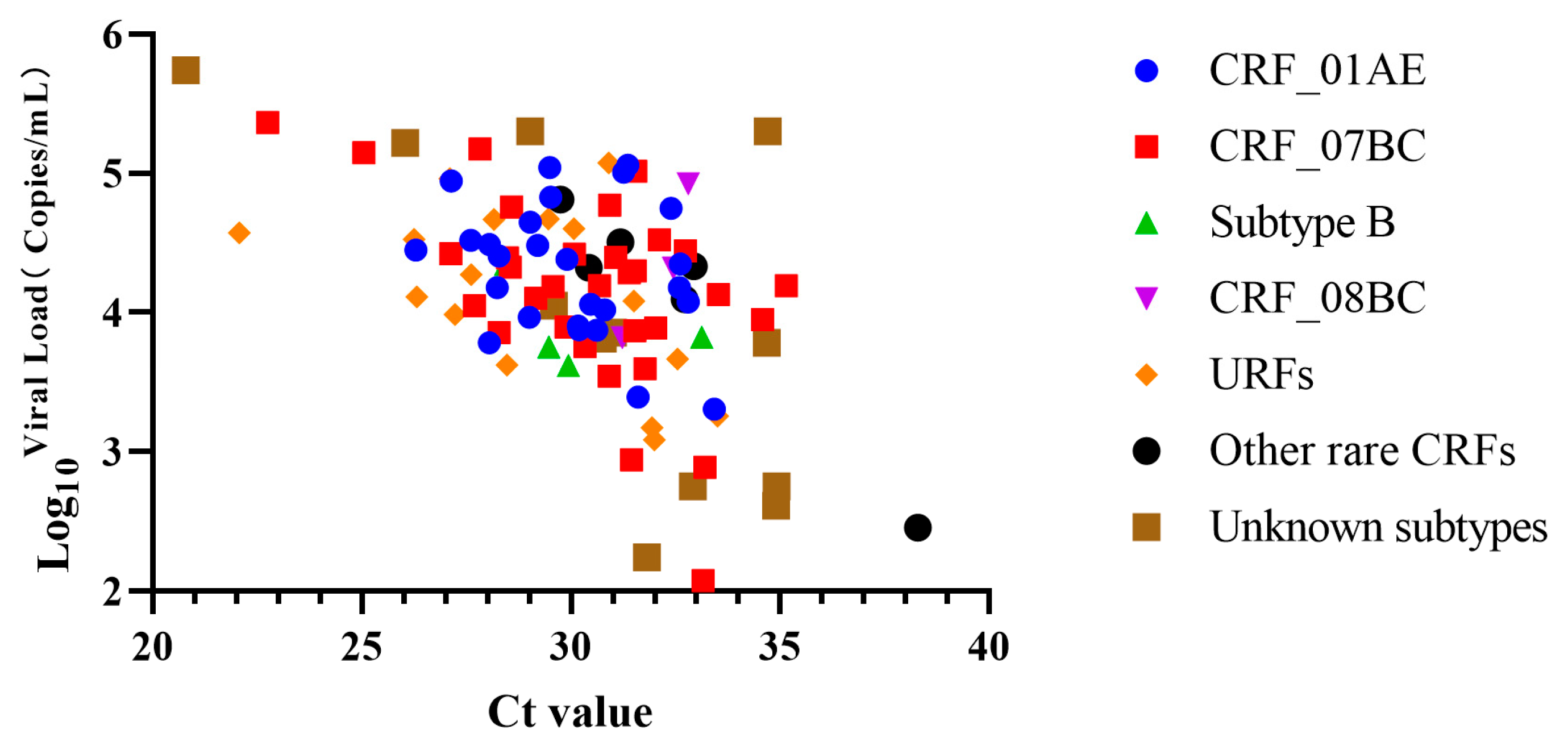
3.3. Viral Loads of Clinical Samples
3.4. Clinical Evaluation and Comparison between the RT-RAP and a Commercial qRT-PCR Kit
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. I. Super. Sanita 2010, 46, 5–14. [Google Scholar] [CrossRef]
- Fischer, W.; Giorgi, E.E.; Chakraborty, S.; Nguyen, K.; Bhattacharya, T.; Theiler, J.; Goloboff, P.A.; Yoon, H.; Abfalterer, W.; Foley, B.T.; et al. HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens. Cell Host Microbe 2021, 29, 1093–1110. [Google Scholar] [CrossRef] [PubMed]
- McCutchan, F.E. Global epidemiology of HIV. J. Med. Virol. 2006, 78, S7–S12. [Google Scholar] [CrossRef]
- Tee, K.K.; Thomson, M.M.; Hemelaar, J. Editorial: HIV-1 genetic diversity, volume II. Front. Microbiol. 2022, 13, 1007037. [Google Scholar] [CrossRef]
- Alexander, T.S. Human Immunodeficiency Virus Diagnostic Testing: 30 Years of Evolution. Clin. Vaccine Immunol. 2016, 23, 249–253. [Google Scholar] [CrossRef]
- Schüpbach, J. Viral RNA and p24 Antigen as Markers of HIV Disease and Antiretroviral Treatment Success. Int. Arch. Allergy Imm. 2003, 132, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Gao, M.; Zheng, J.; Yan, P.; Wang, W.; Lu, X.; Qiu, Y.; Yan, Y. Prevalence of HIV Indeterminate Western Blot Tests and Follow-up of HIV Antibody Sero-Conversion in Southeastern China. Virol. Sin. 2019, 34, 358–366. [Google Scholar] [CrossRef]
- Niemz, A.; Ferguson, T.M.; Boyle, D.S. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011, 29, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Hou, S.-Y.; Ji, S.-Z.; Cheng, J.; Zhang, M.-U.; He, L.-J.; Ye, X.-Z.; Li, Y.-M.; Zhang, Y.-X. A novel method of multiple nucleic acid detection: Real-time RT-PCR coupled with probe-melting curve analysis. Anal. Biochem. 2017, 537, 50–55. [Google Scholar] [CrossRef]
- Müller, J.; Eis-Hübinger, A.M.; Däumer, M.; Kaiser, R.; Rox, J.M.; Gürtler, L.; Hanfland, P.; Pötzsch, B. A novel internally controlled real-time reverse transcription-PCR assay for HIV-1 RNA targeting the pol integrase genomic region. J. Virol. Methods 2007, 142, 127–135. [Google Scholar] [CrossRef]
- Jagodzinski, L.L.; Weston, H.R.; Liu, Y.; O’Connell, R.J.; Peel, S.A. Efficient Quantification of HIV-1 in Heparin Plasma Spiked with Cultured HIV-1 by the Roche Cobas TaqMan and Abbott RealTime HIV-1 Tests. J. Clin. Microbiol 2012, 50, 2804–2806. [Google Scholar] [CrossRef] [PubMed]
- Schalasta, G.; Börner, A.; Speicher, A.; Enders, N. Comparative evaluation of the Aptima HIV-1 Quant Dx assay and COBAS TaqMan HIV-1 v2.0 assay using the Roche High Pure System for the quantification of HIV-1 RNA in plasma. Clin. Chem. Lab. Med. (CCLM) 2016, 54, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, C.; Jiang, Z.; Tang, T.; Jiang, Y.; Pan, P.L. Comparison of commercial HIV-1 viral load tests by using proficiency test results in China, 2013–2015. Zhonghua Liu Xing Bing Xue Za Zhi 2017, 38, 1231–1235. [Google Scholar] [CrossRef]
- Noorbazargan, H.; Nadji, S.A.; Samiee, S.M.; Paryan, M.; Mohammad-Yeganeh, S. New design, development, and optimization of an in-house quantitative TaqMan Real-time PCR assay for HIV-1 viral load measurement. HIV Clin. Trials 2018, 19, 61–68. [Google Scholar] [CrossRef]
- Tosiano, M.A.; Jacobs, J.L.; Shutt, K.A.; Cyktor, J.C.; Mellors, J.W. A Simpler and More Sensitive Single-Copy HIV-1 RNA Assay for Quantification of Persistent HIV-1 Viremia in Individuals on Suppressive Antiretroviral Therapy. J. Clin. Microbiol. 2019, 57, e01714-18. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.A.; Rudolph, D.L.; Owen, S.M. Rapid detection of HIV-1 by reverse-transcription, loop-mediated isothermal amplification (RT-LAMP). J. Virol. Methods 2008, 151, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.L.; Sullivan, V.; Owen, S.M.; Curtis, K.A. Detection of Acute HIV-1 Infection by RT-LAMP. PLoS ONE 2015, 10, e126609. [Google Scholar] [CrossRef]
- Liu, T.; Choi, G.; Tang, Z.; Kshirsagar, A.; Politza, A.J. Fingerpick Blood-Based Nucleic Acid Testing on A USB Interfaced Device towards HIV self-testing. Biosens. Bioelectron. 2022, 209, 114255. [Google Scholar] [CrossRef]
- Boyle, D.S.; Lehman, D.A.; Lillis, L.; Peterson, D.; Singhal, M.; Armes, N.; Parker, M.; Piepenburg, O.; Overbaugh, J. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. Mbio 2013, 4, e00135-13. [Google Scholar] [CrossRef]
- Crannell, Z.A.; Rohrman, B.; Richards-Kortum, R. Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS ONE 2014, 9, e112146. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ao, C.; Wan, Z.; Dzakah, E.; Liang, Y.; Lin, H.; Wang, H.; Tang, S. A point-of-care rapid HIV-1 test using an isothermal recombinase-aided amplification and CRISPR Cas12a-mediated detection. Virus Res. 2021, 303, 198505. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Zhang, R.; He, X.; Tian, F.; Nie, M.; Shen, X.; Ma, X. RAP: A Novel Approach to the Rapid and Highly Sensitive Detection of Respiratory Viruses. Front. Bioeng. Biotech. 2021, 9, 766411. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.A.; Russell, N.B.; Whelan, M.A. A method for the absolute quantification of cDNA using real-time PCR. J. Immunol. Methods 2003, 278, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, Y.C.; Pan, T.M. Quantification bias caused by plasmid DNA conformation in quantitative real-time PCR assay. PLoS ONE 2011, 6, e29101. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Li, Z.; Wu, J.; Hu, J.; Sheng, Y.; Wu, D.; Lin, Y.; Li, M.; Wang, X.; Wang, S. A wearable microfluidic device for rapid detection of HIV-1 DNA using recombinase polymerase amplification. Talanta 2019, 205, 120155. [Google Scholar] [CrossRef]
- Wang, J.; Cai, K.; He, X.; Shen, X.; Wang, J.; Liu, J.; Xu, J.; Qiu, F.; Lei, W.; Cui, L.; et al. Multiple-centre clinical evaluation of an ultrafast single-tube assay for SARS-CoV-2 RNA. Clin. Microbiol. Infect. 2020, 26, 1076–1081. [Google Scholar] [CrossRef]
- Xue, G.; Li, S.; Zhang, W.; Du, B.; Cui, J.; Yan, C.; Huang, L.; Chen, L.; Zhao, L.; Sun, Y.; et al. Reverse-Transcription Recombinase-Aided Amplification Assay for Rapid Detection of the 2019 Novel Coronavirus (SARS-CoV-2). Anal. Chem. 2020, 92, 9699–9705. [Google Scholar] [CrossRef]
- Bai, X.; Ma, X.; Li, M.; Li, X.; Fan, G.; Zhang, R.; Wang, R.; Duan, Q.; Shen, X.; Xie, Y.; et al. Field applicable detection of hepatitis B virus using internal controlled duplex recombinase-aided amplification assay and lateral flow dipstick assay. J. Med. Virol. 2020, 92, 3344–3353. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X.; Pan, M.; Qin, Y.; Zhao, H.; Yang, Q.; Yang, Z. Application of a low-cost, specific, and sensitive loop-mediated isothermal amplification (LAMP) assay to detect Plasmodium falciparum imported from Africa. Mol. Biochem. Parasit. 2022, 252, 111529. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Jiang, Y.; Fang, L.; Pan, P.; Gong, S.; Yao, J.; Tang, Y.-W.; Vermund, S.H.; Jia, Y. Early Infant Human Immunodeficiency Virus Type 1 Detection Suitable for Resource-Limited Settings with Multiple Circulating Subtypes by Use of Nested Three-Monoplex DNA PCR and Dried Blood Spots. J. Clin. Microbiol. 2008, 46, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Moragas, M.; Golemba, M.D.; Mangano, A. A new highly sensitive single-tube nested real-time PCR assay: Clinical utility in perinatal HIV-1 diagnosis. J. Virol. Methods 2021, 297, 114273. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Ragupathy, V.; Saga, A.; Hewlett, I. High sensitivity detection of HIV-1 using two genomic targets compared with single target PCR. J. Med. Virol. 2016, 88, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Seok, Y.; Yin, Q.; Bai, H.; Bau, H.H. Sensitive, Single-Pot, Two-Stage Assay for Hepatitis Viruses. Anal. Chem. 2022, 94, 1778–1786. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Primer/Probe | Sequence (5′—3′) | Source | Position a |
|---|---|---|---|---|
| Gag | HIV-RAA-F | GAAGTAATACCCATGTTTTCAGCATTATCA | This paper | 1288–1317 |
| HIV-RAA-R | TGCAGCTTCCTCATTGATGGTTTCTTTTAAC | This paper | 1419–1389 | |
| HIV-qPCR-F | CATGTTTTCAGCATTATCAGAAGG | [9] | 1299–1322 | |
| HIV-qPCR-R | GCTTCCTCATTGATGGTCTCTTT | [9] | 1415–1393 | |
| HIV-qPCR-P | FAM-CACCCCACAAGATTTAAACACCATGCTAA-BHQ1 | [9] | 1326–1354 | |
| Pol | HIV-RAA-F | ATTTTCGGGTTTATTACAGGGACAGCAGAGA | This paper | 4894–4924 |
| HIV-RAA-R | CACAATCATCACCTGCCATCTGTTTTCCAT | This paper | 5070–5041 | |
| HIV-qPCR-F | GGTTTATTACAGGGACAGCAGAGA | [10] | 4901–4924 | |
| HIV-qPCR-R | ACCTGCCATCTGTTTTCCATA | [10] | 5060–5040 | |
| HIV-qPCR-P | FAM-ACTACTGCCCCTTCACCTTTCCAGAG-BHQ1 | [10] | 4978–4953 |
| Copies/Reaction | No. of Positive Samples Tested by the RT-RAP Assays for HIV-1 | ||||
|---|---|---|---|---|---|
| CRF_01AE | CRF_07BC | Subtype B | Subtype C/BC | CRF_08BC | |
| 1 | 1/8 | 1/8 | 1/8 | 2/8 | 1/8 |
| 101 | 8/8 | 8/8 | 8/8 | 8/8 | 8/8 |
| 102 | 8/8 | 8/8 | 8/8 | 8/8 | 8/8 |
| 103 | 8/8 | 8/8 | 8/8 | 8/8 | 8/8 |
| 104 | 8/8 | 8/8 | 8/8 | 8/8 | 8/8 |
| 105 | 8/8 | 8/8 | 8/8 | 8/8 | 8/8 |
| Detection limit (Probit analysis) | 5.037 | 5.037 | 5.037 | 4.149 | 5.037 |
| Copies/Reaction | No. of Positive Samples | Ct Values of the Commercial qRT-PCR Kits |
|---|---|---|
| 10 | 7/8 | 36.530 |
| 20 | 8/8 | 35.615 |
| 30 | 8/8 | 33.650 |
| 40 | 8/8 | 34.744 |
| 50 | 8/8 | 34.022 |
| 100 | 8/8 | 33.794 |
| Detection limit (Probit analysis) | 13.678 |
| RT-RAP | Commercial qRT-PCR Kits | Total | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Kappa | |
|---|---|---|---|---|---|---|---|---|
| + | − | |||||||
| + | 99 | 1 | 100 | |||||
| − | 0 | 70 | 70 | 100.00 | 98.59 | 99.00 | 100 | 0.988 |
| Total | 99 | 71 | 170 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, F.; Jin, C.; Ji, S.; Tie, Y.; Fan, G.; Zhang, R.; Zheng, Y.; Shen, X.; Ma, X.; Feng, Z. A Rapid and Sensitive Detection of HIV-1 with a One-Pot Two-Stage Reverse Transcription Recombinase Aided Real-Time PCR Assay. Trop. Med. Infect. Dis. 2023, 8, 105. https://doi.org/10.3390/tropicalmed8020105
Tian F, Jin C, Ji S, Tie Y, Fan G, Zhang R, Zheng Y, Shen X, Ma X, Feng Z. A Rapid and Sensitive Detection of HIV-1 with a One-Pot Two-Stage Reverse Transcription Recombinase Aided Real-Time PCR Assay. Tropical Medicine and Infectious Disease. 2023; 8(2):105. https://doi.org/10.3390/tropicalmed8020105
Chicago/Turabian StyleTian, Fengyu, Cong Jin, Shangzhi Ji, Yanqing Tie, Guohao Fan, Ruiqing Zhang, Yehuan Zheng, Xinxin Shen, Xuejun Ma, and Zhishan Feng. 2023. "A Rapid and Sensitive Detection of HIV-1 with a One-Pot Two-Stage Reverse Transcription Recombinase Aided Real-Time PCR Assay" Tropical Medicine and Infectious Disease 8, no. 2: 105. https://doi.org/10.3390/tropicalmed8020105