PKC Proteins and Muscular Dystrophy

 , , , ,
, , , , {kind=link}
Abstract
:1. Introduction
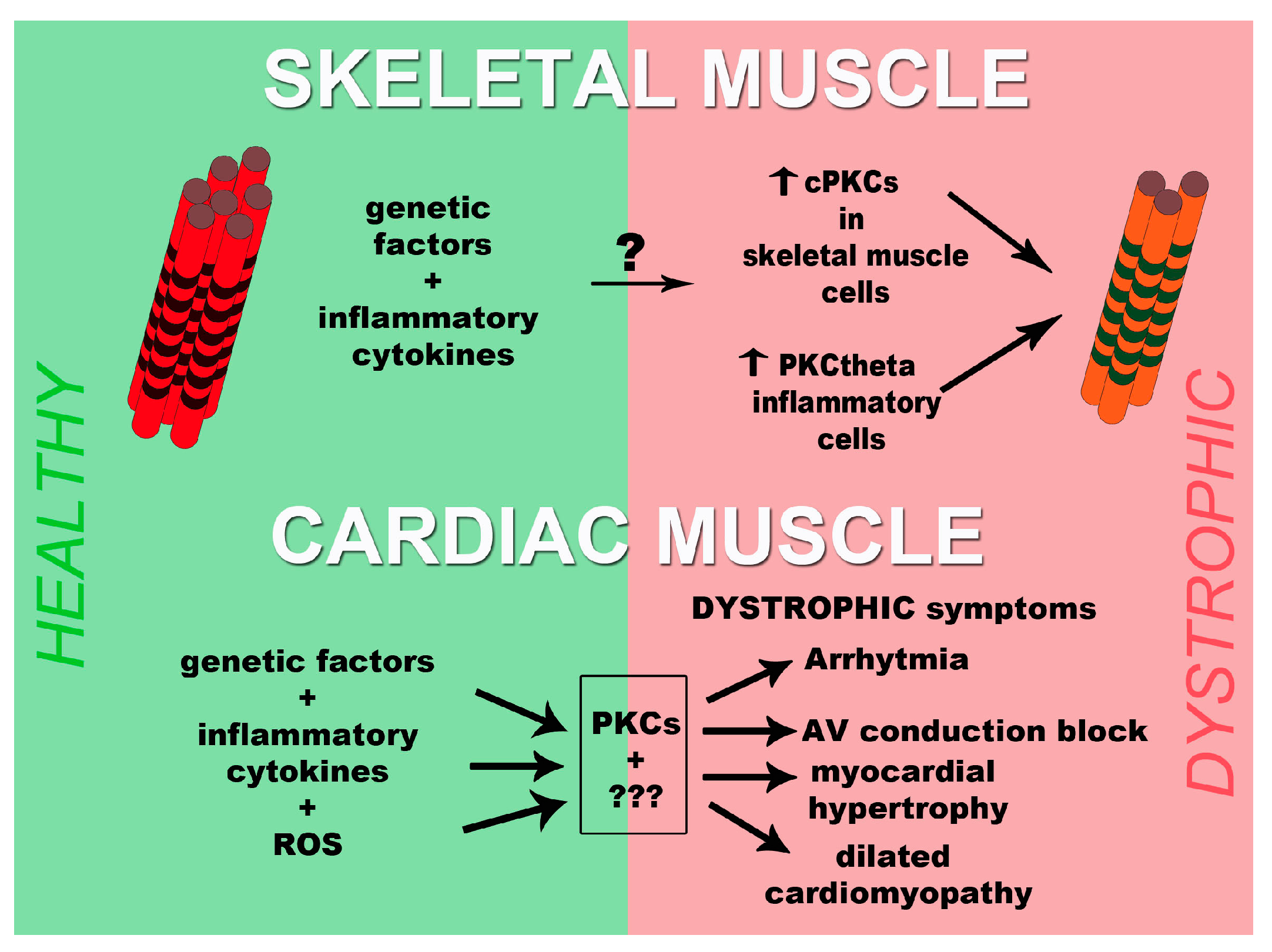
Muscular Dystrophies and Signal Transduction
2. Protein Kinase C (PKC) Protein Family
- (i)
- conventional PKCs (α, β1, β2, γ) that are activated by diacylglycerol (DAG) and calcium (Ca2+);
- (ii)
- novel PKCs (δ, ε, θ, η) that are activated only by diacylglycerol (DAG);
- (iii)
- atypical PKCs (ξ, ι/λ) that are independent of second messengers.
3. PKCs and Muscular Dystrophies
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and Becker muscular dystrophies: A review of animal models, clinical end points, and biomarker quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L. Origins and early descriptions of “Duchenne muscular dystrophy”. Muscle Nerve 2003, 28, 402–422. [Google Scholar] [CrossRef] [PubMed]
- Romitti, P.A.; Zhu, Y.; Puzhankara, S.; James, K.A.; Nabukera, S.K.; Zamba, G.K.; Ciafaloni, E.; Cunniff, C.; Druschel, C.M.; Mathews, K.D.; et al. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics 2015, 135, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Razak, H.; Malerba, A.; Dickson, G. Advances in gene therapy for muscular dystrophies. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Saada, Y.B.; Dib, C.; Lipinski, M.; Vassetzky, Y.S. Genome- and cell-based strategies in therapy of muscular dystrophies. Biochemistry 2016, 81, 678–690. [Google Scholar] [PubMed]
- Mah, J.K. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 2016, 12, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Harper, P.S.; van Engelen, B.G.; Eymard, B.; Rogers, M.; Wilcox, D. 99th ENMC international workshop: Myotonic dystrophy: Present management, future therapy. Neuromuscul. Disord. 2002, 12, 596–599. [Google Scholar] [CrossRef]
- Yamashita, Y.; Matsuura, T.; Kurosaki, T.; Amakusa, Y.; Kinoshita, M.; Ibi, T.; Sahashi, K.; Ohno, K. LDB3 splicing abnormalities are specific to skeletal muscles of patients with myotonic dystrophy type 1 and alter its PKC binding affinity. Neurobiol. Dis. 2014, 69, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Osborne, R.J.; Thornton, C.A. RNA-dominant diseases. Hum. Mol. Genet. 2006, 15, R162–R169. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A.; Wang, E.; Carrell, E.M. Myotonic dystrophy: Approach to therapy. Curr. Opin. Genet. Dev. 2017, 44, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Selma-Soriano, E.; Rapisarda, A.S.; Fernandez-Costa, J.M.; Perez-Alonso, M.; Artero, R. Myotonic dystrophy: Candidate small molecule therapeutics. Drug Discov. Today 2017, 22, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Kumar, A. Therapeutic targeting of signaling pathways in muscular dystrophy. J. Mol. Med. 2010, 88, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Rahimov, F.; Kunkel, L.M. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Models Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kay, D.I.; Rudra, R.T.; Chen, B.M.; Hsu, N.; Izumiya, Y.; Martinez, L.; Spencer, M.J.; Walsh, K.; Grinnell, A.D.; et al. Myogenic Akt signaling attenuates muscular degeneration, promotes myofiber regeneration and improves muscle function indystrophin-deficient mdx mice. Hum. Mol. Genet. 2011, 20, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.S.; Casar, J.C.; Motohashi, N.; Vieira, N.M.; Eisenberg, I.; Marshall, J.L.; Gasperini, M.J.; Lek, A.; Myers, J.A.; Estrella, E.A.; et al. MicroRNA-486-dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy-associatedsymptoms. J. Clin. Investig. 2014, 124, 2651–2667. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.A.; Yutzey, K.E. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev. Biol. 2004, 266, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ravel-Chapuis, A.; Bélanger, G.; Côté, J.; Michel, R.N.; Jasmin, B.J. Misregulation ofcalcium-handling proteins promotes hyperactivation of calcineurin-NFAT signaling in skeletal muscle of DM1 mice. Hum. Mol. Genet. 2017, 26, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Wehling-Henricks, M. Nitric oxide synthase deficiency and the pathophysiology of muscular dystrophy. J. Physiol. 2014, 592, 4627–4638. [Google Scholar] [CrossRef] [PubMed]
- Hofhuis, J.; Bersch, K.; Büssenschütt, R.; Drzymalski, M.; Liebetanz, D.; Nikolaev, V.O.; Wagner, S.; Maier, L.S.; Gärtner, J.; Klinge, L.; et al. Dysferlin mediates membrane tubulation and links T-tubule biogenesis to muscular dystrophy. J. Cell Sci. 2017, 130, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, S.M.; Walsh, G.S.; Balazsi, K.; Seale, P.; Sandoz, J.; Hierlihy, A.M.; Rudnicki, M.A.; Chamberlain, J.S.; Miller, F.D.; Megeney, L.A. Activation of JNK1 contributes to dystrophic muscle pathogenesis. Curr. Biol. 2001, 11, 1278–1282. [Google Scholar] [CrossRef]
- Nakamura, A.; Harrod, G.V.; Davies, K.E. Activation of calcineurin and stress activated protein kinase/p38-mitogen activated protein kinase in hearts of utrophin-dystrophin knockout mice. Neuromuscul. Disord. 2001, 11, 251–259. [Google Scholar] [CrossRef]
- Igumenova, T.I. Dynamics and membrane interactions of protein kinase C. Biochemistry 2015, 54, 4953–4968. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.W.; Pula, G.; Hers, I.; Crosby, D.; Jones, M.L. PKC-interacting proteins: From function to pharmacology. Trends Pharmacol. Sci. 2004, 25, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, G.; Mirandola, P.; Carubbi, C.; Galli, D.; Vitale, M. Protein kinase C ε in hematopoiesis: Conductor or selector? Semin. Thromb. Hemost. 2013, 39, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.S.; Sutton, C.R.; Rao, S. Protein kinase C in the immune system: From signalling to chromatin regulation. Immunology 2015, 146, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Leppänen, T.; Tuominen, R.K.; Moilanen, E. Protein kinase C and its inhibitors in the regulation of inflammation: Inducible nitric oxide synthase as an example. Basic Clin. Pharmacol. Toxicol. 2014, 114, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, A.; Balça-Silva, J.; Matias, D.; Lopes, M.C. PKC signaling in glioblastoma. Cancer Biol. Ther. 2013, 14, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Kuyumcu-Martinez, M.N.; Sarma, S.; Mathur, N.; Wehrens, X.H.; Cooper, T.A. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Investig. 2009, 119, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Abulizi, A.; Perry, R.J.; Camporez, J.P.G.; Jurczak, M.J.; Petersen, K.F.; Aspichueta, P.; Shulman, G.I. A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J. 2017, 31, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Akinci, G.; Topaloglu, H.; Demir, T.; Danyeli, A.E.; Talim, B.; Keskin, F.E.; Kadioglu, P.; Talip, E.; Altay, C.; Yaylali, G.F.; et al. Clinical spectra of neuromuscular manifestations in patients with lipodystrophy: A multicenter study. Neuromuscul. Disord. 2017, 27, 923–930. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, P.A.; McArdle, A.; Edwards, R.H. Acute effects of phorbol esters on the protein-synthetic rate and carbohydrate metabolism of normal and mdx mouse muscles. Biochem. J. 1991, 275, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.V.; Shanmugasundaram, J.; Sundaram, C.; Anandaraj, M.P. Activity of novel protein kinase C and distribution of protein kinase C theta in subcellular fractions of normal and Duchenne muscular dystrophic muscle. Indian J. Biochem. Biophys. 2002, 39, 377–381. [Google Scholar] [PubMed]
- Madaro, L.; Pelle, A.; Nicoletti, C.; Crupi, A.; Marrocco, V.; Bossi, G.; Soddu, S.; Bouché, M. PKC theta ablation improves healing in a mouse model of muscular dystrophy. PLoS ONE 2012, 7, e31515. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, V.; Fiore, P.; Madaro, L.; Crupi, A.; Lozanoska-Ochser, B.; Bouché, M. Targeting PKCθ in skeletal muscle and muscle diseases: Good or bad? Biochem. Soc. Trans. 2014, 42, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Juretić, N.; Jorquera, G.; Caviedes, P.; Jaimovich, E.; Riveros, N. Electrical stimulation induces calcium-dependent up-regulation of neuregulin-1β in dystrophic skeletal muscle cell lines. Cell. Physiol. Biochem. 2012, 29, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Di Marcantonio, D.; Galli, D.; Carubbi, C.; Gobbi, G.; Queirolo, V.; Martini, S.; Merighi, S.; Vaccarezza, M.; Maffulli, N.; Sykes, S.M.; et al. PKCε as a novel promoter of skeletal muscle differentiation and regeneration. Exp. Cell Res. 2015, 339, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, N.J.; Ismail, H.; Zimetbaum, P.; Raynor, E.M. Cardiac involvement in the muscular dystrophies. Muscle Nerve 2017. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Ishikawa, K. Cardiac involvement in progressive muscular dystrophy of the Duchenne type. Jpn. Heart J. 1997, 38, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Limongelli, F.M.; Nigro, V.; de Rimini, M.L.; Giugliano, M.A.; Petretta, V.R.; Passamano, L.; Restucci, B.; et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve 1995, 18, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Connuck, D.M.; Sleeper, L.A.; Colan, S.D.; Cox, G.F.; Towbin, J.A.; Lowe, A.M.; Wilkinson, J.D.; Orav, E.J.; Cuniberti, L.; Salbert, B.A.; et al. Pediatric cardiomyopathy registry study group. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: A comparative study from the pediatric cardiomyopathy registry. Am. Heart J. 2008, 155, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Bassez, G.; Lazarus, A.; Desguerre, I.; Varin, J.; Laforêt, P.; Bécane, H.M.; Meune, C.; Arne-Bes, M.C.; Ounnoughene, Z.; Radvanyi, H.; et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology 2004, 63, 1939–1941. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Meune, C.; Bécane, H.M.; Laforêt, P.; Bassez, G.; Lazarus, A.; Radvanyi-Hoffman, H.; Eymard, B.; Duboc, D. Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: A case control study. Neuromuscul. Disord. 2009, 19, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: A long-term longitudinal study. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Melacini, P.; Boito, C.; Pegoraro, E.; Angelini, C. LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscul. Disord. 2003, 13, 303–309. [Google Scholar] [CrossRef]
- Melacini, P.; Fanin, M.; Duggan, D.J.; Freda, M.P.; Berardinelli, A.; Danieli, G.A.; Barchitta, A.; Hoffman, E.P.; DallaVolta, S.; Angelini, C. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve 1999, 22, 473–479. [Google Scholar] [CrossRef]
- Singh, R.M.; Cummings, E.; Pantos, C.; Singh, J. Protein kinase C and cardiac dysfunction: A review. Heart Fail Rev. 2017, 22, 843–859. [Google Scholar] [CrossRef] [PubMed]
- Simonis, G.; Briem, S.K.; Schoen, S.P.; Bock, M.; Marquetant, R.; Strasser, R.H. Protein kinase C in the human heart: Differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol. Cell. Biochem. 2007, 305, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cohen, M.V.; Downey, J.M. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. 2010, 24, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Budas, G.R.; Churchill, E.N.; Mochly-Rosen, D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol. Res. 2007, 55, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Mochly-Rosen, D.; Boutjdir, M. Regulation of cardiac excitability by protein kinase C isozymes. Front. Biosci. (Scholar Ed.) 2012, 4, 532–546. [Google Scholar] [CrossRef]
- Palaniyandi, S.S.; Sun, L.; Ferreira, J.C.; Mochly-Rosen, D. Protein kinase C in heart failure: A therapeutic target? Cardiovasc. Res. 2009, 82, 229–239. [Google Scholar] [CrossRef]
- Inagaki, K.; Hahn, H.S.; Dorn, G.W., II; Mochly-Rosen, D. Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation 2003, 108, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Alstrom, J.S.; Stroemlund, L.W.; Nielsen, M.S.; MacAulay, N. Protein kinase C-dependent regulation of connexin43 gap junctions and hemichannels. Biochem. Soc. Trans. 2015, 43, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Sag, C.M.; Wagner, S.; Maier, L.S. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic. Biol. Med. 2013, 63, 338–349. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gobbi, G.; Galli, D.; Carubbi, C.; Neri, L.M.; Masselli, E.; Pozzi, G.; Vitale, M.; Mirandola, P. PKC Proteins and Muscular Dystrophy. J. Funct. Morphol. Kinesiol. 2018, 3, 12. https://doi.org/10.3390/jfmk3010012
Gobbi G, Galli D, Carubbi C, Neri LM, Masselli E, Pozzi G, Vitale M, Mirandola P. PKC Proteins and Muscular Dystrophy. Journal of Functional Morphology and Kinesiology. 2018; 3(1):12. https://doi.org/10.3390/jfmk3010012
Chicago/Turabian StyleGobbi, Giuliana, Daniela Galli, Cecilia Carubbi, Luca Maria Neri, Elena Masselli, Giulia Pozzi, Marco Vitale, and Prisco Mirandola. 2018. "PKC Proteins and Muscular Dystrophy" Journal of Functional Morphology and Kinesiology 3, no. 1: 12. https://doi.org/10.3390/jfmk3010012